

Abstract
CCM3 mutations give rise to cerebral cavernous malformations (CCMs) of the vasculature through a mechanism that remains unclear. Interaction of CCM3 with the germinal center kinase III (GCKIII) subfamily of Sterile 20 protein kinases, MST4, STK24, and STK25, has been implicated in cardiovascular development in the zebrafish, raising the possibility that dysregulated GCKIII function may contribute to the etiology of CCM disease. Here, we show that the amino-terminal region of CCM3 is necessary and sufficient to bind directly to the C-terminal tail region of GCKIII proteins. This same region of CCM3 was shown previously to mediate homodimerization through the formation of an interdigitated α-helical domain. Sequence conservation and binding studies suggest that CCM3 may preferentially heterodimerize with GCKIII proteins through a manner structurally analogous to that employed for CCM3 homodimerization.
Keywords: Golgi, Protein Conformation, Protein Domains, Protein Structure, Protein-Protein Interactions, Cerebral Cavernous Malformations, Pdcd10, Ste20 kinase
Introduction
Cerebral cavernous malformations (CCMs)5 are vascular abnormalities in the brain characterized by focal dilations of cranial vasculature that can progress to hemorrhages and stroke (OMIM ID 116860). Mutations have been identified in three distinct genes, denoted CCM1–3, that are causative for the formation of most familial CCM lesions (1, 2). CCM3, also named PDCD10, is a 212 amino acid protein conserved among metazoans (3, 4). Knockdown of CCM3 in zebrafish causes an enlarged heart phenotype (5), whereas targeted deletion of CCM3 in the mouse results in defects of early angiogenesis and early embryonic lethality, a phenotype also observed following tissue-specific deletion in the vascular endothelium (6). A non-cell autonomous role for CCM3 in neuroglia on the vasculature has also been uncovered in mouse recently, indicating that CCMs may arise in the central nervous system by the loss of CCM3 signaling in neural as well as endothelial populations (7).
CCM3 has been detected in complex with CCM1 and CCM2 proteins, suggesting that the three proteins may share a common biochemical function (8–10). Yeast two-hybrid, co-immunoprecipitation, and GST pulldown experiments from cell lysates demonstrated that CCM3 also readily interacts with MST4, STK24, and STK25, a grouping of protein kinases termed the germinal center kinase class III (GCKIII) family (10–13). CCM3 and the GCKIII proteins have also been detected as part of a large multiprotein complex termed STRIPAK (striatin-interacting phosphatase and kinase; see Refs. 14, 15). The knockdown of GCKIII proteins in zebrafish gives rise to the same cardiovascular defects as CCM3 knockdown, suggesting the CCM3-GCKIII protein interaction is important for proper CCM3 function (5).6
The GCKIII proteins (STK24, STK25, and MST4) are members of the larger Sterile 20 kinase family and are characterized by highly conserved catalytic domains and a 100–120 residue carboxyl-terminal tail, whose function is not currently known. The closely related GCKII proteins MST1 and MST2 possess completely distinct C-terminal tails that mediate homotypic and heterotypic interactions (16), raising the possibility that an analogous function might be served by the tail region of GCKIII proteins, albeit through an unrelated structural mechanism.
Crystal structures of the CCM3 protein revealed an architecture consisting of two distinct structural domains (17, 18). The N-terminal helical domain of CCM3 mediates homodimerization. The C-terminal four-helix bundle, termed the focal adhesion targeting (FAT) homology domain (17), functions as a linear peptide binding module that mediates direct interactions with CCM2, paxillin, and the striatin component of STRIPAK (17).7 Of note, the N-terminal region of CCM3 has also been implicated in the interaction with GCKIII proteins in cells and model organisms (5, 13, 19). Given the critical role for CCM3 and GCKIII proteins in maintaining vascular integrity, we have probed the basis for their interaction in close detail. The results presented here demonstrate that the amino terminus of CCM3 interacts directly with the C-terminal regions of GCKIII proteins. Based on sequence similarity between the interacting regions of CCM3 and GCKIII proteins, we propose that heterodimerization of the two proteins is achieved through an analogous structural mechanism to that reported for the homodimerization for CCM3 and present data indicating that heterodimerization may be favored over homodimerization.
EXPERIMENTAL PROCEDURES
Cloning of Expression Constructs
PDCD10/CCM3, STK25/SOK1, MST4, STK4/MST1, and STRN3 were amplified by PCR and cloned into the modified bacterial expression vectors pGEX-TEV and/or ProEX-TEV. CCM3 point mutations were generated by PCR using standard techniques, and all clones were sequence verified. The N-terminal mutant of CCM3 (LAIL-4D) comprises L44D,A47D,I66D,L67D and the C-terminal mutant (K4A) comprises K132A,K139A, K172A,K179A. CCM3 point mutants were subcloned into the pcDNA5-FRT-GFP vector (20) for expression in mammalian cells. pcDNA3-FLAG-MST4 was reported previously (14).
Protein Expression and Purification
BL21-Codon+ cells (Agilent Technologies) were transformed and grown to A600 of 0.8 and induced by addition of 0.25 mm isopropyl 1-thio-β-d-galactopyranoside for 12–18 h at 18 °C. Bacterial cell pellets were harvested and stored at −20 °C. Full-length STK25 and MST4 were also cloned into pFastBAC GST-TEV (Invitrogen), and recombinant baculoviruses were generated for expression in SF9 cells. Infection of SF9 monolayer cells with a multiplicity of infection of >5 were performed for 72 h, followed by harvesting of cell pellets and storage at −80 °C.
Bacterial or SF9 cell pellets of His-tagged proteins were thawed and resuspended in nickel-loading buffer containing 20 mm HEPES, pH 7.5, 400 mm NaCl, 5 mm imidazole, and 5 mm β-mercaptoethanol. Cells were lysed in the presence of 1 mm phenylmethylsulfonyl fluoride by passage through a cell homogenizer (Avestin, Inc.). Supernatant following centrifugation at 20,000 × g was applied to a nickel-chelating column (GE Healthcare) and eluted over a gradient to 300 mm imidazole. Fractions containing the protein of interest were incubated overnight with an aliquot of tobacco etch virus (TEV) protease and 1 mm DTT. Protein was dialyzed into nickel-loading buffer and flowed over a 1-ml nickel chelating column to remove uncleaved protein, concentrated, and injected onto a 120-ml S-75 size exclusion column (GE Healthcare) in 20 mm HEPES, pH 7.5, 100 mm NaCl, and β-mercaptoethanol. Fractions containing purified protein were concentrated, flash frozen, and stored at −80 °C. Bacterial pellets of GST-tagged proteins were lysed in 20 mm HEPES, pH 7.5, 400 mm NaCl, and 5 mm β-mercaptoethanol, and the supernatant was applied to glutathione-Sepharose resin (GE Healthcare). The protein of interest was separated from the GST affinity tag following overnight incubation with TEV protease and further purified by size exclusion chromatography as described above. The heterodimeric CCM3 and MST4 complex was obtained by mixing bacterial lysates expressing GST-MST4 and His-CCM3 proteins. Protein eluted from a nickel-chelating column was applied directly to glutathione Sepharose resin and subsequently purified as described above for GST-tagged proteins.
In Vitro Binding Studies
Bacterial or SF9 cell lysates containing GST fusion proteins were prepared as above, however the supernatant was applied to glutathione-Sepharose resin (GE Healthcare) in GST-loading buffer containing 20 mm HEPES, pH 7.5, 400 mm NaCl, and 5 mm β-mercaptoethanol and washed extensively. The protein-bound affinity resin was equilibrated with interaction buffer (20 mm HEPES, pH 7.5, 150 mm NaCl, and 5 mm β-mercaptoethanol). Previously purified binding partner proteins were added to the protein-bound affinity resin and incubated on ice with gentle mixing for 20 min. The affinity resin was then washed three times with 500 μl of interaction buffer and aliquots of the binding reactions were separated by SDS-PAGE with proteins visualized following Coomassie staining.
Light Scattering
Size exclusion chromatography-multiangle light scattering (SEC-MALS) was performed with 200 μm protein samples injected onto a 24-ml S200 Superdex column and measured using a three-angle (45, 90, and 135°) miniDawn light-scattering instrument equipped with a 690-nm laser and an Optilab rEX differential refractometer (Wyatt Technologies, Inc.) as described in Ref. 21. Molecular weights were calculated by using ASTRA software (Wyatt Technologies, Inc.) based on Zimm plot analysis and by using a protein refractive index increment, dn dc−1 = 0.185 liters g−1.
Analytical Ultracentrifugation
Sedimentation equilibrium experiments were carried out on samples containing CCM3 alone, MST4 alone, and a CCM3-MST4 complex obtained by dual affinity tag purification. Samples were loaded at concentrations yielding initial A280 values of 0.2, 0.4, and 0.8. Ultracentrifugation was performed at 25 °C in 20 mm sodium phosphate buffer, pH 7.0, with 100 mm NaCl and 5 mm β-mercaptoethanol using an Optima XL-I analytical ultracentrifuge (Beckman Instruments, Palo Alto, CA) with a AN50-Ti rotor, quartz windows, and standard six-sector charcoal-filled Epon centerpieces. Absorbance profiles at 280 nm were collected at spin speeds of 3,000, 10,000, 15,000, 20,000, and 25,000 rpm after 24 h of equilibration at each spin speed. Data were analyzed by nonlinear least-squares fitting using Origin software (version 7.0, OriginLab Corp., Northampton, MA). Global fits were obtained to Equation 1, representing a single-species model. Data from CCM3 and MST4 samples were also fit to Equation 2, representing a monomer-homodimer equilibrium model.
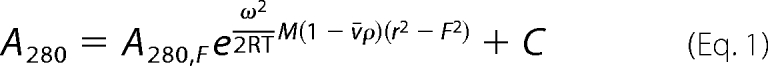 |
 |
In the equations shown above, ω is the spin speed, ϵmon is the 280-nm molar extinction coefficient of a monomer, Cmon,F is the concentration of monomers at the reference radius (F), r is the radius from the spin axis, v̄ is the partial specific volume of the protein, ρ is the density of the solvent, R is the gas constant, T is the temperature, l is the optical path length, C is a baseline correction constant, and Mmon is the sequence molecular mass of a protein monomer. Values of v̄ and ρ were predicted using SEDNTERP software (John Philo, Thousand Oaks, CA). Extinction coefficients of 10,430 m−1 cm−1 and 39,670 m−1 cm−1 were used for CCM3 and MST4, respectively.
Mammalian Cell Culture and Immunoprecipitation
Transient transfection of HEK293T cells and immunoprecipitation followed by immunoblotting was performed essentially as described (14).
RESULTS
CCM3 N Terminus Interacts Directly with C Terminus of GCKIII Proteins
To examine whether binding between CCM3 and GCKIII proteins is direct and to localize the determinants for binding, we tested bacterial- or baculovirus-expressed and purified proteins for interaction in vitro (see Fig. 1A for schematic of constructs). In glutathione-Sepharose pulldown experiments visualized by Coomassie staining (Fig. 1B), GST fusions of full-length STK25 and MST4 interacted robustly with full-length CCM3, whereas the minimal kinase domain of MST4 lacking the C-terminal tail region did not. These results demonstrated that binding is direct and that the C-terminal region of the GCKIII proteins is a key determinant for CCM3 recognition.
FIGURE 1.

Purified CCM3 and GCKIII proteins interact directly. A, domain organization of CCM3 and GCKIII proteins. ND, N-terminal dimerization region of CCM3. B, in vitro interaction between GST fusions of STK25 and MST4 with CCM3. The minimal MST4 kinase domain does not bind CCM3. C, CCM3 interacts with the C-terminal tail regions of MST4 and STK25 but not MST1. The CCM3 C-terminal FAT domain does not interact with the kinases. D, the amino terminus of CCM3 (CCM3-ND) is sufficient for interaction with full length MST4. E, CCM3-ND is sufficient for interaction with the C-terminal tail region of STK25. F, CCM3 binds tightly to the GST-STK25 tail but not to GST-CCM3.
The C-terminal tails of MST4 and STK25, when expressed as GST fusion proteins, were sufficient for robust binding to full length CCM3 (Fig. 1C). Interestingly, neither GST kinase tail fusion was able to bind to the isolated FAT domain of CCM3, suggesting that the N-terminal dimerization domain of CCM3 provides the determinant for GCKIII protein binding. We also demonstrated the interaction is specific to GCKIII protein tails because no interaction was detected between full-length CCM3 and the tail region of MST1, a GCKII protein possessing a divergent C-terminal tail (Fig. 1C).
We next tested whether the N terminus of CCM3 is sufficient for binding to GCKIII proteins. As shown in Fig. 1D, the N terminus of CCM3 but not the FAT domain displayed robust binding to full-length MST4. We reconstituted a robust interaction between the N-terminal tail of CCM3 fused to GST and the free C-terminal tail of STK25, demonstrating that the minimal delineated regions in CCM3 and the GCKIII proteins are fully sufficient for the interaction (Fig. 1E). Together, these results define a model in which the binding of GCKIII proteins with CCM3 is mediated entirely by their C- and N-terminal tail regions, respectively. Interestingly, although full-length CCM3 interacted robustly with the GST-STK25 kinase tail, binding of CCM3 full-length to GST-CCM3 full-length and to the GST-CCM3 N terminus was barely detectable (Fig. 1F). This result raised the question of how precisely the respective N- and C-terminal tail regions of CCM3 and GCKIII proteins mediate complex formation and how this interaction is affected by the ability of the CCM3 N-terminal tail region to form homodimers.
The N-terminal tail region of CCM3 facilitates homodimerization through the formation of a six-helix cluster (17). We reasoned that CCM3 might employ this dimer structure to bind the GCKIII protein tail. Alternatively, CCM3 might bind to GCKIII proteins with a 1:1 stoichiometry that displaces the CCM3 dimer. Supporting the latter model, examination of the primary sequence of the CCM3 N terminus revealed striking similarity to the C-terminal tail region of the GCKIII proteins (Fig. 2A) (19). Of 39 total residues comprising the dimer contact surface of the CCM3 homodimer structure (Fig. 2B), 10 are identical, and an additional nine are similar in nature to residues in MST4 (Fig. 2A). This conservation suggested that CCM3 might form a heterodimeric complex with the C-terminal tail region of GCKIII proteins using the same interaction mode observed in the homodimeric CCM3 crystal structure (Fig. 2B) (17).
FIGURE 2.

Sequence conservation between CCM3 and GCKIII proteins. A, sequence alignment of CCM3 N-terminal and GCKIII protein C-terminal tails regions. h, human; dr, Danio rerio; e, C. elegans; mb, Monosiga brevicollis; and dd, Dictyostelium discoidium sequences of CCM3 and GCKIII proteins (STK24, MST4, STK25, and severin) are shown. Conserved hydrophobic, acidic, basic, and proline/glycine residues are highlighted in yellow, red, blue, and green, respectively. Residues comprising the CCM3 homodimer interface are indicated by circles (●) at the top of the alignment. The CCM3 residues (Leu-44, Ala047, Ile-66, and Leu-67) mutated in this study are highlighted with red circles. B, schematic of the N-terminal dimerization region of CCM3 (Protein Data Bank code 3L8I). Protomer chains are colored yellow and green with residues mutated in this study shown in stick representation. Secondary structure elements are labeled.
CCM3 and MST4 Form Heterodimers in Solution
To differentiate between the two possible models of CCM3-GCKIII protein interaction, we performed SEC-MALS analysis on purified CCM3 and MST4 proteins and their complexes. As demonstrated previously (22), full-length CCM3 eluted as a single 46-kDa species consistent with the expected size of a homodimer (Table 1 and Fig. 3A). Interestingly, full-length MST4 kinase also eluted as dimeric species with a molecular mass of 94 kDa (Fig. 3B). The ability of MST4 to form dimers was dependent on the C-terminal tail region as the isolated kinase domain eluted as a single monomeric species of 38 kDa (Fig. 3C). Thus, the C-terminal tail regions of GCKIII proteins, which are similar in sequence to the N-terminal tail region of CCM3, can also function as homodimerization domains.
TABLE 1.
Summary of SEC-MALS data
| Figure panel | Proteins injected | Expected monomeric molecular mass | Expected dimeric molecular mass | MALS averaged molecular mass | Molecular state |
|---|---|---|---|---|---|
| kDa | kDa | kDa | |||
| 3A | CCM3 | 24.7 | 49.4 | 45.8 ± 4.4 | CCM3 dimer |
| 3B | MST4 | 46.7 | 93.4 | 94.0 ± 8.2 | MST4 dimer |
| 3C | MST4 kinase domain | 34.0 | 68.0 | 37.9 ± 1.6 | MST4 monomer |
| 3D | CCM3+MST4 (co-purified) | 24.7, 46.7 | 49.4, 93.4 (heterodimer, 71.4) | 61.7 ± 2.0 | CCM3-MST4 heterodimer |
| 4A | CCM3 LAIL-4D | 24.7 | 49.4 | 25.2 ± 2.9 | CCM3 monomer |
| 4B | CCM3 LAIL-4D+MST4 | 24.7, 46.7 | 49.4, 93.4 | 20.4 ± 1.0, 104.6 ± 4.9 | CCM3 monomer, MST4 dimer |
FIGURE 3.

Size exclusion chromatography and multiangle light scattering of purified CCM3 and MST4 proteins. The left vertical axis denotes molecular weight of eluting species. The right vertical axis denotes absorbance measurement of eluent. The area of the peak integrated for analysis of molecular mass is indicated by a black line. An SDS-PAGE analysis of the corresponding eluted proteins is shown below each chromatogram. A, CCM3 alone. B, MST4 full-length alone (the asterisk indicates a likely MST4 degradation product). C, MST4 kinase domain (residues 1–312). D, co-purified CCM3-MST4 complex.
SEC-MALS analysis of a CCM3-MST4 protein complex obtained by co-purification utilizing successive affinity tag purification steps yielded a single species with molecular mass of 62 kDa consistent with a 1:1 heterodimer (Fig. 3D). Under these conditions, no evidence of CCM3 or MST4 homodimers was observed. This result indicated that CCM3 and MST4 form a stable heterodimer whose stability may exceed that of the CCM3 and MST4 homodimers.
CCM3 N-terminal Mutation Disrupts Heterodimer Formation with MST4
If the six-helix cluster observed in the crystal structure of the CCM3 homodimer also reflects the mode of CCM3-GCKIII protein heterodimerization, then we expected that mutations that disrupt homodimerization of CCM3 might similarly abolish heterodimerization with GCKIII proteins. Mutation of the four hydrophobic residues, Leu-44, Ala-47, Ile-66, and Leu-67 to aspartic acid residues (mutant denoted LAIL-4D) within the CCM3 dimerization interface abolished homodimerization, as reflected by the transition of CCM3 to a 25-kDa monomeric species in SEC-MALS analysis (Fig. 4A). The four-site mutant, unlike wild type CCM3, was also compromised for its ability to interact with wild type MST4, as assessed by SEC-MALS (Fig. 4B), and to interact with the dimerization mediating tails of either MST4 or STK25 in pulldown experiments (Fig. 4C). These results were consistent with the notion that CCM3 and GCKIII proteins form heterodimers through a mechanism analogous to that employed in the CCM3 homodimer structure (17).
FIGURE 4.

Mutations in the amino terminus of CCM3 disrupt the CCM3-GCKIII heterodimer interaction in vitro and in vivo. A, the N-terminal CCM3 mutant (LAIL-4D) elutes as a monomer, as assessed by SEC-MALS. mAU, milli absorbance units. B, the N-terminal CCM3 mutant (LAIL-4D) does not co-elute with full-length wild type MST4, as assessed by SEC-MALS. C, the N-terminal CCM3 mutant (LAIL-4D) does not interact with MST4 or STK25 tail regions in a GST pulldown assay. D, FLAG-MST4 interaction with GFP-CCM3 in transiently transfected HEK293T cells is disrupted by mutations in the N-terminal region of CCM3 (LAIL-4D) but not by mutations within the FAT domain (K4A) of CCM3. FLAG-tagged PP4C was used as a negative binding control. Right panels show expression in the cell lysate; left panels show immunoprecipitated proteins. Top panels are blotted with anti-GFP antibody; bottom panels have been reprobed with anti-FLAG. E, FLAG-CCM3 and FLAG-MST4 interact strongly with GFP-MST4 and GFP-CCM3, respectively, in HEK293T cells. A weaker interaction of GFP-MST4 with FLAG-MST4 was detected. (Note that this interaction may be mediated via dimerization of other STRIPAK components.) No interaction of FLAG-CCM3 with GFP-CCM3 was detected. To eliminate detection of indirect interactions between GFP-CCM3 and FLAG-CCM3 arising from bridging interactions with a dimeric STRIPAK complex, we employed a four site mutant within the FAT domain of CCM3 (in the context of the GFP-CCM3 construct) that abolishes interaction with STRIPAK. FLAG-tagged PP4C was used as a negative binding control. Right panels show expression in the cell lysate; left panels show immunoprecipitated proteins. Top panels are blotted with anti-GFP antibody; bottom panels have been reprobed with anti-FLAG. vol., volume.
To investigate whether the CCM3-GCKIII protein heterodimers exist in vivo through the formation of a CCM3-like dimer structure, we tested wild type CCM3 and the four site CCM3 mutant for their ability to interact with GCKIII proteins by immunoprecipitation from HEK293T cells co-transfected with GFP-tagged CCM3 and FLAG-tagged MST4 kinase. Wild type GFP-tagged CCM3 was readily recovered in immunoprecipitates of FLAG-MST4, but not in immunoprecipitations of FLAG-PP4C, used here as a negative control (Fig. 4D). This interaction was abolished by the four-site mutation within the CCM3 dimerization domain (Fig. 4D; proteins expressed at similar levels). In contrast, a CCM3 mutation within the C-terminal FAT domain of CCM3 (K4A), which prevents interaction with STRIPAK, CCM2, and paxillin (17),7 had no effect on the interaction with MST4 (Fig. 4D). This result confirmed the role of the N terminus of CCM3 in mediating GCKIII protein interactions in vivo and further supported the notion that heterodimerization is mediated by a mechanism analogous to the CCM3 homodimerization mechanism (17).
We also used the HEK293T cell system to further investigate the homo- and heterodimerization properties of MST4 and CCM3. FLAG-tagged CCM3, MST4, or PP4C (negative control) were co-transfected with GFP-tagged CCM3 (K4A), MST4, or GFP alone. CCM3 (K4A) was employed for this assay to prevent interactions which would be mediated by interaction with the striatin component of STRIPAK, which also homo- and heterodimerizes.7 As seen in Fig. 4D, strong heterodimerization of FLAG-MST4 and GFP-CCM3 (K4A) was observed; the reciprocal interaction between GFP-MST4 and FLAG-CCM3 was also readily detected (Fig. 4E). By contrast, the recovery of GFP-MST4 with FLAG-MST4 was much weaker (note that this could potentially be mediated by striatin dimerization), and no detectable homodimerization of CCM3 was observed (Fig. 4E). These results further hinted that the heterodimer state between CCM3 and MST4 might be a preferred dimer conformation. To test whether this was in fact the case, we sought to quantify the binding affinity for homo- versus heterodimerization by analytical ultracentrifugation.
We performed equilibrium analytical ultracentrifugation analysis on CCM3 and MST4 proteins in isolation and on a CCM3-MST4 complex obtained by co-purification using dual affinity tag purification. Under the protein concentrations tested (see “Experimental Procedures”), CCM3 and MST4 profiles were best fit using a monomer-dimer equilibrium model with an extracted Kd of 2.5 and 2.7 μm, respectively (Fig. 5, A and B). In contrast, the CCM3-MST4 complex profiles were best fit as a single species model corresponding to a tight heterodimer (Fig. 5C). Based on the linearity of ln(A280) versus radius2 plots (Fig. 5C, bottom panel), which revealed no evidence of alternate monomeric or homodimeric species, we estimated a binding constant for heterodimeration of <0.1 μm, which is thus minimally 25-fold tighter than the CCM3 and MST4 homotypic inteactions. From these data, we concluded that the heterodimer state between CCM3 and MST4 is greatly favored over either of the two homodimer states.
FIGURE 5.

Analytical ultracentrifugation analysis of CCM3 and MST4 dimerization potential. A, sedimentation equilibrium analysis of CCM3. Sedimentation equilibrium ultracentrifugation was performed at 25 °C and spin speeds of 10,000, 15,000, 20,000, and 25,000 rpm. Global fits to a dimer-monomer equilibrium model performed on 12 data sets (top panel), which were acquired at four spin speeds and three spin radii. This model yielded a good fit and a measured Kd value of 2.7 ± 0.8 μm. Residuals to the monomer-dimer equilibrium fits are shown in the middle panel. Fit to a single-species dimer model (lower panel) yielded poor agreement. B, sedimentation equilibrium analysis of MST4. Sedimentation equilibrium ultracentrifugation was performed at 25 °C and spin speeds of 10,000, 15,000, and 20,000 rpm. Global fits to a dimer-monomer equilibrium model performed on six data sets (top panel), which were acquired at three spin speeds and two spin radii. This model yielded a good fit and a measured KD value of 2.5 ± 0.4 μm. Residuals to the monomer-dimer equilibrium fits are shown in middle panel. Fit to a single-species dimer model (lower panel) yielded poor agreement. C, sedimentation equilibrium analysis of an equimolar CCM3-MST4 complex. Sedimentation equilibrium ultracentrifugation was performed at 25 °C and spin speeds of 10,000, 15,000, 20,000, and 25,000 rpm. Global fits to a single-species model performed on eight data sets (top panel) acquired at four spin speeds and two spin radii. With the mass of the heterodimer input as an initial parameter, this model yielded a good fit. Residuals to the monomer-dimer equilibrium fits are shown in middle panel. Plot of the natural logarithm of the A280 value versus the square of the spin radius (lower panel). All spin speeds yield straight lines, consistent with only one species present at detectable levels at equilibrium.
DISCUSSION
In this study, we mapped the determinants of a direct interaction between CCM3 and GCKIII proteins to the N-terminal region of CCM3 and the C-terminal tail region of GCKIII proteins. These elements of both protein families are highly related in amino acid sequence, suggesting a common folded structure and binding function (Fig. 2A). Our data lead us to propose that CCM3-MST4 complex formation is achieved through the adoption of a heterodimeric helical structure analogous to that revealed by the CCM3 homodimer crystal structure (17). We also confirmed the existence of CCM3 homodimers in solution and that mutations within the N-terminal region of CCM3 disrupt both homodimerization and heterodimerization with purified GCKIII proteins (Fig. 4). Dimerization of STE20 family kinases mediated by conserved auxiliary domains has now been observed for GCKIII proteins (this study), the GCKII proteins (16), and the p21 activated kinases (23, 24). We reason that conserved regions flanking the kinase domains of other STE20 family kinases might serve analogous interaction functions albeit through the adoption of distinct structures.
The uncovered binding mode between CCM3 and GCKIII proteins helps to explain the following biological observations. Depletion of CCM3 led to the destabilization of STK25/SOK1 in cells (13) demonstrating an interdependence of protein function. A mutant in exon 5 of CCM3 that results in deletion of residues 33–50 within the N-terminal region (3) failed to bind the GCKIII proteins MST4, STK24, and STK25 (5, 19). This observation is consistent with our finding that point mutations within the N-terminal region of CCM3 disrupt binding to GCKIII proteins both in vitro and in cells (Fig. 4). Exon 5-deleted CCM3 also failed to rescue the cardiac phenotype of zebrafish, further demonstrating the biological importance of the CCM3-GCKIII protein interaction (5, 19). Because these regions of CCM3 and GCKIII proteins mediate both hetero- and homotypic dimerization, the relative importance of each state in the etiology of CCM disease needs to be explored further.
The interacting tail regions of GCKIII proteins and CCM3 are similar in sequence across the three α-helices that mediate CCM3 homodimerization (Fig. 2) (19). Although highly similar, the observed differences (29 of 39 contact residues are not identical) likely account for the tendency of CCM3 and MST4 kinases to preferentially heterodimerize versus homodimerize. Because the dimerization-mediating tails of the two other GCKIII proteins, STK24 and STK25, are more similar to MST4 than to CCM3 (Fig. 2A), we predict that they too will preferentially heterodimerize with CCM3. This, however, remains to be tested experimentally.
The CCM3-interacting region of GCKIII proteins is unrelated in sequence to the C-terminal SARAH domain of GCKII kinase MST1, an element that does not interact with CCM3 (Fig. 1C). Further database searches with the dimerization sequences of CCM3 and GCKIII proteins did not reveal other proteins in the human genome that might interact with CCM3 or GCKIII through a related structural mechanism. Interestingly, the dimerization regions of CCM3 and GCKIII proteins are well conserved throughout evolution, even in more distantly-related species, such as Caenorhabditis elegans and Monosiga brevicollis, the latter being the most distantly related choanoflagellate of metazoan origin sequenced to date (Fig. 2A) (25). This conserved evolution of CCM3 (formerly called DUF1241) and GCKIII proteins further supports the functional relevance of their observed interaction. A GCKIII-related kinase called severin exists in the slime mold Dictyostelium discoideum; however, a CCM3 homologue has not been identified in this organism suggesting that GCKIII proteins may retain functions independent of a CCM3 protein.
Acknowledgments
We thank P. Stathopulos, Le Zheng, and M. Ikura for useful discussions regarding SEC-MALS analysis.
This work was supported in part by Canadian Institutes of Health Research Grants MOP-36399 (to F. S.) and MOP-84314 (to A.-C. G.).
This article was selected as a Paper of the Week.
B. Yoruk, B. S. Gillers, N. C. Chi, and Ian C. Scott, submitted for publication.
M. J. Kean, D. F. Ceccarelli, M. Goudreault, S. Tate, B. Larsen, M. Sanches, L. C. D. Gibson, W. B. Derry, I. C. Scott, L. Pelletier, G. S. Baillie, F. Sicheri, and A.-C. Gingras, submitted for publication.
- CCM
- cerebral cavernous malformation
- GCKIII
- germinal center kinase group III
- STRIPAK
- striatin-interacting phosphatase and kinase
- TEV
- tobacco etch virus
- SEC-MALS
- size exclusion chromatography-multiangle light scattering
- FAT
- focal adhesion targeting
- PP4C
- catalytic subunit of the serine/threonine protein phosphatase 4
- mAU
- milli absorbance units.
REFERENCES
- 1. Labauge P., Denier C., Bergametti F., Tournier-Lasserve E. (2007) Lancet Neurol. 6, 237–244 [DOI] [PubMed] [Google Scholar]
- 2. Revencu N., Vikkula M. (2006) J. Med. Genet. 43, 716–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bergametti F., Denier C., Labauge P., Arnoult M., Boetto S., Clanet M., Coubes P., Echenne B., Ibrahim R., Irthum B., Jacquet G., Lonjon M., Moreau J. J., Neau J. P., Parker F., Tremoulet M., Tournier-Lasserve E. (2005) Am. J. Hum. Genet. 76, 42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guclu B., Ozturk A. K., Pricola K. L., Bilguvar K., Shin D., O'Roak B. J., Gunel M. (2005) Neurosurgery 57, 1008–1013 [DOI] [PubMed] [Google Scholar]
- 5. Zheng X., Xu C., Di Lorenzo A., Kleaveland B., Zou Z., Seiler C., Chen M., Cheng L., Xiao J., He J., Pack M. A., Sessa W. C., Kahn M. L. (2010) J. Clin. Invest. 120, 2795–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He Y., Zhang H., Yu L., Gunel M., Boggon T. J., Chen H., Min W. (2010) Sci. Signal 3, ra26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Louvi A., Chen L., Two A. M., Zhang H., Min W., Günel M. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 3737–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hilder T. L., Malone M. H., Bencharit S., Colicelli J., Haystead T. A., Johnson G. L., Wu C. C. (2007) J. Proteome Res. 6, 4343–4355 [DOI] [PubMed] [Google Scholar]
- 9. Voss K., Stahl S., Schleider E., Ullrich S., Nickel J., Mueller T. D., Felbor U. (2007) Neurogenetics 8, 249–256 [DOI] [PubMed] [Google Scholar]
- 10. Stahl S., Gaetzner S., Voss K., Brackertz B., Schleider E., Sürücü O., Kunze E., Netzer C., Korenke C., Finckh U., Habek M., Poljakovic Z., Elbracht M., Rudnik-Schöneborn S., Bertalanffy H., Sure U., Felbor U. (2008) Hum. Mut. 29, 709–717 [DOI] [PubMed] [Google Scholar]
- 11. Rual J. F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G. F., Gibbons F. D., Dreze M., Ayivi-Guedehoussou N., Klitgord N., Simon C., Boxem M., Milstein S., Rosenberg J., Goldberg D. S., Zhang L. V., Wong S. L., Franklin G., Li S., Albala J. S., Lim J., Fraughton C., Llamosas E., Cevik S., Bex C., Lamesch P., Sikorski R. S., Vandenhaute J., Zoghbi H. Y., Smolyar A., Bosak S., Sequerra R., Doucette-Stamm L., Cusick M. E., Hill D. E., Roth F. P., Vidal M. (2005) Nature 437, 1173–1178 [DOI] [PubMed] [Google Scholar]
- 12. Ma X., Zhao H., Shan J., Long F., Chen Y., Chen Y., Zhang Y., Han X., Ma D. (2007) Mol. Biol. Cell 18, 1965–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fidalgo M., Fraile M., Pires A., Force T., Pombo C., Zalvide J. (2010) J. Cell Sci. 123, 1274–1284 [DOI] [PubMed] [Google Scholar]
- 14. Goudreault M., D'Ambrosio L. M., Kean M. J., Mullin M. J., Larsen B. G., Sanchez A., Chaudhry S., Chen G. I., Sicheri F., Nesvizhskii A. I., Aebersold R., Raught B., Gingras A. C. (2009) Mol. Cell Proteomics 8, 157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Glatter T., Wepf A., Aebersold R., Gstaiger M. (2009) Mol. Syst. Biol. 5, 237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hwang E., Ryu K. S., Pääkkönen K., Güntert P., Cheong H. K., Lim D. S., Lee J. O., Jeon Y. H., Cheong C. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 9236–9241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li X., Zhang R., Zhang H., He Y., Ji W., Min W., Boggon T. J. (2010) J. Biol. Chem. 285, 24099–24107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ding J., Wang X., Li D. F., Hu Y., Zhang Y., Wang D. C. (2010) Biochem. Biophys. Res. Commun. 399, 587–592 [DOI] [PubMed] [Google Scholar]
- 19. Voss K., Stahl S., Hogan B. M., Reinders J., Schleider E., Schulte-Merker S., Felbor U. (2009) Hum. Mut. 30, 1003–1011 [DOI] [PubMed] [Google Scholar]
- 20. Skarra D. V., Goudreault M., Choi H., Mullin M., Nesvizhskii A. I., Gingras A. C., Honkanen R. E. (2011) Proteomics 11, 1508–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stathopulos P. B., Zheng L., Li G. Y., Plevin M. J., Ikura M. (2008) Cell 135, 110–122 [DOI] [PubMed] [Google Scholar]
- 22. Dibble C. F., Horst J. A., Malone M. H., Park K., Temple B., Cheeseman H., Barbaro J. R., Johnson G. L., Bencharit S. (2010) PLoS One 5, e11740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lei M., Lu W., Meng W., Parrini M. C., Eck M. J., Mayer B. J., Harrison S. C. (2000) Cell 102, 387–397 [DOI] [PubMed] [Google Scholar]
- 24. Parrini M. C., Lei M., Harrison S. C., Mayer B. J. (2002) Molecular cell 9, 73–83 [DOI] [PubMed] [Google Scholar]
- 25. King N., Westbrook M. J., Young S. L., Kuo A., Abedin M., Chapman J., Fairclough S., Hellsten U., Isogai Y., Letunic I., Marr M., Pincus D., Putnam N., Rokas A., Wright K. J., Zuzow R., Dirks W., Good M., Goodstein D., Lemons D., Li W., Lyons J. B., Morris A., Nichols S., Richter D. J., Salamov A., Sequencing J. G., Bork P., Lim W. A., Manning G., Miller W. T., McGinnis W., Shapiro H., Tjian R., Grigoriev I. V., Rokhsar D. (2008) Nature 451, 783–788 [DOI] [PMC free article] [PubMed] [Google Scholar]