

Abstract
Mcl-1, a pro-survival member of the Bcl-2 family located at the mitochondrial outer membrane, is subject to constitutive ubiquitylation by the Bcl-2 homology 3-only E3 ligase, Mule/Lasu1, resulting in rapid steady-state degradation via the proteasome. Insertion of newly synthesized Mcl-1 into the mitochondrial outer membrane is dependent on its C-terminal transmembrane segment, but once inserted, the N terminus of a portion of the Mcl-1 molecules can be subject to proteolytic processing. Remarkably, this processing requires an intact electrochemical potential across the inner membrane. Three lines of evidence directed at the endogenous protein, however, indicate that the resulting Mcl-1ΔN isoform resides in the outer membrane: (i) full-length Mcl-1 and Mcl-1ΔN resist extraction by alkali but are accessible to exogenous protease; (ii) almost the entire populations of Mcl-1 and Mcl-1ΔN are accessible to the membrane-impermeant Cys-reactive agent 4-acetamido-4′-[(iodoacetyl)amino]stilbene-2,2′-disulfonic acid; and (iii) Mcl-1 and Mcl-1ΔN exhibit equivalent chemical cross-linking to Bak in intact mitochondria, an Mcl-1 binding partner located in the outer membrane. In addition to the Mule Bcl-2 homology 3 domain, we show that interaction between Mcl-1 and Mule also requires the extreme N terminus of Mcl-1, which is lacking in Mcl-1ΔN. Thus, Mcl-1ΔN does not interact with Mule, exhibits reduced steady-state ubiquitylation, evades the hyper-rapid steady-state degradation that is observed for full-length Mcl-1 in response to treatments that limit global protein synthesis, and confers resistance to UV stress-induced cell death.
Keywords: Apoptosis, Membrane Proteins, Mitochondria, Proteasome, Protein Degradation, Mcl-1, Mitochondrial Potential, Mule/Lasu1, Proteasome
Introduction
A plethora of cell-intrinsic pathways can activate apoptosis, commensurate with the diverse roles that programmed cell death plays in development, immunity, and disease. Many of these pathways are both executed and regulated by the Bcl-2 family of proteins. The family is comprised of three groups, defined according to their function and content of Bcl-2 homology (BH)9 domains (1–3): the pro-apoptotic effectors, including Bax and Bak, contain BH1, BH2, and BH3 domains, as well as a C-terminal transmembrane segment that selectively targets these proteins to the membranes of mitochondria and endoplasmic reticulum; the pro-survival members Bcl-2, Mcl-1, Bcl-XL, Bcl-w, Bcl-B, and Bcl-A1 have the same overall architecture as Bax and Bak but in addition contain a BH4 domain located toward their N termini; and finally, a large and diverse group within this family contains only a single BH3 domain. The latter, the BH3-only, proteins couple upstream death stimuli to downstream regulation of the multiple BH domain members. Considerable work has begun to provide an understanding of the regulation and function of specific Bcl-2 family members, which has helped to elucidate a number of important cell death pathways.
Mcl-1 was discovered based on its increased expression during cell commitment to differentiation in a human myeloid leukemia cell line (4). It has since been shown to be essential in the early stages of embryogenesis as Mcl-1 knock-out mice display peri-implantation lethality (5). Furthermore, it is necessary for the development and maintenance of B and T lymphocytes and for the survival of hematopoietic stem cells (6, 7). Similar to other pro-survival Bcl-2 family members, elevated levels of Mcl-1 have also been shown to play a critical role in the initiation and progression of certain cancers. In particular, Mcl-1 is overexpressed in a number of human hematological cancers, including B cell lymphoma and chronic lymphocytic leukemia, as well as in solid tumor malignancies such as hepatocellular, pancreatic, and cervical carcinomas (8–12).
Mcl-1 is uniquely subject to extensive regulation (13–15). This includes the regulation of rapid Mcl-1 protein turnover (16, 17), in which degradation by the 26 S proteasome system is mediated by at least three E3 ubiquitin ligases: Mule/Lasu1, a BH3-only protein that selectively binds Mcl-1 among Bcl-2 family members and contributes to constitutive Mcl-1 turnover (18, 19); βTrCP, which controls Mcl-1 ubiquitylation and degradation in response to Mcl-1 phosphorylation by GSK-3β (20, 21); and the tumor suppressor SCF(FBW7), which is lost in a number of cancers (22). Additionally, there may be other ubiquitylation pathways, as well as ubiquitin-independent mechanisms for degrading Mcl-1 (23). As mentioned above, there is selective pressure across many cancer indications to increase Mcl-1 expression (24), and this includes enhanced Mcl-1 stabilization because of elevated expression of the de-ubiquitinase USP9X (25). Alternatively, N-terminal truncation of Mcl-1 is associated with stabilization and abundant expression in many tumor cells (26), but the underlying mechanisms remain to be determined.
Here, we show that following insertion of Mcl-1 into the mitochondrial outer membrane, the N terminus of Mcl-1 can be processed by a mechanism that depends on an intact electrochemical potential across the inner membrane. We show that although both isoforms localize to the outer mitochondrial membrane and inhibit apoptosis following cellular stress, the N-terminally truncated isoform evades interaction with Mule/Lasu1, exhibits reduced ubiquitylation, and resists the hyper-rapid degradation associated with full-length Mcl-1. As a result, Mcl-1ΔN retains its capacity to resist cell death in response to stresses such as UV irradiation that inhibit global protein synthesis.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
The following commercial antibodies were used: α-FLAG M2 monoclonal antibody, monoclonal α-tubulin, and α-HA monoclonal antibody (Sigma); rabbit α-Bak antibody (Upstate Biotechnology, Inc.); mouse α-actin monoclonal antibody (ICN Pharmaceuticals); rabbit α-mouse Mcl-1 antibody (Rockland); rabbit-human Mcl-1 antibody (StressGen); mouse anti-bovine ubiquitin (P4D1) (Santa Cruz Biotechnology); and rabbit anti-manganese superoxide dismutase (Assay Design). The Mcl-1 (N-terminal) antibody was generated by immunizing rabbits with the biotin-NH-MFGLRRNAVIGLNLYCGGASLGAGGGSPAG-NH2 peptide, corresponding to the N-terminal 30 aa of mouse Mcl-1. All other antibodies were developed in house and have been characterized previously. MG132 was from Calbiochem, and Polybrene and cycloheximide were from Sigma. [35S]Methionine/cysteine labeling mixture was from PerkinElmer Life Sciences.
Vector Construction
Full-length and N-terminally truncated mMcl-1 and hMcl-1 cDNAs were generated by PCR and cloned into pcDNA3. Mouse Mcl-1 G30E (GGG→GAG) was obtained by random mutation. For Biacore binding studies, Mcl-1ΔTM(1–327) was generated by PCR and cloned into a pET29b vector modified to encode an N-terminal FLAG epitope along with the existing C-terminal polyhistidine (His6) tag for purification. Murine stem cell virus-based vectors were introduced by retroviral infections as described previously by utilizing the Phoenix packaging system (27).
Cell Lines
All nonprimary cell lines were maintained in growth media (DMEM supplemented with 10% fetal bovine serum). Wild-type (WT) Mcl-1 and Mcl-1−/− MEFs were kindly provided by Joseph T. Opferman (St. Jude Children's Research Hospital). Tsc2−/−p53−/− MEFs were kindly provided by David Kwiatkowski (Brigham and Women's Hospital).
Treatments
For studies with UV treatment, cells were plated at a density of 3 × 105 cells per well in 6-well dishes. The following day, media were removed, and cells were washed twice in cold PBS and then treated with the indicated dose of UV light using a UV Stratalinker 2400 (Stratagene); the media were then replaced, and cells were analyzed at the indicated time points. Unless indicated otherwise, cycloheximide was used at 100 μg/ml in distilled H2O, and MG-132 was added at a concentration of 10 μm.
Viability Measurements
Following treatment, floating cells were collected and pooled with trypsinized adherent cells. The cells were then resuspended in 500 μl of staining buffer (PBS containing 2% FBS) containing 10 μl of PI (50 μg/ml). Cells were analyzed on a FACScan instrument (BD Biosciences).
Biacore 3000 Surface Plasmon Resonance
25-Amino acid synthetic peptides corresponding to the BH3 domain of Lasu1/Mule (CGVMTQEVGQLLQDMGDDVYQQYRS), Bim (CMRPEIWIAQELRRIGDEFNAYYAR), Puma (CEQWAREIGAQLRRMADDLNAQYER), Bak (CSSTMGQVGRQLAIIGDDINRRYDS), Bid (CEDIIRNIARHLAQVGDSMDRSIPP), Bid (CMEGSDALALRLACIGDEMDVSLRA), Noxa (CAELEVECATQLRRFGDKLNFRQKL), and mtLASU1/Mule (CGVMTQEVGQGGQDMGDDVYQQYRS) were immobilized (∼300 arbitrary response units) on Biacore CM-5 sensorchip surfaces at a flow rate of 5 μl/min using standard amine coupling via the N terminus of each peptide, except for Noxa in which thiol coupling via an N-terminal cysteine was used. For amine coupling, a 35-μl injection of a mixture of 0.05 m N-hydroxysuccinimide (NHS) and 0.2 m N-ethyl-N′-(3-diethylaminopropyl) carbodiimide hydrochloride was followed by the manual injection of ∼40 μm BH3 domain peptide and 1 m NaCl in 10 mm HEPES, pH 7.5, until the desired immobilization level was reached. The remaining activated surface groups were inactivated by a 35-μl injection of 1 m ethanolamine. For thiol coupling of the NOXA peptide, 60 μl of the N-hydroxysuccinimide/N-ethyl-N′-(3-diethylaminopropyl) carbodiimide hydrochloride solution was followed by 120 μl of 80 mm 2-(2-pyridinyldithio)ethaneamine in 0.1 m sodium borate, pH 8.5. NOXA peptide (∼40 μm) in 10 mm NaOAc, pH 5, was then manually injected over the surface until ∼300 arbitrary response units was reached. The remaining activated surface groups were quenched by injecting 120 μl of 50 mm cysteine and 1 m NaCl in 0.1 m sodium acetate, pH 4.0. Matching blank control surfaces for each peptide were created as above except for the omission of the manual peptide injection. For binding analysis, recombinant FLAG-Mcl-1ΔTM-His6 was simultaneously injected over the sample and blank control surfaces at the concentrations indicated. Sensograms derived from each concentration series were double referenced, and the KD value was determined by global analysis fit to a 1:1 Langmuir binding model using BIAEvaluation 3.2 software. Three independent runs were used to obtain the average KD value and standard deviation.
Transient Transfection and Immunoprecipitations
For transfections, HeLa and NIH3T3 cells were plated to 20% confluency in growth media. Cells were transfected by LipofectamineTM (Invitrogen) 24 h later according to the manufacturer's protocol. For immunoprecipitations, cells were washed twice in PBS 48 h later and lysed at 4 °C for 20 min in 1 ml of lysis buffer (50 mm HEPES, pH 7.7, 150 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40, 0.1% Triton X-100, 1 μg/ml aprotinin, 1 μg/μl leupeptin, 1 μg/μl pepstatin, and 1 mm PMSF). The cell lysate was centrifuged at 12,000 rpm to remove membranes and insoluble debris. The cell lysate was pre-cleared with protein G-Sepharose beads (Amersham Biosciences), and the cleared cell lysate was incubated for 4 h at 4 °C with the indicated antibody. Protein G-Sepharose was then added and incubated for 1.5 h. After incubation, the samples were pelleted and washed three times with lysis buffer and boiled in SDS sample buffer. After the samples were resolved by SDS-PAGE and transferred to nitrocellulose, immunoblotting was performed by standard procedures with the indicated antibodies, and blots were visualized with enhanced chemiluminescence. For densitometric analysis, ImageJ software (National Institutes of Health) was used. Following background subtraction, immunoprecipitation band intensities were divided by the corresponding lysate band intensities.
Biotin Pulldowns
HeLa cells were collected and lysed as described above and precleared for 1 h with streptavidin beads. Biotinylated synthetic peptide corresponding to the first 30 aa of mMcl-1 (Biotin-MFGLRRNAVIGLNLYCGGASLGAGGGSPAG-NH2) or a control biotinylated peptide (Biotin-PPRPRTPGRPLSSYGMDSRPPMAIFEL-OH) were added at a concentration of 50 ng/mg of lysate and incubated for 3 h. Streptavidin beads were then added for 1 h, and samples were pelleted and washed as described above. Bound material was eluted by boiling in SDS-PAGE sample buffer.
Mcl-1 Pulse Labeling
MEFs were incubated in cysteine-free growth media containing twice-dialyzed FBS and then incubated for 15 min in the same fresh media. Media were then replaced with 4 ml of the same media containing 250 μCi/ml [35S]methionine/cysteine labeling mixture. Cells were labeled for 45 min. Labeling was terminated with the addition of cycloheximide (100 μg/ml) and harvested at the indicated times. Cells were washed three times in ice-cold PBS and lysed in 500 μl of lysis buffer (50 mm HEPES, pH 7.7, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 0.1% Triton X-100, 1 μg/ml aprotinin, 1 μg/μl leupeptin, 1 μg/μl pepstatin, and 1 mm PMSF). 500 μg of protein from each lysate was pre-cleared with rabbit IgG antibody. The cleared lysates were then incubated with 2 μg of anti-Mcl-1 antibody and recovered with protein A-Sepharose. Mcl-1 was resolved on SDS-PAGE and transferred to a PVDF membrane. Radioactive signal was detected with autoradiographic film. The PVDF membrane was then immunoblotted with the indicated antibodies.
Subcellular Fractionation
Cells (∼5 × 107 cells) were collected and homogenized in 500 μl of 200 mm mannitol, 70 mm sucrose, 10 mm HEPES, pH 7.5, 1 mm EGTA using a Teflon glass homogenizer maintained in ice water. The total homogenate was centrifuged in a microcentrifuge for 10 min at 4 °C at 1800 rpm (600 × g). The supernatant was designated S1 (containing mitochondria). S1 was centrifuged for 10 min at 4 °C at 9000 rpm to yield a pellet (P9) enriched in mitochondria; the supernatant was designated S9. Mitochondria containing fraction P9 was treated with exogenous trypsin (0.125 μg/ml) for 30 min at 4 °C with the addition of trypsin inhibitor (1.25 μg/ml) either at time 0 or after 30 min. Alkaline extraction was carried out by extracting P9 with 0.1 m Na2CO3, pH 11.5. Chemical labeling of the proteins in the P9 fraction with 4-acetamido-4′-[(iodoacetyl)amino]stilbene-2,2′-disulfonic acid (IASD) (Molecular Probes) was carried out exactly as described previously (28). Cross-linking of Mcl-1 and Bak using LC-succinimidyl 4(N-maleimidomethyl)cyclohexane-1-carboxy-(6-amidocaproate) was performed exactly as described previously (29) except that the post-nuclear S1 fraction was used.
Mitochondrial Protein Import
For an in vitro import reaction, mMcl-1 mRNA was in vitro translated (IVT) in rabbit reticulocyte lysate for 15 min at 30° after which time S1 or S9 was added at the ratio of IVT:S1/S9 of 1:4. The import reactions were carried out with the indicated fractions at 30 °C for at least 6 h. When examined, 0.1 μm CCCP or 5 μm MG132 was added to S1 prior to the addition of IVT.
Immunofluorescence Microscopy
Mcl-1 null MEFs were transiently transfected with cDNA expressing murine Mcl-1 or Mcl-1ΔN2-30 as described above. 48 h post-transfection, cells were fixed in 4% paraformaldehyde and probed with Mcl-1 or cytochrome c antibody. Alexa 488-conjugated anti-rabbit IgG or Alexa 594-conjugated anti-mouse IgG (Molecular Probes) were used to decorate the cells, and fluorescence was visualized by Zeiss fluorescence microscopy.
RESULTS AND DISCUSSION
To investigate Mcl-1 expression, we utilized SV40-transformed MEFs derived from WT and Mcl-1−/− mice (30). High resolution gel electrophoresis and combined immunoprecipitation and immunoblot with an antibody that recognizes either the extreme N terminus or internal epitopes revealed the expression of both full-length Mcl-1 (∼40 kDa) and a truncated Mcl-1ΔN isoform (∼36 kDa) (Fig. 1, A and B). The latter roughly co-migrated with recombinant Mcl-1ΔN lacking aa 2–30, whereas the former co-migrated with the recombinant primary in vitro translation product of full-length Mcl-1 (Fig. 1C). In certain gels, an apparent intermediate size band was observed (e.g. see Fig. 2A and discussed below). Mcl-1 and Mcl-1ΔN were recovered in a heavy membrane fraction enriched in mitochondria (Fig. 1D) and exhibited equivalent co-localization with mitochondria as judged by immunofluorescence microscopy (Fig. 1E). Both isoforms co-immunoprecipitated with the established Mcl-1 outer membrane binding partner Bak (supplemental Fig. S1) (4, 15), indicating that they both have functional BH3-binding modules. As expected (13), however, the two isoforms responded very differently to a blockade of global protein synthesis (conferred by the translation inhibitor CHX or treatment with UV irradiation). As shown in Fig. 1F, full-length Mcl-1 but not Mcl-1ΔN rapidly disappeared in response to these treatments. Furthermore, the proteasome inhibitor MG132 inhibited this loss of full-length Mcl-1, resulting in its accumulation, but it had little effect on Mcl-1ΔN. Recombinant murine (supplemental Fig. S2) and human Mcl-1ΔN2–30 (supplemental Fig. S3) also exhibited relative stability in transfected cells following treatment with CHX or UV light. Importantly, transfected recombinant Mcl-1ΔN2–30 retained the ability to confer resistance to cell death in response to irradiation to an extent similar to that observed for full-length Mcl-1 (Fig. 1G).
FIGURE 1.

Differential steady-state stability of Mcl-1 and Mcl-1ΔN. A, Mcl-1−/− and WT MEF whole cell lysates were analyzed by immunoblotting with the indicated antibodies. B, MEF cell lysates were subjected to immunoprecipitation (IP) with either α-mMcl-1 antibody or preimmune IgG, and the precipitates were analyzed by immunoblotting with α-mMcl-1 antibody against the C-terminal region. The immunoblot was then stripped and reprobed with α-mMcl-1(N-term). C, immunoblot of in vitro translated product of mMcl-1 and mMcl-1Δ2–30 and MEF cell lysate. White line denotes lanes from the same blot that are not shown. The lanes are from the same blot and the same exposure. D, MEF cell lysate was collected and fractionated into heavy membrane enriched in mitochondria, light membrane enriched in endoplasmic reticulum, and cytosol (Cyto) and subjected to immunoblot with the indicated antibodies. Tom20 and Bap31 are markers for mitochondria and endoplasmic reticulum, respectively. E, Mcl-1−/− MEFs were transfected with vectors expressing mMcl-1 or mMcl-1ΔN30, and the expression of Mcl-1 proteins was visualized with immunofluorescence microscopy, as indicated. Endogenous cytochrome c was used as marker for mitochondria. White arrows indicate mitochondrial localization. F, MEFs were treated with either 100 μg/ml CHX or 20 mJ/cm2 UV irradiation in the presence or absence of MG132 (100 μm) and analyzed by immunoblot at the indicated time points. G, hMcl-1ΔN2–30 confers resistance to UV-induced cell death. G, panel i, Tsc2−/− MEFs were transduced with the indicated vectors. Cells were then UV-irradiated (20 mJ/cm2), and both expression and stability of exogenous hMcl-1 were analyzed 2 h later by immunoblotting. G, panel ii, Tsc2−/− MEFs expressing constructs as in G, panel I, were treated with UV irradiation (16 h), and cell death was determined.
FIGURE 2.

Mcl-1ΔN isoform is present at the surface of the outer mitochondrial membrane. A, mitochondria isolated from MEFs were treated either with exogenous trypsin or extracted with Na2CO3, pH 11. 5, as described under “Experimental Procedures,” and analyzed by immunoblot with the indicated antibodies. Cyt c, cytochrome c. Mitos, mitochondria. B, as in A, except mitochondria were isolated following a 0- or 2-h pulse of MEFs with CHX to enrich for mMcl-1 ΔN, and the mitochondria were treated with IASD (see “Experimental Procedures”). C, as in B, except that untreated (UT) mitochondria were subjected to membrane disruption by sonication or 0.5% Triton prior to incubation with IASD, and probed with anti- manganese superoxide dismutase (MnSOD) (left panel) or anti-Bak (right panel). D, as in B, except mitochondria were isolated from MEFs following a 0- or 3.5-h pulse with CHX and were subjected to cross-linking with LC-succinimidyl 4(N-maleimidomethyl)cyclohexane-1-carboxy-(6-amidocaproate). After immunoprecipitation (IP) with anti-mMcl-1, cross-linked Bak-Mcl-1 adducts (denoted by black dots) were identified by immunoblot with anti-Bak (right panel). Upper left panel shows immunoblot of total cell lysate; lower left panel shows immunoblot of the anti-Mcl-1 immunoprecipitate probed with anti-Mcl-1. WB, Western blot.
Mcl-1 and Mcl-1ΔN Are Located in the Mitochondrial Outer Membrane
A recent study based on siRNA knockdown suggested that the N terminus of Mcl-1 is subject to mitochondrial processing by the matrix-localized processing peptidase and concluded that this was due to import of Mcl-1 into the interior of the organelle, leaving Mcl-1ΔN within an internal compartment (31). In contrast, we find that Mcl-1ΔN is anchored in the outer membrane and exposed to the cytoplasm, consistent with the earlier findings that the Mcl-1 C-terminal transmembrane (TM) segment is both necessary and sufficient to target and insert Mcl-1 into the outer membrane lipid bilayer (32). Thus, removal of the N-terminal 2–30 aa of Mcl-1 did not interfere with the ability of ectopic expressed Mcl-1ΔN2–30 to target and localize to mitochondria in transfected cells (Fig. 1E). In isolated intact mitochondria, endogenous Mcl-1 and Mcl-1ΔN were both found to be resistant to alkaline extraction, pH 11.5, indicative of membrane integration, and both were sensitive to exogenous trypsin, indicating that they are surface-exposed; in contrast, cytochrome c, an intermembrane space protein, was extracted by alkali and resisted trypsin degradation (Fig. 2A). To extend this analysis, we probed Mcl-1 and Mcl-1ΔN in isolated mitochondria with the thiol-conjugating agent IASD, which cannot enter or cross membranes and has previously been used to deduce the topology of surface versus membrane-integrated Cys-containing domains of Bcl-2 in the mitochondrial outer membrane (28). Conjugation to surface-exposed Cys residues could be conveniently assayed because it resulted in a mobility shift in Bcl-2 following gel electrophoresis (28). Similarly, exposure of isolated intact mitochondria to IASD caused a quantitative mobility shift of both Mcl-1 (contains 2 Cys) and Mcl-1ΔN (contains one Cys) following gel electrophoresis (Fig. 2B), consistent with a location of Mcl-1 and Mcl-1ΔN in the outer membrane. Of note, for this assay, cells had been briefly pulsed with CHX to enrich for Mcl-1ΔN prior to the isolation of mitochondria. A similar quantitative shift following IASD treatment was observed for Bak (Fig. 2B). In contrast, the intra-mitochondrial protein manganese superoxide dismutase exhibited a mobility shift in response to IASD only if the membranes were disrupted either by detergent or sonication, whereas outer membrane Bak was unaffected by detergent (Fig. 2C). Thus, the two known forms of Mcl-1 and Bak appear to be primarily associated to the outer membrane of the organelle. Therefore, as expected, endogenous Mcl-1 and Mcl-1ΔN were both observed to physically interact with Bak under conditions where the outer membrane remains intact, based on chemical cross-linking with LC-succinimidyl 4(N-maleimidomethyl)cyclohexane-1-carboxy-(6-amidocaproate) in intact mitochondria (Fig. 2D) (29). In this experiment, one sample of cells was pulsed with CHX prior to isolation of mitochondria to generate mitochondria primarily containing Mcl-1ΔN (Fig. 2D, top left panel, 2nd lane, and bottom left panel, 4th lane). As seen in Fig. 2D (right panel, lane 6), this sample containing primarily Mcl-1ΔN clearly exhibited cross-linking to Bak. Collectively, therefore, the evidence indicates that endogenous Mcl-1ΔN is functional and is located in the mitochondrial outer membrane.
Processing of Mcl-1 Occurs at the Mitochondrial Outer Membrane and Requires an Intact Mitochondrial Δψ
Transfection of cDNA encoding full-length Mcl-1 into Mcl-1-null MEFs generated the same isoform pattern as expressed in WT MEFs (Fig. 3A), indicating that these isoforms are generated from the same mRNA. However, whereas the possibility exists for translation to initiate at noncanonical translation start sites within codons at the N terminus (15), extensive mutagenesis excluded this possibility (data not shown). Remarkably, however, a single point mutation at codon 30, in which a glycine is changed to glutamic acid (G30E), strongly decreased the generation of the Mcl-1ΔN isoform (Fig. 3A), suggesting that processing requires either a sequence or a structural requirement present in Mcl-1. Importantly, ectopically expressed Mcl-1G30E co-localized with mitochondria (Fig. 3B), indicating that mitochondrial targeting and subsequent processing can be disconnected. This is consistent with the prior findings that the Mcl-1 TM segment is both necessary and sufficient for targeting Mcl-1 to the mitochondria (31). Moreover, ectopic expression of Mcl-1ΔTM (full-length Mcl-1 lacking the TM segment) in Mcl-1−/− MEFs exhibited a complete lack of processing and therefore expression of Mcl-1ΔN(ΔTM) (Fig. 3C). We therefore conclude that processing of Mcl-1 to Mcl-1ΔN occurs primarily at the mitochondrial outer membrane.
FIGURE 3.

Mitochondrial processing of Mcl-1. A, Mcl-1−/− MEFs were transfected with vector or vector expressing mMcl-1, mMcl-1ΔN30, or mMcl-1G30E, and together with cell extract from WT, MEFs were analyzed by immunoblot with mMcl-1 antibody. B, Mcl-1G30E transiently expressed in Mcl-1 null MEFs was analyzed by immunofluorescence microscopy in parallel with endogenous cytochrome c (Cyto c) White arrows indicate mitochondrial localization. C, Mcl-1−/− MEFs were transfected with vector expressing mMcl-1ΔTM (aa 1–312), and together with mMcl-1, IVT was analyzed by immunoblot with anti-mMcl-1 directed toward a C-terminal region of the protein. The blot was stripped and re-probed with anti-mMcl-1 specific for the mMcl-1 N terminus. D, WT MEFs were radiolabeled with 35S-Met/Cys for 45 min and then chased by the addition of CHX. Mcl-1 was immunoprecipitated (IP) and resolved by SDS-PAGE and transferred to PVDF. 35S-Mcl-1 was detected with autoradiography for 24 h, and subsequently total mMcl-1 was detected by immunoblotting. The arrowheads depict full-length and processed Mcl-1. E, mMcl-1 IVT product was incubated with S1 or S9 fractions from Mcl-1-null MEFs under protein import conditions at the indicated temperatures. Import mixtures together with input IVT and WT MEF cell lysate were analyzed by immunoblot. F, as in E, except import was conducted in the presence or absence of 5 μm MG132 or 0.1 μm CCCP. G, as in F, except that following import, mitochondria (mito) (P9) were recovered prior to analysis by immunoblot.
Pulse labeling of 35S-Mcl-1 followed by chase in the presence of CHX suggested a conversion of Mcl-1 to Mcl-1ΔN (Fig. 3D). Furthermore, analysis of the Mcl-1 N terminus by MITOPROT suggested a potential sequence similarity to mitochondrial inner membrane/matrix targeting signals (supplemental Fig. S4) (30). Such sequences include positively charged amino acids and an enrichment of smaller size amino acids, with an intolerance for negatively charged amino acids (33). Interestingly, introduction of the negatively charged Glu at position 30, which interfered with processing (Fig. 3A), would also be expected to be incompatible with a matrix targeting function. Furthermore, siRNA-mediated knockdown of the matrix-localized pre-protein processing peptidase was shown to interfere with the generation of Mcl-1ΔN (31). Finally, processing was also shown to be dependent on mitochondrial Δψ, which is restricted to the inner membrane of the mitochondria. This was demonstrated by incubating the Mcl-1 primary translation product in rabbit reticulocyte lysate containing an S1 post-nuclear cell extract (contains intact mitochondria) or an S9 post-mitochondrial cell extract (lacks mitochondria) from Mcl-1−/− MEFs under conditions of protein import (34). Mcl-1 processing was achieved by S1 at 37 °C but not at 4 °C, whereas the S9 fraction was inactive (Fig. 3E). Employing the uncoupler CCCP, which collapses mitochondrial inner membrane Δψ, processing of Mcl-1 was abolished (Fig. 3F). Unlike CCCP, the proteasome inhibitor MG132 had no effect on processing (Fig. 3F). The processed product generated by S1 remained with mitochondria and was subsequently recovered in the P9 mitochondrion-enriched pellet fraction (Fig. 3G). Of note, however, based on co-migration of import products with endogenous Mcl-1 and Mcl-1ΔN, the in vitro import assay appeared to generate the intermediate size product rather than the fully processed Mcl-1ΔN (Fig. 3, E and G), despite significant alterations to the import reaction in an attempt to achieve faithful processing to the in vivo isoform.
There are at least two explanations to account for processing of Mcl-1 to Mcl-1ΔN at the mitochondrial outer membrane. Either the long flexible N-terminal region of Mcl-1 (35) can gain Δψ-dependent transient (i.e. reversible) access to the matrix processing machinery, although the protein is still anchored at the outer membrane, or processing at the outer membrane is dependent on a system that is sensitive to inner membrane Δψ.
Processing of Mcl-1 to Mcl-1ΔN Prevents Its Interaction with Mule/Lasu
Mule/Lasu1 appears to be responsible mainly for constitutive steady-state degradation of Mcl-1 (18, 19); consequently, efficient siRNA knockdown of Mule results in increased levels of Mcl-1 (19). Like most proteins that interact with pro-survival members of the Bcl-2 family, Mule contains a BH3 domain (18, 19). Notably, its BH3 module exhibits differential selectivity for binding to Mcl-1. Thus, efficient Mule-dependent ubiquitylation requires that Mule effectively competes with other Mcl-1 binding partners (e.g. Bim or Bak) to maintain efficient steady-state degradation of Mcl-1 (19). One possibility is that the Mcl-1 N terminus, in addition to the Mule BH3 domain, contributes to Mcl-1/Mule interactions, allowing Mule to effectively compete with other Mcl-1 binding partners. The affinities of the interaction of full-length Mcl-1 lacking the C-terminal membrane anchor with 25 amino acid peptides corresponding to the BH3 domains of multiple known Mcl-1 binding partners (Mule/Lasu1, Bak, Bim, Bid, Puma, and Noxa) (18, 19, 36) were first analyzed by surface plasmon resonance. Interestingly, the Mule BH3 peptide exhibited the weakest binding affinity (65 nm) among this group (Fig. 4, A and B). However, it was also found that Mule interacted with full-length Mcl-1 but not with Mcl-1ΔN as revealed by co-immunoprecipitation (Fig. 4C). Deletion mutagenesis indicated that a region corresponding to aa 20–30 may make an important contribution to this interaction (Fig. 4D).
FIGURE 4.

N terminus of Mcl-1 promotes increased association with Mule. A and B, synthetic peptides corresponding to the BH3 domain of the indicated BH3-containing proteins were immobilized on Biacore CM-5 sensorchip surfaces via the N terminus of each peptide. Recombinant FLAG-Mcl-1ΔTM (Mcl-1 aa 1–347) was injected over the surface at the concentrations indicated. Sensorgrams derived from each concentration series were double referenced, and the KD value was determined by global analysis fit to a 1:1 Langmuir binding model using BIAEvaluation 3.2 software. Three independent runs were used to obtain the average KD value and standard deviations. A representative sensorgram of immobilized BidBH3 is shown in A. RU, response units. C, mMcl-1 cDNA was transfected into HeLa cells, and whole cell lysates were immunoprecipitated (IP) with α-Mule antibody or preimmune IgG. Immunoprecipitates were immunoblotted with murine-specific mMcl-1 antibody. D, HeLa cells were transiently transfected with mMcl-1 lacking the N-terminal aa 2–10, 2–20, or 2–30, and cell lysates were immunoprecipitated with α-Mule antibody or preimmune IgG. To confirm a reduced interaction between Mcl-1Δ2–30 and Mule, densitometric analysis was performed with ImageJ software (National Institutes of Health). Values represent the immunoprecipitated band intensities (background subtracted) divided by the corresponding lysate band intensities. NS, nonspecific. E, HeLa cell lysates were precleared with streptavidin beads, incubated with biotin-N30 synthetic peptide corresponding to the first 30 aa of mMcl-1 or a control biotinylated synthetic peptide at a concentration of 50 ng/mg lysate protein, and biotin was precipitated using streptavidin beads.
To determine whether the N terminus of Mcl-1 can independently associate with Mule, we constructed a biotinylated peptide comprising the first 30 amino acids of Mcl-1 (N30). Endogenous Mule was recovered from HeLa cell extracts with streptavidin beads bound with biotin-N30 peptide but not with a biotinylated control peptide (Fig. 4E). Thus, there exist (at least) two points of interaction between Mule and Mcl-1 as follows: docking of the Mule BH3 domain into the BH3 binding groove of Mcl-1 (18, 19) and binding of the Mcl-1 N terminus to Mule. This bipartite interaction presumably permits Mule to be an effective competitor for Mcl-1 interaction. Mcl-1ΔN, on the other hand, would evade such effective competitive binding to Mule. If so, it would be expected that Mcl-1ΔN exhibits reduced steady-state ubiquitylation relative to full-length Mcl-1. This was demonstrated by ectopically expressing similar levels of Mcl-1 and Mcl-1ΔN in HeLa cells (Fig. 5, bottom panel, lanes 3 and 5) and probing with anti-ubiquitin following a brief treatment of cells with MG132 and immunoprecipitation of Mcl-1. Although no quantitative differences were noted in the ubiquitylation profile of total cellular proteins (Fig. 5, middle panel), there was a clear reduction in ubiquitylation of transfected Mcl-1ΔN compared with ubiquitylation of transfected Mcl-1 (Fig. 5, top panel).
FIGURE 5.
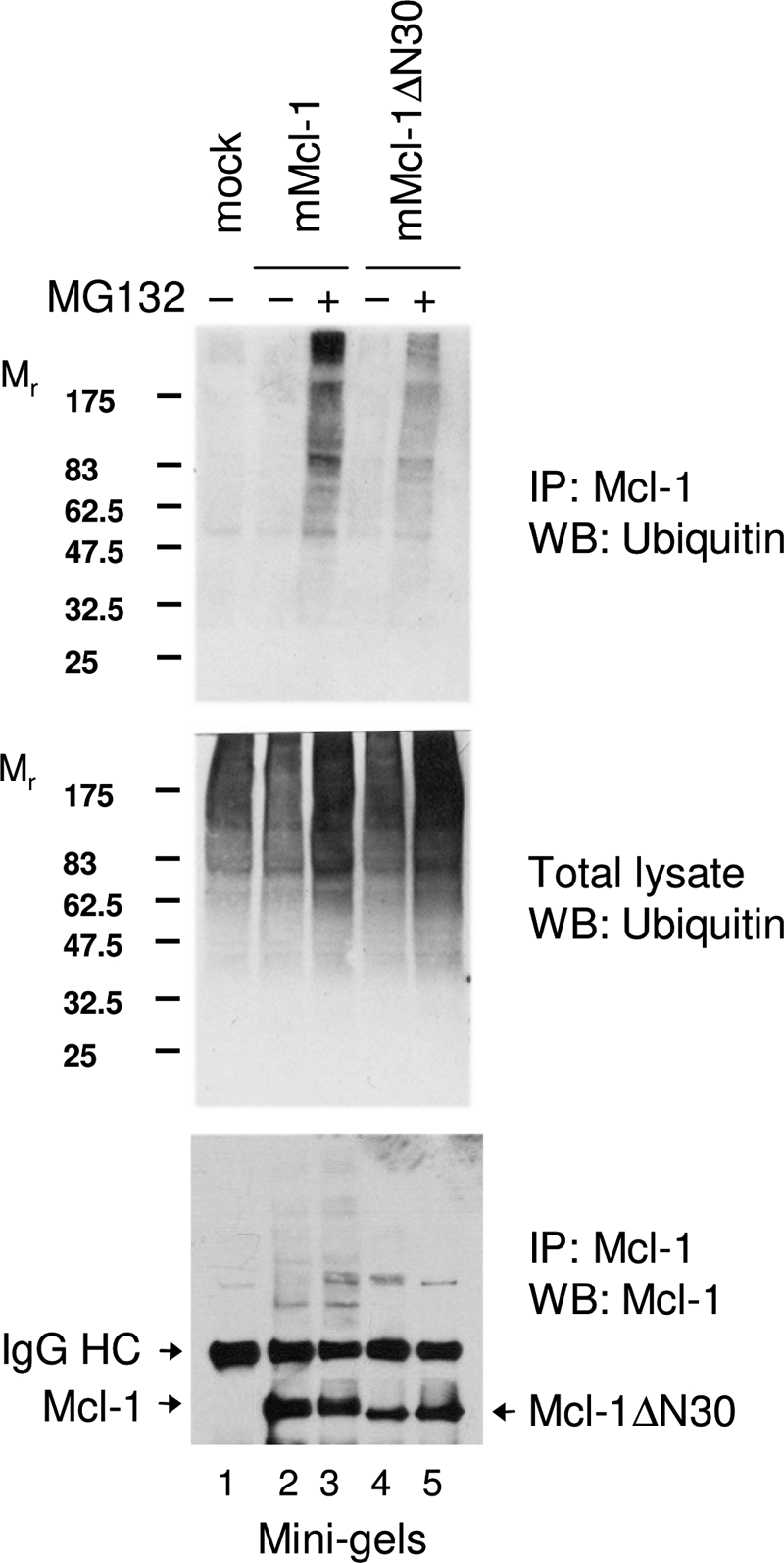
N terminus of Mcl-1 promotes ubiquitylation at steady state. HeLa cells were transfected with vectors expressing mouse Mcl-1 or mouse Mcl-1ΔN30. 48 h post-transfection, cells were treated with or without MG132 for 3 h. Cell lysates were prepared and subjected to immunoprecipitation (IP) using anti-mouse Mcl-1 antibody. Total cellular proteins or the immunoprecipitates were subjected to immunoblot with anti-ubiquitin or anti-mMcl-1 antibodies. Note: mini-gel electrophoresis was employed, accounting for the close migration of Mcl-1 and Mcl-1ΔN30. WB, Western blot.
Concluding Remarks
The extent of Mcl-1 turnover at the mitochondrial outer membrane depends on the relative frequency of its interaction with proteins that govern its degradation. One such key regulator that appears responsible for the rapid constitutive degradation of Mcl-1 is the E3 ligase Mule/Lasu1. Efficient degradation demands therefore that Mule can effectively compete with other Mcl-1 binding partners. Thus, for example, overexpression of the constitutive binding partner Bim results in elevated levels of full-length Mcl-1 (19). Here, we show that the interaction between Mcl-1 and Mule depends on the presence of the Mcl-1 N terminus, in addition to the Mule BH3 domain. Our results further demonstrate that Mcl-1 can be processed at its N terminus, thereby rendering Mcl-1ΔN unable to interact with Mule and resulting in relative stabilization of Mcl-1 compared with the hyper-rapid degradation that is characteristic of full-length Mcl-1. It remains to be determined if the N terminus of Mcl-1 likewise influences its interactions with other regulators of Mcl-1 stability. Several lines of evidence show that such processing occurs only after newly synthesized Mcl-1 reaches the mitochondrion and is dependent on its presence at the mitochondrion. Despite the fact that manipulations that interfere with inner membrane Δψ inhibit Mcl-1 processing, both full-length and processed Mcl-1 appear to reside integrated via their TM at the outer membrane. Future studies will be required to determine the precise details associated with the mechanism of this processing and whether it is associated with reversible import of the Mcl-1 N terminus into the organelle or on Δψ-dependent events that indirectly influence processing activity at the mitochondrial surface. By whatever mechanism, however, a processing pathway has evolved to allow Mcl-1 to escape rapid steady-state loss in response to antagonists of global protein synthesis. Enhanced mitochondrial processing of Mcl-1 to its more stable Mcl-1ΔN isoform could represent a control point for Mcl-1 stabilization and thus a potential mechanism for a cell survival advantage in the face of stress stimuli that influence global protein synthesis.
Supplementary Material
Acknowledgments
We thank Mark Watson for helpful discussions and recombinant FLAG-Mcl-1ΔTMHis6; Denis Paquette for assistance in vector constructions; Joseph Opferman for providing Mcl-1Δ/null MEFs; David Kwiatkowski for Tsc2−/−p53−/− MEFs, Anders Dydensborg for reagents; and Julie St-Pierre for advice.
This work was supported in part by research grants from the Canadian Institutes of Health Research (to G. C. S., J. P., J. C. Y., and L. P.), the National Cancer Institute of Canada (to G. C. S. and J. P.), and the National Research Council, Canada (to M. O. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4 and an additional reference.
- BH
- Bcl-2 homology
- TM
- transmembrane
- aa
- amino acid
- IASD
- 4-acetamido-4′-[(iodoacetyl)amino]stilbene-2,2′-disulfonic acid
- CHX
- cycloheximide
- m
- mouse
- h
- human
- IVT
- in vitro translated
- CCCP
- carbonyl cyanide 3-chlorophenylhydrazone
- MEF
- mouse embryo fibroblast.
REFERENCES
- 1. Breckenridge D. G., Germain M., Mathai J. P., Nguyen M., Shore G. C. (2003) Oncogene 22, 8608–8618 [DOI] [PubMed] [Google Scholar]
- 2. Danial N. N., Korsmeyer S. J. (2004) Cell 116, 205–219 [DOI] [PubMed] [Google Scholar]
- 3. Youle R. J., Strasser A. (2008) Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 4. Kozopas K. M., Yang T., Buchan H. L., Zhou P., Craig R. W. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 3516–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rinkenberger J. L., Horning S., Klocke B., Roth K., Korsmeyer S. J. (2000) Genes Dev. 14, 23–27 [PMC free article] [PubMed] [Google Scholar]
- 6. Opferman J. T., Iwasaki H., Ong C. C., Suh H., Mizuno S., Akashi K., Korsmeyer S. J. (2005) Science 307, 1101–1104 [DOI] [PubMed] [Google Scholar]
- 7. Opferman J. T., Letai A., Beard C., Sorcinelli M. D., Ong C. C., Korsmeyer S. J. (2003) Nature 426, 671–676 [DOI] [PubMed] [Google Scholar]
- 8. Cho-Vega J. H., Rassidakis G. Z., Admirand J. H., Oyarzo M., Ramalingam P., Paraguya A., McDonnell T. J., Amin H. M., Medeiros L. J. (2004) Hum. Pathol. 35, 1095–1100 [DOI] [PubMed] [Google Scholar]
- 9. Kitada S., Andersen J., Akar S., Zapata J. M., Takayama S., Krajewski S., Wang H. G., Zhang X., Bullrich F., Croce C. M., Rai K., Hines J., Reed J. C. (1998) Blood 91, 3379–3389 [PubMed] [Google Scholar]
- 10. Chung T. K., Cheung T. H., Lo W. K., Yim S. F., Yu M. Y., Krajewski S., Reed J. C., Wong Y. F. (2002) Cancer Lett. 180, 63–68 [DOI] [PubMed] [Google Scholar]
- 11. Miyamoto Y., Hosotani R., Wada M., Lee J. U., Koshiba T., Fujimoto K., Tsuji S., Nakajima S., Doi R., Kato M., Shimada Y., Imamura M. (1999) Oncology 56, 73–82 [DOI] [PubMed] [Google Scholar]
- 12. Sieghart W., Losert D., Strommer S., Cejka D., Schmid K., Rasoul-Rockenschaub S., Bodingbauer M., Crevenna R., Monia B. P., Peck-Radosavljevic M., Wacheck V. (2006) J. Hepatol. 44, 151–157 [DOI] [PubMed] [Google Scholar]
- 13. Craig R. W. (2002) Leukemia 16, 444–454 [DOI] [PubMed] [Google Scholar]
- 14. Opferman J. T. (2006) Cell Death Differ. 13, 1260–1262 [DOI] [PubMed] [Google Scholar]
- 15. Warr M. R., Shore G. C. (2008) Curr. Mol. Med. 8, 138–147 [DOI] [PubMed] [Google Scholar]
- 16. Cuconati A., Mukherjee C., Perez D., White E. (2003) Genes Dev. 17, 2922–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nijhawan D., Fang M., Traer E., Zhong Q., Gao W., Du F., Wang X. (2003) Genes Dev. 17, 1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhong Q., Gao W., Du F., Wang X. (2005) Cell 121, 1085–1095 [DOI] [PubMed] [Google Scholar]
- 19. Warr M. R., Acoca S., Liu Z., Germain M., Watson M., Blanchette M., Wing S. S., Shore G. C. (2005) FEBS Lett. 579, 5603–5608 [DOI] [PubMed] [Google Scholar]
- 20. Maurer U., Charvet C., Wagman A. S., Dejardin E., Green D. R. (2006) Mol. Cell 21, 749–760 [DOI] [PubMed] [Google Scholar]
- 21. Ding Q., He X., Hsu J. M., Xia W., Chen C. T., Li L. Y., Lee D. F., Liu J. C., Zhong Q., Wang X., Hung M. C. (2007) Mol. Cell. Biol. 27, 4006–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Inuzuka H., Shaik S., Onoyama I., Gao D., Tseng A., Maser R. S., Zhai B., Wan L., Gutierrez A., Lau A. W., Xiao Y., Christie A. L., Aster J., Settleman J., Gygi S. P., Kung A. L., Look T., Nakayama K. I., DePinho R. A., Wei W. (2011) Nature 471, 104–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stewart D. P., Koss B., Bathina M., Perciavalle R. M., Bisanz K., Opferman J. T. (2010) Mol. Cell. Biol. 30, 3099–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beroukhim R., Mermel C. H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J. S., Dobson J., Urashima M., Mc Henry K. T., Pinchback R. M., Ligon A. H., Cho Y. J., Haery L., Greulich H., Reich M., Winckler W., Lawrence M. S., Weir B. A., Tanaka K. E., Chiang D. Y., Bass A. J., Loo A., Hoffman C., Prensner J., Liefeld T., Gao Q., Yecies D., Signoretti S., Maher E., Kaye F. J., Sasaki H., Tepper J. E., Fletcher J. A., Tabernero J., Baselga J., Tsao M. S., Demichelis F., Rubin M. A., Janne P. A., Daly M. J., Nucera C., Levine R. L., Ebert B. L., Gabriel S., Rustgi A. K., Antonescu C. R., Ladanyi M., Letai A., Garraway L. A., Loda M., Beer D. G., True L. D., Okamoto A., Pomeroy S. L., Singer S., Golub T. R., Lander E. S., Getz G., Sellers W. R., Meyerson M. (2010) Nature 463, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schwickart M., Huang X., Lill J. R., Liu J., Ferrando R., French D. M., Maecker H., O'Rourke K., Bazan F., Eastham-Anderson J., Yue P., Dornan D., Huang D. C., Dixit V. M. (2010) Nature 463, 103–107 [DOI] [PubMed] [Google Scholar]
- 26. De Biasio A., Vrana J. A., Zhou P., Qian L., Bieszczad C. K., Braley K. E., Domina A. M., Weintraub S. J., Neveu J. M., Lane W. S., Craig R. W. (2007) J. Biol. Chem. 282, 23919–23936 [DOI] [PubMed] [Google Scholar]
- 27. Mills J. R., Hippo Y., Robert F., Chen S. M., Malina A., Lin C. J., Trojahn U., Wendel H. G., Charest A., Bronson R. T., Kogan S. C., Nadon R., Housman D. E., Lowe S. W., Pelletier J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10853–10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim P. K., Annis M. G., Dlugosz P. J., Leber B., Andrews D. W. (2004) Mol. Cell 14, 523–529 [DOI] [PubMed] [Google Scholar]
- 29. Nguyen M., Marcellus R. C., Roulston A., Watson M., Serfass L., Murthy Madiraju S. R., Goulet D., Viallet J., Bélec L., Billot X., Acoca S., Purisima E., Wiegmans A., Cluse L., Johnstone R. W., Beauparlant P., Shore G. C. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 19512–19517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Willis S. N., Chen L., Dewson G., Wei A., Naik E., Fletcher J. I., Adams J. M., Huang D. C. (2005) Genes Dev. 19, 1294–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang C. R., Yang-Yen H. F. (2010) FEBS Lett. 584, 3323–3330 [DOI] [PubMed] [Google Scholar]
- 32. Chou C. H., Lee R. S., Yang-Yen H. F. (2006) Mol. Biol. Cell 17, 3952–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neupert W., Herrmann J. M. (2007) Annu. Rev. Biochem. 76, 723–749 [DOI] [PubMed] [Google Scholar]
- 34. Goping I. S., Gross A., Lavoie J. N., Nguyen M., Jemmerson R., Roth K., Korsmeyer S. J., Shore G. C. (1998) J. Cell Biol. 143, 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Q., Gehring K. (2010) J. Biol. Chem. 285, 41202–41210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen L., Willis S. N., Wei A., Smith B. J., Fletcher J. I., Hinds M. G., Colman P. M., Day C. L., Adams J. M., Huang D. C. (2005) Mol. Cell 17, 393–403 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.