

Abstract
The dopamine D2 receptor (D2R) plays a crucial role in the regulation of diverse key physiological functions, including motor control, reward, learning, and memory. This receptor is present in vivo in two isoforms, D2L and D2S, generated from the same gene by alternative pre-mRNA splicing. Each isoform has a specific role in vivo, underlining the importance of a strict control of its synthesis, yet the molecular mechanism modulating alternative D2R pre-mRNA splicing has not been completely elucidated. Here, we identify heterogeneous nuclear ribonucleoprotein M (hnRNP M) as a key molecule controlling D2R splicing. We show that binding of hnRNP M to exon 6 inhibited the inclusion of this exon in the mRNA. Importantly, the splicing factor Nova-1 counteracted hnRNP M effects on D2R pre-mRNA splicing. Indeed, mutations of the putative Nova-1-binding site on exon 6 disrupted Nova-1 RNA assembly and diminished the inhibitory effect of Nova-1 on hnRNP M-dependent exon 6 exclusion. These results identify Nova-1 and hnRNP M as D2R pre-mRNA-binding proteins and show their antagonistic role in the alternative splicing of D2R pre-mRNA.
Keywords: Neurons, Neurotransmitter Receptors, RNA-binding Protein, RNA Splicing, RNA-Protein Interaction, Dopamine D2 Receptor (D2R), Nova-1, Alternative Pre-mRNA Splicing, hnRNP M
Introduction
Dopamine is a key regulator of mammalian central nervous system functions. Dysfunctions of the dopaminergic system are linked to neurological and neuropsychiatric disorders and to pituitary tumors (1). Dopamine action is mediated through activation of dopamine receptors. Among them, the dopamine D2 receptor (D2R)4 is a major target of pharmacological interventions for the treatment of Parkinson disease, schizophrenia, depression, and pituitary tumors. The generation of D2R knock-out mice has highlighted its key role in the control of motor functions (2), regulation of reward circuitries (3, 4), and pituitary physiology (5, 6). D2R also acts as the main dopaminergic autoreceptor (7–9). Two isoforms of D2R, long (D2L) and short (D2S), are produced from the same gene by alternative pre-mRNA splicing (10). D2L differs from D2S by an additional 29 amino acids encoded by exon 6, inserted within the third cytoplasmic loop of the receptor, the region interacting with G proteins. This insertion likely accounts for a differential interaction of D2L and D2S with G proteins (11), activation of distinct downstream signaling pathways (6, 12), and function (12, 13). Indeed, analyses of D2L knock-out mice suggest that D2L is involved mainly in the control of postsynaptic functions, whereas D2S is involved in the control of dopamine neuron firing and dopamine release (8, 13). Thus, control of the synthesis of D2L relative to D2S is critical for proper neuronal activities (14). However, the mechanisms controlling the production of D2L relative to D2S are not yet known.
Alternative pre-mRNA splicing is a powerful and versatile regulatory mechanism that produces protein diversity from a single gene, which contributes to the control of almost every cellular process (15). Tissue- or developmental stage-specific alternative RNA splicing including neuronal splicing can be explained by various combinations of ubiquitously expressed and tissue-specific splicing factors (16). For example, SRp30c enhances SMN (survival of motor neurons) exon 7 inclusion, and its association with exon 7 is mediated by a direct interaction with hTra2β1 (17). In addition, inclusion of the neuron-specific c-src N1 exon during alternative splicing is facilitated by the neuron-enriched KSRP protein (18).
In this study, we identify heterogeneous nuclear ribonucleoprotein M (hnRNP M) and neuro-oncological ventral antigen-1 (Nova-1) as splicing regulators that bind to D2R exon 6 and show their functional relevance to the molecular mechanism generating the D2L and D2S isoforms of the D2R gene. In transfected cells, hnRNP M enhanced D2R exon 6 excision, leading to D2S mRNA production, whereas Nova-1 antagonized it, leading to D2L mRNA production. When the binding sequence of Nova-1 was mutated, the inhibitory effect of Nova-1 on hnRNP M was decreased. These results demonstrate that hnRNP M and Nova-1 regulate alternative D2R pre-mRNA splicing in an antagonistic manner.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
All materials for cell culture were obtained from Invitrogen. Other chemicals, if not specified, were purchased from Sigma. Anti-His antibody was purchased from Cell Signaling Technology (Beverly, MA), anti-actin antibody from Chemicon, anti-Nova-1 antibody from Upstate Cell Signaling Solutions (Lake Placid, NY), anti-hnRNP M antibody from Novus Biologicals (Littleton, CO), and anti-FLAG M2 affinity gel from Sigma.
Oligonucleotides for PCR and Plasmid Constructions
To generate the D2R expression plasmid for the RT-PCR that determined the splicing efficiencies of D2R pre-mRNA, D2L cDNA that had been cloned into the eukaryotic expression vector pTL1, named pTL1-D2L (11), was digested with PstI and subsequently self-ligated. Next, the intron 5- and intron 6-containing minigene (pTL1-D2R) was inserted into PstI-digested pTL1-D2L between the XmaI and AflII sites. Then, it was digested with EcoRI and KpnI and ligated to pEGFP vector, generating pEGFP-D2R. The primers used for RT-PCR were as follows: D2R-up, 5′-CCTTCATCGTCACCCTGCTGG-3′; and D2R-dn, 5′-CTCCATTTCCAGCTCCTGAG-3′. The 87 bp of exon 6 were subdivided in two fragments by PCR amplification using four specific oligonucleotides. The two PCR products were cloned in pBS(+) that had been digested with SacI and KpnI. Template constructs for mutant riboprobes (E6-m1, -m2, -m3, -m4, and -m5) were cloned by PCR into the pGEM-T Easy vector (Promega, Madison, WI). D2R-m2 and D2R-m5 were generated by the site-directed mutagenesis method using PCR with oligomers. All oligomers used to construct plasmids are listed in Table 1.
TABLE 1.
Primer sequences for plasmid constructs
| Name | Primer sequence |
|---|---|
| T7-E6 | |
| Upstream | 5′-TAATACGACTCACTATAGGGGGCAACTGTACCCACCCT-3′ |
| Downstream | 5′-CATTCTCCGCCTGTTCA-3′ |
| E6-1 | |
| Upstream | 5′-ATGCGAGCTCGGCAACTGTACCCACCC-3′ |
| Downstream | 5′-ATGCGGTACCAACGGTGCAGAGTTTCA-3′ |
| E6-2 | |
| Upstream | 5′-ATGCGAGCTCATCATGAAGTCTAATGG-3′ |
| Downstream | 5′-ATGCGGTACCCATTCTCCGCCTGTTCA-3′ |
| T7-E6-m1 | |
| Upstream | 5′-TAATACGACTCACTATAGGGGGCAACTGTACCTTCCCTG-3′ |
| T7-E6-m2 | |
| Downstream | 5′-CATTCTCCGCCTGTTCACTGGGAAACTCCCATTAGACTTCAAAATAAC-3′ |
| T7-E6-m3 | |
| Downstream | 5′-CATTCTCCGCCTGTTCACAAGGAAA-3′ |
| T7-E6-m4 | |
| Upstream | 5′-TAATACGACTCACTATAGGGGGTTACTGTACCCACCCTG-3′ |
| T7-E6-m5 | |
| Upstream | 5′-TAATACGACTCACTATAGGGGGCAACTGTACCCACCCTGAGGATTTGAAAC-3′ |
| D2R-m2 | |
| Upstream | 5′-CTCCACAGGGCAACTGTACCTTCCCTGAGGACATGAAACTC-3′ |
| Downstream | 5′-GAGTTTCATGTCCTCAGGGAAGGTACAGTTGCCCTGTGGAG-3′ |
| D2R-m5 | |
| Upstream | 5′-CATGAAACTCTGCACCGTTATTTTGAAGTCTAATGGGAGTTTCC-3′ |
| Downstream | 5′-GGAAACTCCCATTAGACTTCAAAATAACGGTGCAGAGTTTCATG-3′ |
Cell Culture and Transient Transfection
COS-1 cells and NIH3T3 cells were maintained in DMEM supplemented with 4 mm glutamine, 1 mm sodium pyruvate, 100 units/ml penicillin/streptomycin, and 10% FBS; MMQ rat pituitary tumor cells were maintained in RPMI 1640 medium containing 10% horse serum under a humidifying atmosphere at 5% CO2 at 37 °C. NIH3T3 cells were plated into 6-well plates and grown to 60–80% confluence for 1 day. For transfection experiments, cells were washed twice with Dulbecco's PBS, and the medium was then changed to serum- and antibiotic-free DMEM before transfection. Plasmids for transfection experiments were purified using Qiagen columns according to the manufacturer's instructions and dissolved in 1 mm Tris (pH 8.0) and 0.1 mm EDTA. The cells were transfected with plasmid DNAs using Lipofectamine PLUS reagent (Invitrogen), and excess DNA complexes were washed away with Dulbecco's PBS the next day, after which regular medium was added. After 48 h of incubation in regular medium, cells were harvested. Lysates were subjected to RT-PCR and immunoprecipitation.
RT-PCR Analysis
Total cell RNA was prepared. To eliminate possible DNA contamination, 3 μg of RNA was further treated with 10 units of DNase I (Takara Bio Inc.) for 30 min at 37 °C. DNase-treated RNA was heated for 10 min at 75 °C, The RNA was reverse-transcribed using random hexamers, and the resulting cDNA was amplified by PCR.
UV Cross-linking Assay
RNA-protein binding reactions were performed in a reaction mixture with 0.4 mm ATP, 20 mm creatine phosphate, 3 mm MgCl2, 20 units of ribonuclease inhibitor, 5 μg of yeast tRNA, 32P-labeled RNA probe, and purified proteins for 30 min at 30 °C according to a previously described method with minor modifications (19). UV cross-linking was performed on ice, 4.5 cm away from a 1.2-J UV source (Stratagene, La Jolla, CA). Each sample was then incubated with 200 units of RNase T1 and 200 units of RNase A for 10 min at 37 °C. The resulting RNA-protein complexes were denatured and resolved by SDS-PAGE. Gels were dried and autoradiographed at −70 °C for 1 day.
EMSA
EMSAs were carried out as described previously (19). RNA-protein binding reactions were performed in a 30-μl reaction mixture containing 0.4 mm ATP, 20 mm creatine phosphate, 3 mm MgCl2, 20 units of ribonuclease inhibitor, 5 μg of yeast tRNA, 32P-labeled RNA probe, and the indicated GST-tagged recombinant protein for 30 min at 30 °C. In the case of supershift experiments, 200 ng of anti-Nova-1 antibody was added and incubated for another 30 min. After incubation, 6 μl of glycerol was added to each reaction mixture. Mixtures were immediately separated on a 5% nondenaturing polyacrylamide gel. Thereafter, the gel was dried and exposed to x-ray film (Fuji) at −70 °C for 1 day.
Immunoprecipitation
Cell pellets from 60-mm dishes of transfected NIH3T3 cells were suspended in 1 ml of lysis buffer (20 mm Tris-HCl (pH 7.4), 100 mm NaCl, 1.5 mm MgCl2, and 0.1% (v/v) Nonidet P-40) containing protease inhibitors. Lysates were incubated in the presence or absence of RNase A (125 μg/ml) for 10 min on ice and then incubated with 20 μl of anti-FLAG M2 affinity gel for 1 h at 4 °C. After centrifugation, the pelleted resin was washed three times with 1 ml of lysis buffer and resuspended in 30 ml of SDS lysis buffer (40 mm Tris-HCl (pH 6.8), 10% glycerol, and 1% SDS). After centrifugation, the supernatant was transferred to new tube and mixed with 2× SDS loading buffer (80 mm Tris-HCl (pH 6.8), 20% glycerol, 2% SDS, 4% β-mercaptoethanol and 0.02% bromphenol blue). Samples were boiled for 5 min, resolved by SDS-PAGE, and analyzed by immunoblotting.
Western Blot Analysis
For immunoblotting, proteins were resolved by 8% SDS-PAGE and transferred onto PVDF membranes (Millipore, Bedford, MA). After blocking with 10% nonfat milk in TBS (10 mm Tris (pH 7.5) and 150 mm NaCl) with 0.3% Tween 20 (TBS/Tween), primary antibody (anti-His at a final dilution of 1:2000, anti-β-actin at 1:5000, anti-Nova-1 at 1:2000, anti-FLAG at 1:5000, and anti-hnRNP M at 1:1000) was applied to the membrane and incubated overnight at 4 °C. Thereafter, blots were washed five times with TBS/Tween and incubated with horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature. After five washes with TBS/Tween, immunoreactive bands were visualized using SuperSignal West Pico chemiluminescent Substrate (Pierce) following the manufacturer's instructions.
Bimolecular Fluorescence Complementation and Immunocytochemistry
Bimolecular fluorescence complementation assays were performed as described previously (20). The partial sequences encoding amino acid residues 1–173 and 155–238 of the enhanced variant of yellow fluorescent protein (Venus) were fused with hnRNP M (VN-hnRNP M) and Nova-1 (VC-Nova-1), respectively. HeLa cells transfected with the indicated constructs were incubated at 37 °C for 24 h. For immunocytochemistry, cells were fixed with 3.75% paraformaldehyde in PBS, and anti-FLAG and anti-GFP antibodies were applied to visualize hnRNP M and Nova-1, respectively.
RESULTS
hnRNP M Directly Interacts with D2R Exon 6
To investigate cis-acting elements and trans-acting factors that regulate alternative D2R pre-mRNA splicing, we prepared two RNA probes by subdividing D2R exon 6 (E6) into two fragments (E6-1 and E6-2) (Fig. 1A). Each of these RNA probes or an unrelated sequence (polylinker sequence) was utilized in EMSA that employed nuclear extracts from the lactotroph cell line MMQ. MMQ expresses D2R, and as in most brain regions, D2L mRNA is more abundant than D2S mRNA (21). The electrophoretic mobilities of all three RNA probes were shifted; however, only E6-1 RNA-protein and E6-2 RNA-protein complexes were decreased by an unlabeled molar excess of each probe (Fig. 1B). These results demonstrated that trans-acting factor(s) that can bind D2R E6 RNA specifically exist. To identify protein(s) that bind to D2R E6 RNA, we prepared nuclear extracts from HeLa cells, followed by fractionation by ammonium sulfate precipitation. EMSA was performed with four different fractions (20, 40, 60, and 75% ammonium sulfate concentrations). An E6-1 RNA-protein complex was found in the 60% ammonium sulfate fraction, whereas E6-2 associated with proteins in the 40% fraction (data not shown). Proteins contained in the 60 and 40% fractions were further purified by affinity chromatography using SP-Sepharose Fast Flow and then tested by EMSA using the E6-1 or E6-2 RNA probe. We found an E6-1 RNA-protein complex in the 80 mm NaCl fraction, whereas E6-2 failed to form a complex in any fraction (data not shown). The 60% ammonium sulfate SP fraction was then separated on by 10% SDS-PAGE. This procedure identified a protein of ∼75 kDa in the 80 mm NaCl SP fraction that was absent in other SP fractions (data not shown). These experiments were repeated several times with the same result. The identity of the 75-kDa protein was obtained by MALDI-TOF MS, which identified hnRNP M as a candidate protein that interacts with E6-1 RNA. hnRNP M was identified by nine unique peptides (p < 0.05) (supplemental Fig. S1).
FIGURE 1.

Identification of D2R exon 6-interacting protein. A, schematic diagram of E6-1 and E6-2 (upper) and sequences of E6-1 and E6-2 (lower). B, EMSA using MMQ nuclear extracts was performed with radiolabeled E6-1, E6-2, or a linker RNA together with an unlabeled molar excess of E6-1, E6-2, or linker RNA (Comp). Arrowheads indicate the interacting complex with each probe. C, COS-1 cells were transiently transfected with a His-tagged hnRNP M expression vector. His-tagged hnRNP M protein was purified using His-binding resin. UV cross-linking assays using purified His-tagged hnRNP M protein were performed with radiolabeled E6-1 and E6-2 riboprobes (upper panel). Expression of transfected His-tagged hnRNP M was confirmed by immunoblotting (IB) using anti-His antibody (lower panel).
To confirm the interaction between hnRNP M and E6-1, we purified His6-tagged recombinant hnRNP M protein from COS-1 cells transfected with pTL1-His-hnRNP M plasmid, and this protein was used in UV cross-linking assays. As shown in Fig. 1C, hnRNP M interacted with the E6-1 RNA sequence directly, confirming that hnRNP M recognizes a specific cis-acting element(s) that is present within the proximal region of D2R exon 6. Transfections of mutants generated by substitution of partial regions of D2R exon 6 (supplemental Fig. S2A) showed that the distal region of E6-1 (substituted region of the D2R-B mutant) is crucial for D2S mRNA production (supplemental Fig. S2, A and B). Importantly, transfection of hnRNP M was inefficient in increasing the D2S/D2L mRNA ratio when this region was mutated (supplemental Fig. S2C).
hnRNP M Enhances D2S mRNA Production
To assess the role of hnRNP M in the alternative splicing of D2R pre-mRNA, increasing amounts of the hnRNP M expression vector were cotransfected with a D2R minigene in cultured cells, and the resulting D2S/D2L mRNA ratio was calculated by RT-PCR. As shown in Fig. 2, hnRNP M overexpression increased the D2S/D2L mRNA ratio in an hnRNP M dose-dependent manner. The specific role of hnRNP M overexpression in D2S mRNA production was further supported by results obtained by cotransfection of an antisense hnRNP M expression vector (Fig. 2). On the contrary, cotransfection of the D2R minigene with 9G8 or ASF/SF2, two members of the SR protein family involved in alternative RNA splicing mechanisms, did not influence the D2S/D2L ratio (supplemental Fig. S3A). To test the specificity of hnRNP M in promoting D2S mRNA production, we tested its ability in the splicing of lymphotoxin α pre-mRNA. These experiments showed that although D2S mRNA production was enhanced in the presence of hnRNP M, the RNA splicing of the lymphotoxin α minigene was not (supplemental Fig. S3B).
FIGURE 2.

hnRNP M enhances exon 6 exclusion. His-tagged hnRNP M was overexpressed in the presence or absence of an antisense hnRNP M-expressing vector (AS-hnRNP M) in NIH3T3 cells together with the pEGFP-D2R minigene. The ratio of D2S to D2L mRNA was examined by RT-PCR. The arrow and arrowhead indicate D2L and D2S mRNA PCR products, respectively. Error bars represent the mean ± S.E. (n = 3). *, p < 0.05; **, p < 0.01 versus mock-transfected cells. HPRT, hypoxanthine-guanine phosphoribosyltransferase.
Nova-1 Protein Specifically Binds to D2R Exon 6
Alternative splicing, to include or exclude a cassette exon, is regulated by dynamic interactions of particular splicing factors (22). Because region-specific mutations within D2R exon 6 resulted in both increased and decreased ratios of D2S/D2L mRNA expression (supplemental Fig. S2), we examined the possibility that there might be other proteins that bind to this exon and that affect the mRNA ratio. To screen for this unidentified protein(s), in silico analyses were performed on the mouse D2R exon 6 sequences to identify putative splicing factor-binding sites. Indeed, in this region, we found YCAY element-like sequences (Fig. 3A), which are known as Nova-1-binding sites (23). To validate Nova-1 as a potential splicing factor of exon 6 of the D2R pre-mRNA, we performed UV cross-linking assays and EMSAs. These experiments were done using purified GST-tagged recombinant Nova-1 protein and a radioactive exon 6 riboprobe (Fig. 3, B and C). These assays revealed that Nova-1 bound directly to exon 6; in agreement with this, anti-Nova-1 antibodies evoked a supershift in EMSA (Fig. 3C).
FIGURE 3.

Nova-1 protein interacts with exon 6 of D2R. A, sequence of D2R exon 6. The putative Nova-1-binding site is underlined. B, UV cross-linking using GST-tagged purified Nova-1 protein (300 nm in a 15-μl reaction mixture) was performed with radiolabeled D2R exon 6 (E6) RNA probe. C, supershift experiments were carried out in the presence of anti-Nova antibody (200 ng). Arrows indicate the specific binding of Nova-1 to exon 6, and arrowheads indicate the super-shifted tertiary complexes.
To determine the sequences within exon 6 that bind Nova-1, we converted several CA dinucleotides to UU (m1, m2, m3, m4, and m5) (Fig. 4A). The resulting mutated probes were used for binding assays with the Nova-1 protein. As shown in Fig. 4B, only the m5 mutant failed to form an RNA-protein complex, whereas the other mutated probes were electrophoretically shifted in the presence of the Nova-1 protein. These results are consistent with data indicating that Nova-1 binds directly to the YCAY tetramer (24). Moreover, this sequence is highly conserved among mammalian species, including mouse, rat, human, chimpanzee, dog, and cow (supplemental Fig. S4). To determine the splicing efficiencies of transcripts generated from the D2R mutants that contained the m2 or m5 mutation (D2R-m2 or D2R-m5), we performed RT-PCR using total RNA from transfected cells. We observed that the m5 mutation increased the D2S/D2L mRNA ratio compared with the corresponding wild-type sequence, whereas the m2 mutation did not (Fig. 4C). These results indicate that Nova-1 recognizes UCAU sequences within the m5 region of D2R exon 6 and critically regulates D2L mRNA formation.
FIGURE 4.

Nova-1 protein recognizes a specific sequence of D2R exon 6. A, sequence of D2R exon 6. Mutated sequences are underlined, and each “CA” was changed to “TT” in each mutant DNA. B, EMSA using Nova-1 (300 nm) was performed with various point mutant exon 6 RNA probes (m1, m2, m3, m4, and m5). The arrow indicates the specific binding of Nova-1 to RNA probes. C, the pEGFP-D2R-WT, pEGFP-D2R-m2, or pEGFP-D2R-m5 minigene was introduced into NIH3T3 cells. The D2S/D2L mRNA ratio was examined by RT-PCR. Error bars represent the mean ± S.E. (n = 3). *, p < 0.05 versus WT. HPRT, hypoxanthine-guanine phosphoribosyltransferase.
Nova-1 Blocks the Effect of hnRNP M on D2S mRNA Production via Exon 6 Binding
To investigate whether Nova-1 can affect the hnRNP M-mediated regulation of alternative D2R pre-mRNA splicing, we transiently transfected NIH3T3 cells with hnRNP M and/or Nova-1 expression vectors together with the D2R minigene. Interestingly, although the D2S/D2L mRNA ratio was increased by hnRNP M in a dose-dependent manner, as expected, this effect was dose-dependently offset by Nova-1 expression (Fig. 5). Importantly, these same effects were also observed on the endogenous D2S/D2L mRNA ratio, as assessed by transfecting MMQ cells with hnRNP M and/or Nova-1 expression vectors. Also in this case, the D2S/D2L mRNA ratio was increased by hnRNP M expression and decreased by Nova-1 expression in a dose-dependent manner (supplemental Fig. S5).
FIGURE 5.

Nova-1 and hnRNP M regulate alternative D2R pre-mRNA splicing antagonistically. NIH3T3 cells were transiently transfected with His-tagged hnRNP M expression plasmid and/or FLAG-tagged Nova-1 expression plasmid. Western blotting (WB) was performed using anti-hnRNP M, anti-FLAG, or anti-β-actin antibody to detect hnRNP M, Nova-1, or β-actin, respectively (upper panels). The ratio of D2S to D2L mRNA was analyzed by RT-PCR (lower panels). Error bars represent the mean ± S.E. (n = 3). *, p < 0.05; **, p < 0.01 versus mock-transfected cells. HPRT, hypoxanthine-guanine phosphoribosyltransferase.
Next, we assessed whether binding of Nova-1 to exon 6 is required to elicit its inhibitory effect on the role of hnRNP M in D2R pre-mRNA splicing. NIH3T3 cells were cotransfected with hnRNP M and/or Nova-1 expression vectors and the D2R-WT, -m2, or -m5 minigene. RT-PCR analyses showed that the inhibitory effect of Nova-1 on hnRNP M activity in alternative D2R pre-mRNA splicing was blunted with the D2R-m5 mutation (Fig. 6, right panel), in agreement with the impaired Nova-1 binding of this mutant. Conversely, transfection of the D2R-m2 minigene provided results similar to those obtained using the wild-type minigene (Fig. 6, middle panel). These results indicate that Nova-1 counteracts hnRNP M activity in D2S mRNA production via direct binding to exon 6.
FIGURE 6.

Nova-1 regulates alternative D2R pre-mRNA splicing via binding to D2R exon 6 directly. D2R-WT and mutant minigenes that express D2R-m2 or D2R-m5 were introduced together with hnRNP M and/or Nova-1 protein expression plasmids into NIH3T3 cells. Arrows and arrowheads indicate D2L and D2S mRNAs, respectively. The D2S/D2L mRNA ratio was analyzed by RT-PCR. Error bars represent the mean ± S.E. (n = 3). *, p < 0.05; **, p < 0.01 versus hnRNP M-transfected cells. HPRT, hypoxanthine-guanine phosphoribosyltransferase.
hnRNP M and Nova-1 Physically Interact with Each Other
Because both hnRNP M and Nova-1 proteins bind to D2R exon 6, we used EMSA to investigate the effect and pattern of binding of both proteins to this exon. In extracts containing both proteins, the band amount of the complex formed by Nova-1 and the exon 6 riboprobe (E6) was drastically decreased, and we observed the appearance of supershifted bands that migrated above this complex (Fig. 7A). To rule out the possibility that hnRNP M may affect the binding affinity of Nova-1 to exon 6, we performed UV cross-linking assays. Our data showed that increasing the amount of hnRNP M protein did not change the intensity of Nova-1 protein binding to E6 (Fig. 7B). Next, we examined whether hnRNP M interacts with Nova-1 by performing co-immunoprecipitations after transient transfection of NIH3T3 cells with hnRNP M and/or Nova-1 expression vectors. Cell extracts were immunoprecipitated using anti-FLAG resin in the presence or absence of RNase A, and the interactions were analyzed by immunoblotting using anti-hnRNP M or anti-FLAG antibody. As shown in Fig. 7C, Nova-1 interacted with hnRNP M in an RNase A-insensitive manner. Interactions between hnRNP M and Nova-1 were further analyzed by the bimolecular fluorescence complementation assay. Coexpression of the N terminus of Venus (VN)-tagged hnRNP M and the C terminus of Venus (VC)-tagged Nova-1 exhibited a yellow fluorescence signal, showing an interaction between these proteins (Fig. 7D). These results suggest that hnRNP M and Nova-1 interact physically and that this interaction does not affect the binding affinity of Nova-1 for exon 6.
FIGURE 7.

Nova-1 interacts with hnRNP M. A, EMSA using Nova-1 (300 nm) and/or hnRNP M (300 nm) was carried out using radiolabeled E6. The arrow and arrowhead indicate specific binding of Nova-1 and hnRNP M to E6, respectively. B, UV cross-linking assays using Nova-1 (150 nm) and various amounts of hnRNP M (150 nm in the third lane and 75, 150, and 300 nm in the fourth, fifth, and sixth lanes, respectively) were performed on the D2R exon 6 probe. Cross-linked proteins were analyzed by SDS-PAGE, followed by autoradiography (upper panel). The band intensity of Nova-1 or hnRNP M was quantified using ImageQuant software (lower panel). Error bars represent the mean ± S.E. (n = 3). C, NIH3T3 cells were transiently transfected with FLAG-tagged Nova-1 and/or His-tagged hnRNP M. Total cell lysate was immunoprecipitated (IP) using anti-FLAG M2 affinity gel in the presence (+) or absence (−) of RNase A. Western blotting was performed with anti-hnRNP M, anti-FLAG, or anti-β-actin antibody to detect His-hnRNP M, FLAG-Nova-1, or β-actin, respectively (upper panels). GAPDH mRNA was analyzed using RT-PCR to demonstrate that RNase A digestion was complete (lower panel). D, HeLa cells were transiently transfected with VN-hnRNP M and VC-Nova-1. Twenty-four hours after transfection, the cells were fixed with paraformaldehyde; stained with anti-FLAG or anti-GFP antibody to visualize hnRNP M or Nova-1, respectively; and observed by confocal microscopy. Representative confocal images of cells are shown. BiFC, bimolecular fluorescence complementation; ICC, immunocytochemistry.
DISCUSSION
Dopamine plays a crucial role in the regulation of a variety of physiological functions. Among dopamine receptors, D2R has important functions in both dopamine signaling and dopaminergic system homeostasis (10). This receptor exists in two isoforms, D2L and D2S, which are produced by alternative pre-mRNA splicing. Importantly, splicing of D2R is a highly conserved feature present in all mammals. Studies performed in vitro and most importantly on genetically engineered animals have shown that D2L and D2S signaling differentially affects physiological responses, and therefore, these isoforms do not have redundant functions. An altered D2S/D2L ratio has recently been found in schizophrenic patients (14), and importantly, most antipsychotics have D2R as a major target. Thus, control of alternative D2R pre-mRNA splicing might have major consequences on dopamine-mediated responses. D2L plays an important role in postsynaptic activity and striatal signaling (12, 13), whereas D2S appears to have major autoreceptor function that modulates dopamine release at presynaptic sites (9, 13). This shows that each D2R isoform has distinct functions in vivo. However, the molecular regulatory mechanism of alternative D2R pre-mRNA splicing is not fully elucidated.
In this study, we have revealed that alternative D2R pre-mRNA splicing is controlled by two splicing regulators, hnRNP M and Nova-1. hnRNP M recognizes specific elements within D2R exon 6, and it enhances exon 6 excision during alternative pre-mRNA splicing. In contrast, Nova-1 antagonizes this effect via direct binding to both exon 6 and hnRNP M.
The identification and characterization of the factors binding to the cis-acting regulatory elements are important in unraveling mechanisms that modulate gene expression by affecting alternative pre-mRNA splicing. We have identified hnRNP M as a D2R exon 6-binding protein using EMSA, followed by MALDI-TOF MS and UV cross-linking. hnRNP M was first identified in 1993 by Swanson and co-workers (25). hnRNP M is a member of a family of RNA-binding proteins; it contains three RNA recognition motifs. hnRNPs bind to RNA via RNA recognition motifs and influence diverse cellular events such as transcription; telomere maintenance; RNA splicing; polyadenylation; and mRNA localization, translation, and decay (26, 27). hnRNPs are known to be negative splicing regulators by directly antagonizing the recognition of splice sites or interfering with the binding of splicing regulators to splicing enhancer sequences (28). More recently, Drosophila hnRNP M, HRP59, has been shown to bind to its own mRNA, inhibiting HRP59-mediated exon 3 inclusion (29, 30). In addition, hnRNP M binds to the intronic splicing enhancer/intronic splicing silencer-3 of FGFR2 (FGF receptor 2) pre-mRNA and promotes FGFR2 exon IIIc skipping and also enhances the excision of preprotachykinin exon 4 (31). hnRNP M affects both 5′- and 3′-alternative splice site choices and activates C4 exon skipping of the calcitonin/dhfr pre-mRNA reporter gene (32). In this study, we have shown that hnRNP M directly binds to D2R exon 6 and inhibits its inclusion, thus favoring D2S mRNA production in a dose-dependent manner (Figs. 1 and 2). This observation is consistent with previous studies implicating hnRNP M as a negative regulator of alternative pre-mRNA splicing.
Although hnRNP M enhances D2S mRNA production, D2L mRNA is dominant in most brain regions. Thus, positive splicing regulators certainly exist that enhance D2R exon 6 inclusion during the D2R pre-mRNA splicing process. Recently, it was reported that PTBP1 (polypyrimidine tract-binding protein 1) enhances exon 6 inclusion (33), but there is no evidence that it binds to pre-mRNA of D2R directly. We analyzed exon 6 sequences using a bioinformatics approach. Remarkably, this search revealed the presence of a putative binding site for brain-enriched Nova-1. Indeed, Nova-1 directly binds to UCAU sequences within D2R exon 6 RNA (Fig. 3). Nova-1 is known as a splicing factor that regulates the inclusion or exclusion of exons depending on the position of Nova binding relative to splice sites (34). When Nova binds to the 3′-region of the cassette exon, it promotes inclusion of this exon (34). Consistent with this, overexpression of Nova-1 dose-dependently increases D2R exon 6 inclusion likely by blocking hnRNP M function (Fig. 6). The antagonistic action of Nova-1 is very likely mediated by its binding to D2R exon 6. Indeed, mutation of a UCAU Nova-1-binding site causes an increase in the D2S/D2L mRNA ratio (Fig. 4C). Thus, it appears that Nova-1 enhances D2L mRNA production via direct binding to a UCAU motif on D2R exon 6, and converting UCAU to UUUU abolishes the Nova-1 effect on D2R RNA. We have thus identified two splicing proteins that, by interacting with each other, contribute to the regulation of alternative D2R pre-mRNA splicing.
Several possible mechanisms might be evoked to explain how D2R pre-mRNA splicing can be regulated by an antagonistic interaction between hnRNP M and Nova-1. One is that hnRNP M competes with the ability of Nova-1 to bind D2R exon 6. However, hnRNP M may bind to the upstream region of exon 6, and it appears to supershift a complex between Nova-1 and RNA (Figs. 1 and 7A and supplemental Fig. S2). Moreover, hnRNP M does not interfere with the ability of Nova-1 to bind to exon 6 (Fig. 7B). A Drosophila hnRNP M homolog, Rumpelstiltskin (Rump), is known to specifically bind to a CGUU sequence (35), and interestingly, D2R exon 6 contains a CGUU motif immediately upstream of the UCAU Nova-1-binding site. This sequence is included in E6-1 and in the region substituted in the D2R-B mutant. This possibility is in line with our data suggesting that hnRNP M and Nova-1 may bind to adjacent sequence within exon 6, rather than competing for the same site.
We favor the possibility that, by binding to exon 6, Nova-1 blocks the binding of hnRNP M to it. Indeed, disruption of Nova-1 binding to exon 6 decreases the inhibitory effect on hnRNP M activity during D2R pre-mRNA splicing (Fig. 7). This is the case for the alternative pre-mRNA splicing of inhibitory glycine receptor α2 exons 3A and 3B, in which Nova-1 binding to an intron that is situated near exon 3A enhances the inclusion of exon 3A rather than exon 3B by a mechanism that is antagonized by brain-enriched polypyrimidine tract-binding protein. Moreover, the latter protein interacts with Nova-1 and binds glycine receptor α2 RNA at a position that is situated 90 nucleotides from the Nova-1-binding site (36). However, blocking Nova-1 assembly on D2R exon 6 by the m5 point mutation does not completely abolish the effect of Nova-1 on hnRNP M function in the process of D2R pre-mRNA splicing (Fig. 6). Thus, we cannot exclude the possibility that other Nova-1 recognition sites exist within D2R pre-mRNA or that Nova-1 may inhibit the function of hnRNP M by direct interaction. Because Nova-1 interacts with hnRNP M RNA-independently, it may block the binding of hnRNP M to exon 6 both by RNA binding and by protein-protein interaction. The simplest model consistent with these data is that Nova-1 binds D2R exon 6 at a site that is adjacent to the hnRNP M-binding motif so as to mediate an antagonistic effect on alternative D2R pre-mRNA splicing via protein-protein interactions (Fig. 8). An additional finding in support of the possible role of Nova-1 in the splicing mechanism regulating the D2R gene is that Nova-1 has been localized in inhibitory neurons. Furthermore, Nova-1-dependent mechanisms have been reported for the localization of glycine receptor α2 and G-protein-activated inwardly rectifying potassium channel subunit 2 (GIRK2) mRNAs in dendrites (37). Interestingly, striatal medium spiny neurons, where D2R is mainly expressed, are GABAergic, and D2R is functionally linked to GIRK2 channel activity. Thus, we speculate that D2R mRNA might be processed in a similar way as glycine receptor α2 and GIRK2.
FIGURE 8.
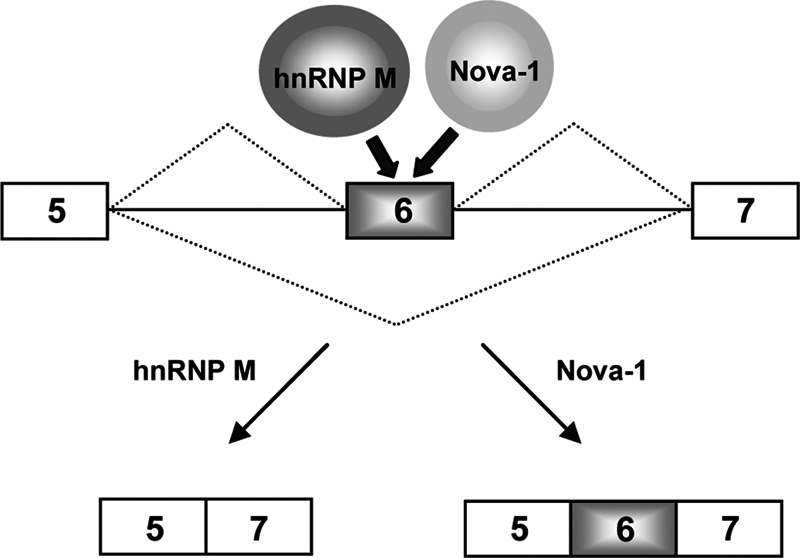
Schematic model of the molecular regulatory mechanisms for the alternative pre-mRNA splicing of D2R by Nova-1 and hnRNP M. Nova-1 and hnRNP M regulate alternative D2R pre-mRNA splicing antagonistically. Both Nova-1 and hnRNP M specifically interact with D2R exon 6, but hnRNP M inhibits the binding of Nova-1 to D2R exon 6. Nova-1 antagonizes the exon 6 exclusion activity of hnRNP M and enhances exon 6 inclusion.
In conclusion, hnRNP M promotes D2R exon 6 excision, leading to production of a D2S mRNA variant during D2R pre-mRNA splicing, whereas Nova-1 protein abolishes hnRNP M function and enhances D2L mRNA expression via RNA-independent interaction with hnRNP M and/or specific binding to D2R RNA. An increased understanding of how alternative D2R pre-mRNA splicing is regulated by the antagonistic functions of hnRNP M and Nova-1 may provide novel insight into key mechanisms that regulate the ratio of D2L and D2S isoforms.
Supplementary Material
Acknowledgments
We thank Drs. Stevenin, Gattoni, Fuchs, and Argentini for materials and discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant DA 024689-02 from the National Institute on Drug Abuse (to E. B.). This work was also supported by a grant from the Brain Research Center of the 21st Century Frontier Research Program in Neuroscience funded by the Ministry of Science and Technology, Republic of Korea (to K. K.), INSERM (to E. B.), and Brain Korea 21 research fellowships from the Korea Ministry of Education and Human Resources (to E. P., J. L., I. K., S. M. B., and G. H. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental “Experimental Procedures” and Figs. S1–S5.
- D2R
- dopamine D2 receptor
- hnRNP M
- heterogeneous nuclear ribonucleoprotein M.
REFERENCES
- 1. Missale C., Nash S. R., Robinson S. W., Jaber M., Caron M. G. (1998) Physiol. Rev. 78, 189–225 [DOI] [PubMed] [Google Scholar]
- 2. Baik J. H., Picetti R., Saiardi A., Thiriet G., Dierich A., Depaulis A., Le Meur M., Borrelli E. (1995) Nature 377, 424–428 [DOI] [PubMed] [Google Scholar]
- 3. Maldonado R., Saiardi A., Valverde O., Samad T. A., Roques B. P., Borrelli E. (1997) Nature 388, 586–589 [DOI] [PubMed] [Google Scholar]
- 4. Welter M., Vallone D., Samad T. A., Meziane H., Usiello A., Borrelli E. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 6840–6845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saiardi A., Bozzi Y., Baik J. H., Borrelli E. (1997) Neuron 19, 115–126 [DOI] [PubMed] [Google Scholar]
- 6. Iaccarino C., Samad T. A., Mathis C., Kercret H., Picetti R., Borrelli E. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 14530–14535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mercuri N. B., Saiardi A., Bonci A., Picetti R., Calabresi P., Bernardi G., Borrelli E. (1997) Neuroscience 79, 323–327 [DOI] [PubMed] [Google Scholar]
- 8. Rouge-Pont F., Usiello A., Benoit-Marand M., Gonon F., Piazza P. V., Borrelli E. (2002) J. Neurosci. 22, 3293–3301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Mei C., Ramos M., Iitaka C., Borrelli E. (2009) Curr. Opin. Pharmacol. 9, 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tirotta E., De Mei C., Litaka C., Ramos M., Holmes D., Borrelli E. (2010) in The Dopamine Receptors (Neve K. A. ed) 2nd Ed., pp. 303–322, Humana Press, New York [Google Scholar]
- 11. Guiramand J., Montmayeur J. P., Ceraline J., Bhatia M., Borrelli E. (1995) J. Biol. Chem. 270, 7354–7358 [DOI] [PubMed] [Google Scholar]
- 12. Lindgren N., Usiello A., Goiny M., Haycock J., Erbs E., Greengard P., Hokfelt T., Borrelli E., Fisone G. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 4305–4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Usiello A., Baik J. H., Rougé-Pont F., Picetti R., Dierich A., LeMeur M., Piazza P. V., Borrelli E. (2000) Nature 408, 199–203 [DOI] [PubMed] [Google Scholar]
- 14. Bertolino A., Fazio L., Di Giorgio A., Blasi G., Romano R., Taurisano P., Caforio G., Sinibaldi L., Ursini G., Popolizio T., Tirotta E., Papp A., Dallapiccola B., Borrelli E., Sadee W. (2009) J. Neurosci. 29, 1224–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lopez A. J. (1998) Annu. Rev. Genet 32, 279–305 [DOI] [PubMed] [Google Scholar]
- 16. Cáceres J. F., Stamm S., Helfman D. M., Krainer A. R. (1994) Science 265, 1706–1709 [DOI] [PubMed] [Google Scholar]
- 17. Young P. J., DiDonato C. J., Hu D., Kothary R., Androphy E. J., Lorson C. L. (2002) Hum. Mol. Genet 11, 577–587 [DOI] [PubMed] [Google Scholar]
- 18. Min H., Turck C. W., Nikolic J. M., Black D. L. (1997) Genes Dev. 11, 1023–1036 [DOI] [PubMed] [Google Scholar]
- 19. Seong J. Y., Han J., Park S., Wuttke W., Jarry H., Kim K. (2002) Mol. Endocrinol. 16, 2426–2438 [DOI] [PubMed] [Google Scholar]
- 20. Hu C. D., Kerppola T. K. (2003) Nat. Biotechnol. 21, 539–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guivarc'h D., Vincent J. D., Vernier P. (1998) Endocrinology 139, 4213–4221 [DOI] [PubMed] [Google Scholar]
- 22. Licatalosi D. D., Darnell R. B. (2006) Neuron 52, 93–101 [DOI] [PubMed] [Google Scholar]
- 23. Dredge B. K., Darnell R. B. (2003) Mol. Cell. Biol. 23, 4687–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ule J., Jensen K. B., Ruggiu M., Mele A., Ule A., Darnell R. B. (2003) Science 302, 1212–1215 [DOI] [PubMed] [Google Scholar]
- 25. Datar K. V., Dreyfuss G., Swanson M. S. (1993) Nucleic Acids Res. 21, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dreyfuss G., Kim V. N., Kataoka N. (2002) Nat. Rev. Mol. Cell Biol. 3, 195–205 [DOI] [PubMed] [Google Scholar]
- 27. Singh R., Valcárcel J. (2005) Nat. Struct. Mol. Biol. 12, 645–653 [DOI] [PubMed] [Google Scholar]
- 28. Martinez-Contreras R., Cloutier P., Shkreta L., Fisette J. F., Revil T., Chabot B. (2007) Adv. Exp. Med. Biol. 623, 123–147 [DOI] [PubMed] [Google Scholar]
- 29. Hase M. E., Yalamanchili P., Visa N. (2006) J. Biol. Chem. 281, 39135–39141 [DOI] [PubMed] [Google Scholar]
- 30. Kiesler E., Hase M. E., Brodin D., Visa N. (2005) J. Cell Biol. 168, 1013–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hovhannisyan R. H., Carstens R. P. (2007) J. Biol. Chem. 282, 36265–36274 [DOI] [PubMed] [Google Scholar]
- 32. Llères D., Denegri M., Biggiogera M., Ajuh P., Lamond A. I. (2010) EMBO Rep. 11, 445–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sasabe T., Futai E., Ishiura S. (2011) J. Neurochem. 116, 76–81 [DOI] [PubMed] [Google Scholar]
- 34. Ule J., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B. J., Darnell R. B. (2006) Nature 444, 580–586 [DOI] [PubMed] [Google Scholar]
- 35. Jain R. A., Gavis E. R. (2008) Development 135, 973–982 [DOI] [PubMed] [Google Scholar]
- 36. Polydorides A. D., Okano H. J., Yang Y. Y., Stefani G., Darnell R. B. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 6350–6355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Racca C., Gardiol A., Eom T., Ule J., Triller A., Darnell R. B. (2010) Front. Neural Circuits 4, 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.