

Abstract
Presenilin (PS), a causative molecule of familial Alzheimer disease, acts as a crucial component of the γ-secretase complex, which is required to cleave type I transmembrane proteins such as amyloid precursor protein and Notch. However, it also functions through γ-secretase-independent pathways. Recent reports suggested that PS could regulate the expression level of cell surface receptors, including the PDGF and EGF receptors, followed by modulating their downstream pathways via γ-secretase-independent mechanisms. The main purpose of this study was to clarify the effect of PS on expression of the insulin receptor (IR) as well as on insulin signaling. Here, we demonstrate that PS inhibited IR transcription and reduced IR expression, and this was followed by down-regulation of insulin signaling. Moreover, we suggest that neither γ-secretase activity nor Wnt/β-catenin signaling can reduce the expression of IR, but a PS-mediated increase in the intracellular Ca2+ level can be associated with it. These results clearly indicate that PS can functionally regulate insulin signaling by controlling IR expression.
Keywords: Alzheimer Disease, Calcium Channels, Glucose Metabolism, Insulin, Presenilin, γ-Secretase, Insulin Receptor
Introduction
Presenilin (PS)2 has been identified as a causative gene of familial Alzheimer disease (AD). Mutations in two homologous PS genes, presenilin 1 (PS1) and presenilin 2 (PS2), account for the vast majority of early onset familial AD (FAD). Although various PS functions have been reported so far, the most intensively examined is the one associated with proteolysis, in other words, the function as the catalytic core of γ-secretase. PS/γ-secretase is an enzymatic complex composed of at least four proteins: PS1 or PS2, nicastrin, Pen2, and Aph1 (1–3). PS/γ-secretase plays important roles not only in AD pathology by producing amyloid β peptides but also in various cellular signaling processes. Cleavage of various type I membrane proteins by PS/γ-secretase leads to nuclear translocation of the intracellular domain of the cleaved proteins, which could modulate the transcription of target proteins. For example, PS/γ-secretase cleaves Notch and releases the Notch intracellular domain (4) from the membrane. The Notch intracellular domain translocates into the nucleus, where it interacts with the DNA-binding protein CSL/RBP-J to regulate the transcription of target genes such as Hes1 (5). This kind of signaling process mediated by intramembranous cleavage of a protein is now well known as regulated intramembrane proteolysis.
In addition to an essential role in regulated intramembrane proteolysis mediated by γ-secretase, PS works through γ-secretase-independent pathways. For example, it has been shown that PS can interact with β-catenin (6) and regulate β-catenin stability (7, 8), thereby modulating β-catenin-mediated cellular signaling processes. Moreover, PS can also act as a regulator of intracellular Ca2+ homeostasis. Recent reports of electrophysiological experiments using reconstituted lipid bilayer and Ca2+ imaging experiments using mouse embryonic fibroblast (MEF) cells from PS1/PS2 double knock-out mice demonstrated that PS functions as a passive endoplasmic reticulum (ER) Ca2+ leak channel that controls steady-state ER Ca2+ levels (9, 10). Interestingly, these reports suggested that several FAD mutants of PS disrupt the activity of transporting calcium ions, which might be associated with AD pathophysiology. Furthermore, PS is also known to regulate the expression of several cell surface receptors as well as their downstream pathways. For instance, it has been reported that expression of the PDGF receptor is decreased in cells genetically deficient in PS (11), whereas that of the EGF receptor is dramatically increased in these cells (12). Both PDGF and EGF receptors belong to a large family of growth factor receptors with intrinsic tyrosine kinase activity, and both regulate common downstream pathways such as PI3K/Akt signaling. Thus, PS itself possesses these functions, independent of γ-secretase activity.
The insulin receptor (IR) is a tetrameric transmembrane protein that acts as a cell surface receptor with intrinsic tyrosine kinase activity (13). Insulin binding to IR at the plasma membrane activates the tyrosine-specific kinase of the intracellular domain of the IR β-subunit. This activation subsequently phosphorylates intracellular substrates, which have roles in cell growth, energy metabolism, cholinergic gene expression, inhibition of oxidative stress, and apoptosis (14). Notably, it has been reported that γ-secretase processes IR along with other substrates (15, 16). Therefore, in this study, we directed our attention to IR, which belongs to the tyrosine kinase family, and hypothesized that IR/insulin signaling may also be regulated by PS.
Here, we demonstrate that PS inhibits IR transcription and reduces IR expression, followed by down-regulation of insulin signaling. In addition, the negative regulation of IR/insulin signaling by PS is not through either a γ-secretase-dependent mechanism or Wnt/β-catenin signaling but is caused by a change in intracellular Ca2+ homeostasis. Our findings suggest the possibility that PS can regulate insulin signaling via modifying IR expression through regulation of intracellular Ca2+ homeostasis.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
Plasmids expressing WT PS1 and a dominant-negative mutant of PS1 that is deficient in γ-secretase activity (D385A PS1) were constructed as described previously (17). The precision of cloning of reading frames was verified by sequencing.
Cell Culture and Transfection
PS1/PS2 double knock-out MEF (−/− MEF) cells were generously donated by Dr. B. DeStrooper (Catholic University, Leuven, Belgium). WT and −/− MEF cells were maintained in DMEM (Sigma) with 10% FBS (Invitrogen). For transient transfection into −/− MEFs, cells were plated in 35-mm dishes, and plasmid DNA was transfected using TransFectin reagent (Bio-Rad) according to the manufacturer's instructions. For β-catenin knockdown, a predesigned siRNA construct was synthesized by Dharmacon and transfected into WT and −/− MEF cells using Lipofectamine 2000 (Invitrogen).
Adipocyte Differentiation and Oil Red O Staining
Adipocyte differentiation of WT and −/− MEF cells was induced with 0.5 mm 3-isobutyl-1-methylxanthine, 0.25 μm dexamethasone, and 10 μg/ml insulin (all from Sigma) for 6 days and maintained with 10% FBS/DMEM to 8 days (18). At 8 days, the cells were washed with PBS and fixed in 4% paraformaldehyde for 1 h. After fixation, the cells were stained with 0.6% Oil Red O solution (60% isopropyl alcohol and 40% water) for 2 h at room temperature. The cells were then washed with water to removed unbound dye.
Antibodies and Chemical Reagents
Mouse anti-IR monoclonal antibody was purchased from NeoMarkers. Rabbit anti-Akt and anti-phospho-Akt (Ser-473) polyclonal antibodies were from Cell Signaling Technology. Mouse anti-β-actin monoclonal and rabbit anti-amyloid precursor protein (APP) polyclonal antibodies were from Sigma. Rabbit anti-PS1 N-terminal fragment polyclonal antibody was from Santa Cruz Biotechnology. Mouse anti-β-catenin monoclonal antibody was from BD Transduction Laboratories. Insulin solution and the γ-secretase inhibitor N-(N-(3,5-difluorophenacetyl)-l-alanyl)-S-phenylglycine t-butyl ester (DAPT) were from Sigma. Glycogen synthase kinase inhibitor VIII, LiCl, dantrolene, thapsigargin, 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester (BAPTA-AM), and ionomycin were from Calbiochem. Me2SO was from Nacalai Tesque.
Western Blotting
Cells were washed twice with PBS and scraped off. Cell pellets were suspended in ice-cold buffer containing 10 mm Tris-HCl (pH 7.8), 150 mm NaCl, 1% Nonidet P-40, and 1 mm EDTA supplemented with protease inhibitor mixture and briefly subjected to sonication. The samples were centrifuged at 14,000 × g for 20 min at 4 °C, and the supernatants were collected to obtain protein samples. The protein concentration was determined using the Bradford assay (19). Protein samples were diluted with sample buffer (125 mm Tris (pH 6.8), 4% SDS, 2% 2-mercaptoethanol, 20% glycerol, and 0.01% bromphenol blue) and denatured at 95 °C for 5 min. Samples containing equal amounts of protein were electrophoresed on polyacrylamide gradient gels (5–20%; Atto) in running buffer (25 mm Tris, 192 mm glycine, and 0.1% SDS). Immunoblotting was carried out by transferring the proteins to polyvinylidene difluoride microporous membrane, blocking this membrane with 5% skimmed milk in 20 mm TBS containing 0.1% Tween 20 (TBS/Tween), and incubating with the primary antibodies in PBS containing 4% BSA (Nacalai Tesque) overnight at 4 °C. The membranes were then washed with TBS/Tween and incubated with horseradish peroxidase-conjugated anti-mouse IgG (GE Healthcare) in TBS/Tween for 1 h at room temperature. The specific reaction was visualized using the ECL method (GE Healthcare).
Real-time PCR Assay
Total RNA was isolated using Isogen (Nippon Gene) according to the manufacturer's protocol. For real-time PCR analysis, 5 μg of total RNA from each sample was used with a cDNA synthesis kit (GE Healthcare). Real-time PCR primers were designed as follows: IR, 5′-TTCGAGAGCTGGGGCAG-3′ and 5′-GGCGGACCACATGATGGCAG-3′; and β-actin, 5′-CACACTGTGCCCATCTAC-3′ and 5′-CTCCTGCTCGAAGTCTAG-3′. For the amplification of IR or β-actin, 5 μl of cDNA was added to SYBR Green Master Mix (Roche Applied Science), and a real-time PCR assay was performed in a Light Cycler 480 (Roche Applied Science).
Calcium Imaging
To visually analyze the semiquantitative concentration of intracellular calcium, we used the Fluo-8/AM system (ABD Bioquest) according to the manufacturer's protocol. Briefly, 5.0 × 104 −/− MEF cells were cultured for 24 h on 35-mm glass-base dishes (Iwaki). After treatment with reagents or transfection, media were replaced with 5 μm Fluo-8/AM in Hanks' buffer. After a 30-min incubation, the buffer was replaced with native Hanks' buffer, and intracellular calcium was visually analyzed using a laser confocal scanning microscope (FV10i-LIV, Olympus).
Statistical Analysis
Analysis of the band density was carried out using NIH ImageJ to compare the relative expression of proteins. All values are given as means ± S.E. Comparisons were performed using an unpaired Student's t test. For comparison of multiparametric analysis, one-way factorial analysis of variance, followed by post hoc analysis by Fisher's protected least significant difference test, was used. p < 0.05 was considered to indicate a significant difference (n = four indicated four independent experiments).
RESULTS
Insulin-mediated Phosphorylation of Akt Is Enhanced in −/− MEF Cells
To investigate the effect of PS on insulin signaling, we treated WT and −/− MEF cells with insulin and statistically analyzed the phosphorylation ratio of Akt, a downstream phosphorylation target of insulin signaling. Because insulin is known to cause rapid signal transmission, we analyzed the phosphorylation ratio of Akt (phosphorylated/total Akt) at several time points within 30 min. Interestingly, the phosphorylation ratio of Akt in −/− MEF cells was significantly larger than that in WT MEF cells after insulin treatment (Fig. 1, A and B), suggesting that PS may down-regulate insulin signaling.
FIGURE 1.

Insulin-mediated phosphorylation of Akt is increased in −/− MEF cells. A, WT and PS1/PS2-null (−/−) MEF cells were treated with 50 nm insulin. After 5, 15, and 30 min, cell lysates were subjected to immunoblotting with anti-phospho-Akt (Ser-473) and anti-Akt antibodies. B, the relative phosphorylation ratio of Akt was quantitatively analyzed (n = 3). C, cell lysates from WT or −/− MEF cells were subjected to immunoblotting with anti-IR antibody. D, the relative band density of IR was quantitatively analyzed (n = 4). The level of IR was normalized to that of β-actin (loading control). E, −/− MEF cells were transfected with WT PS1. pcDNA was used as a negative control. 48 h after transfection, cell lysates were subjected to immunoblotting. F, the relative band density of IR was quantitatively analyzed (n = 3). The level of IR was normalized to that of β-actin.
To determine the mechanism of how PS down-regulates insulin signaling, we next analyzed the expression level of several molecules associated with insulin signaling. Among them, we found that the expression level of IR in −/− MEF cells was significantly increased compared with that in WT MEF cells (Fig. 1, C and D). Moreover, overexpression of WT PS1, which is one type of PS gene, in −/− MEF cells also significantly decreased IR expression (Fig. 1, E and F). Collectively, these results indicate that PS negatively regulates IR expression, which may lead to inhibition of insulin signaling.
IR mRNA Is Increased in −/− MEF Cells
Next, we asked whether PS decreases IR expression levels by inhibition of transcription. For this, we examined the mRNA level of IR by real-time PCR assay. In addition to the expression level, the mRNA level of IR in −/− MEF cells was significantly increased compared with that in WT MEF cells (Fig. 2A). Moreover, transiently expressing PS1 in −/− MEF cells also decreased the mRNA level of IR (Fig. 2B). Thus, these results show that PS negatively regulates IR transcription, followed by reduction of IR expression.
FIGURE 2.
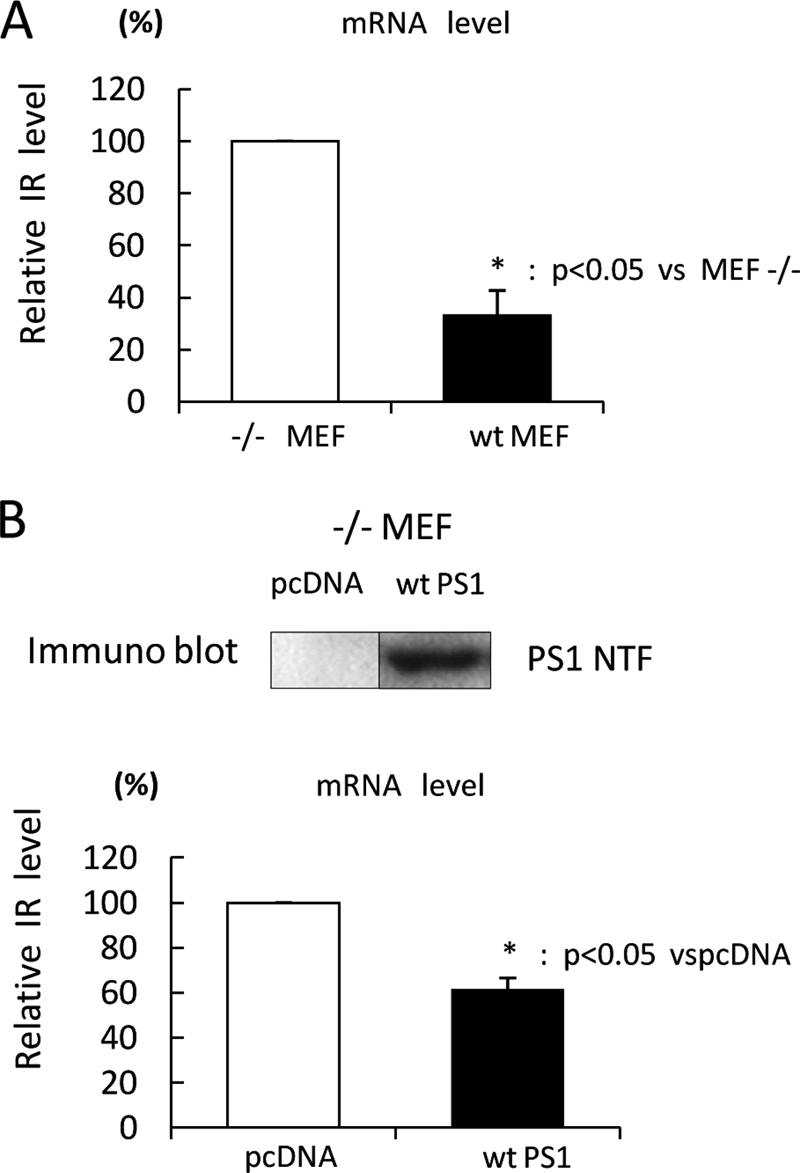
IR mRNA is increased in −/− MEF cells. A, mRNA was extracted from WT or −/− MEF cells. The mRNA level of IR was analyzed by real-time PCR assay (n = 4). The mRNA level of IR was normalized to that of β-actin. B, −/− MEF cells were transfected with WT PS1 or pcDNA. 36 h after transfection, mRNA was extracted, and the mRNA level of IR was analyzed by real-time PCR assay (lower panel). The mRNA level of IR was normalized to that of β-actin. WT PS1 expression was checked by immunoblotting (upper panel).
PS-mediated IR Reduction Is Independent of γ-Secretase Activity
Given that PS down-regulates insulin signaling by inhibiting transcription of IR as well as its expression, we next tried to elucidate the detailed mechanism of IR reduction by PS. According to the previous literature (20), PS carries out at least three major physiological functions: 1) intramembrane cleavage as a component of γ-secretase, 2) regulation of Wnt/β-catenin signaling, and 3) intracellular Ca2+ homeostasis. Because PS is known to cleave IR (15), we wondered if the cleavage of IR may reflect the expression level of IR in the above experiments. Thus, we first examined the effect of γ-secretase activity on the reduction of IR expression mediated by PS using the γ-secretase inhibitor DAPT. The band of the APP C-terminal fragment was detected as a positive control to show the inhibition of γ-secretase activity by DAPT. As shown in Fig. 3A, the APP C-terminal fragment accumulated not only in −/− MEF cells but also in WT MEF cells treated with DAPT. On the other hand, the expression level of IR was significantly higher in −/− MEF cells compared with that in WT MEF cells even after DAPT treatment (Fig. 3, A and B), which was almost unresponsive to DAPT application. Thus, the difference in IR levels between WT and −/− MEF cells is not attributable to γ-secretase activity. This result was confirmed using L-685458, another γ-secretase inhibitor (supplemental Fig. 1). However, the IR level was slightly enhanced in WT MEF cells treated with DAPT, indicating that this enhancement could be caused by the inhibition of IR cleavage by γ-secretase (15).
FIGURE 3.
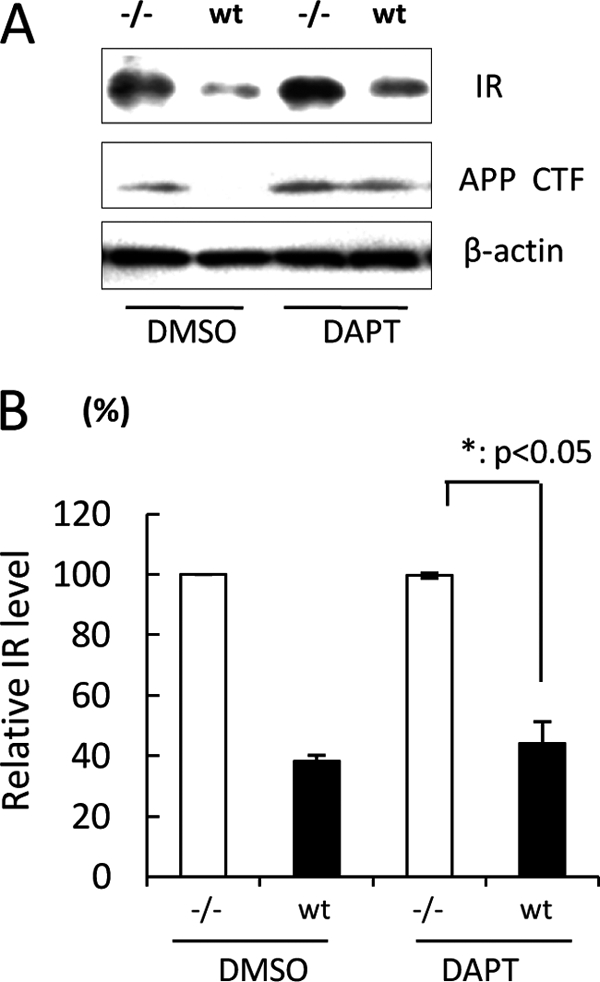
PS-mediated IR reduction is independent of γ-secretase activity. A, WT or −/− MEF cells were treated with 5 μm DAPT for 48 h. Cell lysates were subjected to immunoblotting with anti-IR antibody. The band of the APP C-terminal fragment was detected as a positive control of γ-secretase inhibition. B, the relative band density of IR was quantitatively analyzed (n = 3). The level of IR was normalized to that of β-actin.
PS-mediated IR Reduction Is Independent of Wnt/β-Catenin Signaling
PS is also reported to be associated with β-catenin stability (7, 8), followed by regulation of Wnt/β-catenin signaling. Because Wnt/β-catenin signaling regulates the transcription of several target genes (21), we next analyzed the effect of β-catenin on IR levels in WT and −/− MEF cells. To down-regulate β-catenin activity, we transfected β-catenin siRNA into WT and −/− MEF cells. The base-line expression level of β-catenin in WT MEF cells was lower than that in −/− MEF cells, which is in agreement with previous reports (22). Moreover, the expression level of β-catenin was clearly decreased in siRNA-transfected WT MEF cells as well as in −/− MEF cells compared with control transfected cells. Interestingly, siRNA transfection did not change the IR level in either WT or −/− MEF cells (Fig. 4, A and B). On the other hand, to enhance β-catenin activity, we treated WT and −/− MEF cells with glycogen synthase kinase inhibitors (23). Treatment of cells with glycogen synthase kinase inhibitors did not change the difference in IR levels between WT and −/− MEF cells (Fig. 4, C and D). Collectively, these results suggest that PS-mediated IR reduction is independent of Wnt/β-catenin signaling.
FIGURE 4.

PS-mediated IR reduction is independent of β-catenin activity. A, WT or −/− MEF cells were transfected with β-catenin siRNA. Control cells were transfected with BLOCK-iT Fluorescent Oligo. 48 h after transfection, cell lysates were subjected to immunoblotting with anti-IR and β-catenin antibodies. β-Actin was detected as a loading control. B, the relative band density of IR was quantitatively analyzed (n = 3). The level of IR was normalized to that of β-actin. C, WT or −/− MEF cells were treated with 10 μm glycogen synthase kinase (GSK) inhibitor VIII or 25 mm LiCl for 48 h. Control cells were treated with Me2SO (DMSO). Cell lysates were subjected to immunoblotting with anti-IR and β-catenin antibodies. β-Actin was detected as a loading control. D, the relative band density of IR was quantitatively analyzed (n = 3). The level of IR was normalized to that of β-actin.
PS-mediated Up-regulation of Intracellular Ca2+ Decreases the Level of IR
As mentioned above, PS is also known to regulate intracellular Ca2+ homeostasis by functioning as a Ca2+ leak channel in the ER membrane (9, 10). It has been reported that the intracellular Ca2+ level in the presence of PS is 1.4 times larger than in its absence (9). Therefore, we hypothesized that a PS-mediated increase in the intracellular Ca2+ level may induce IR reduction. To examine this hypothesis, we treated WT or −/− MEF cells with BAPTA-AM or ionomycin to decrease or increase the intracellular Ca2+ level, respectively. Microscopic analysis using the Fluo-8 calcium imaging system confirmed that these reagents actually changed the intracellular Ca2+ state (Fig. 5, A, C, and D). Interestingly, the IR level was increased by BAPTA-AM, whereas ionomycin treatment decreased the IR level in WT and −/− MEF cells (Fig. 5, E and F). Moreover, we treated WT or −/− MEF cells with dantrolene to inhibit Ca2+ leakage via the ryanodine receptor localizing at the ER. Calcium imaging analysis suggested that dantrolene also decreased the intracellular Ca2+ level (Fig. 5B). As expected, dantrolene dose-dependently increased the IR level. Statistical analysis indicated that the IR level in WT MEF cells treated with 10 μm dantrolene was the same as that in untreated −/− MEF cells (Fig. 5, E and F). In addition, we treated WT or −/− MEF cells with thapsigargin to inhibit Ca2+ transport from the cytoplasm to the ER via Ca2+-ATPase (sarco/endoplasmic reticulum Ca2+-ATPase). Thapsigargin clearly decreased the IR level in WT cells as well as in −/− MEF cells (supplemental Fig. 2). These results suggest that enhancement of the intracellular Ca2+ level decreases the level of IR.
FIGURE 5.

Up-regulation of intracellular Ca2+ decreases the IR level. A–D, −/− MEF cells were treated with Me2SO (DMSO), 10 μm dantrolene, 20 μm BAPTA-AM, or 10 μm ionomycin, respectively. 24 h after treatment, intracellular Ca2+ imaging was performed by laser confocal microscopy with Fluo-8/AM. E, WT or −/− MEF cells were treated with dantrolene (5 and 10 μm), 20 μm BAPTA-AM, or 10 μm ionomycin. Control cells were treated with Me2SO. 48 h after treatment, cell lysates were subjected to immunoblotting with anti-IR antibody. β-Actin was detected as a loading control. F, the relative band density of IR was quantitatively analyzed. The level of IR was normalized to that of β-actin. G–I, pcDNA, WT PS1, or D385A PS1 was transfected into −/− MEF cells. 24 h after transfection, intracellular Ca2+ imaging was performed by laser confocal microscopy with Fluo-8/AM. J, −/− MEF cells were transfected with pcDNA, WT PS1, or D385A PS1. 48 h after transfection, cell lysates were subjected to immunoblotting with anti-IR antibody. β-Actin was detected as a loading control. K, the relative band density of IR was quantitatively analyzed. The level of IR was normalized to that of β-actin.
Next, to clarify the contribution of PS to these events, we analyzed the effect of D385A PS1 on the IR level. Although the D385A PS1 construct is frequently used as a dominant-negative construct of γ-secretase (24), which is known to fail to undergo endoproteolysis, it has also been shown to be nonfunctional as an ER Ca2+ leak channel (10). Thus, D385A PS1 fails not only to cleave several substrates but also to pass Ca2+ through the ER membrane. Calcium imaging analysis indicated that the level of intracellular Ca2+ was enhanced in WT PS1-transfected cells compared with pcDNA- or D385A PS1-transfected cells (Fig. 5, G–I), which is in agreement with previous reports (9, 10). Interestingly, the IR level in D385A PS1-transfected cells was significantly larger than that in WT PS1 cells (Fig. 5, J and K). Collectively, these results demonstrate that PS functions as an ER Ca2+ leak channel and enhances the intracellular Ca2+ level, which may lead to inhibition of the expression of IR.
PS Decreases the IR Level in Adipose Cells
To clarify the contribution of these PS-mediated events to insulin target tissues, we analyzed the expression of PS1 in these tissues. Immunoblot analysis clearly indicated that PS1 was expressed in the adipose tissue, skeletal muscle, and liver of mice (supplemental Fig. 3). Because PS1 was expressed at a high level in adipose tissue, we next examined the effect of PS1 on the IR level in adipose cells. After 8 days of incubation with insulin, 3-isobutyl-1-methylxanthine, and dexamethasone, the lipid accumulation in WT and −/− MEF cells was evaluated by Oil Red O staining (Fig. 6A). To examine the expression level of IR in the adipose cells derived from MEFs, we conducted immunoblot analysis. Notably, the IR level in the adipocytes derived from WT MEF cells was lower than that derived from −/− MEF cells (Fig. 6, B and C). These results indicate that PS might decrease the level of IR in adipose cells.
FIGURE 6.

PS decreases the IR level in adipose cells. A, WT and −/− MEF cells were incubated with 0.5 mm 3-isobutyl-1-methylxanthine, 0.25 μm dexamethasone, and 10 μg/ml insulin for 6 days and maintained in 10% FBS/DMEM to 8 days. Differentiated adipocytes were fixed and stained with Oil Red O solution. B, cell lysates from the adipocytes derived from WT or −/− MEF cells were subjected to immunoblotting with anti-IR antibody. β-Actin was detected as a loading control. C, the relative band density of IR was quantitatively analyzed. The level of IR was normalized to that of β-actin.
DISCUSSION
PS was originally identified as a causative gene for FAD and has been clearly demonstrated to work as an essential component of γ-secretase, producing amyloid β from APP (25). In addition, PS/γ-secretase is known to play an important role in many cellular events, including differentiation, proliferation, and apoptosis, via cleavage of various substrates (19, 26). On the other hand, PS also works through γ-secretase-independent pathways (26). Of note, PS is known to be involved in regulation of the expression of cell surface receptors and their downstream pathways, including PDGF and EGF receptors (11, 12). In this study, we directed our attention to IR, a cell surface receptor for insulin signaling, and asked whether PS could regulate IR expression as well as insulin signaling because IR is functionally associated with PS (27). The mechanism of insulin signaling and the biological effects of insulin have been studied mainly in classical insulin target tissues such as skeletal muscle, adipose tissue, and liver with respect to glucose uptake, regulation of cell proliferation, gene expression, and suppression of hepatic glucose production (28). However, recent studies suggested that insulin also has profound effects in the CNS, where it regulates key processes such as energy homeostasis and neuronal survival (29). In addition, insulin signaling is also known to be associated with the pathology setting of AD (30, 31). Therefore, it is essential to clarify the cellular regulatory mechanism of insulin signaling with AD-related proteins.
This study addressed that PS down-regulates insulin signaling by using PS-deficient (−/− MEF) and WT MEF cells. Interestingly, the mRNA level of IR was significantly decreased in the presence of endogenous PS compared with that in cells deficient in PS, which presumably leads to the reduction of IR expression. Moreover, transient transfection of WT PS1 in −/− MEF cells reduced the mRNA level of IR (Fig. 2), indicating that the decrease in the level of IR by PS could be attributed to the reduction of its mRNA. A previous report showed that turnover of the EGF receptor is reduced in the presence of PS, suggesting that PS can negatively regulate EGF receptor degradation (12). On the other hand, PS was reported to reduce the protein and mRNA levels of the PDGF receptor, suggesting that PS also may inhibit its transcription (11). Thus, PS regulates the expression of cell surface receptors in various ways. In the case of IR, we have demonstrated that PS down-regulated IR transcription, which led to a decrease in IR expression.
To clarify the detailed mechanism for this, we examined three possibilities for the down-regulation of IR transcription: 1) regulated intramembrane proteolysis mediated by γ-secretase, 2) Wnt/β-catenin signaling, and 3) intracellular Ca2+ homeostasis. In this study, γ-secretase inhibitors did not change the difference in the IR expression level (Fig. 3), indicating that γ-secretase-mediated regulated intramembrane proteolysis is not associated with the reduction in IR transcription. Moreover, the down- and/or up-regulation of β-catenin also did not change IR expression (Fig. 4), indicating that Wnt/β-catenin signaling is not associated with the down-regulation of IR transcription.
Recent reports suggested that PS functions as a passive ER Ca2+ leak channel (9, 10). However, the readout of this function remains largely unknown. One report showed that FAD mutants of PS1 failed to activate this function, leading to impairment of synaptic plasticity in the pathological setting of AD (10). On the other hand, Ca2+ leak activity could be associated with neurotransmitter release, thereby modulating long-term potentiation and short-term plasticity in the presynaptic domain (32). Therefore, we asked whether IR reduction is associated with the PS-mediated change in intracellular Ca2+ homeostasis. Interestingly, using several compounds that can change the intracellular Ca2+ level, we demonstrated that the decrease in the intracellular Ca2+ level could elevate the level of IR (Fig. 5), indicating that the intracellular Ca2+ level might be associated with the IR level. Moreover, to confirm the contribution of PS to these events, we used the D385A PS1 mutant, which lacks ER Ca2+ leak channel activity (10). Notably, the expression of D385A PS1 elevated the IR level compared with that of WT PS1 (Fig. 5). We concluded that PS can regulate IR expression by changing the intracellular Ca2+ level. Our result may be one of the novel readouts of PS-mediated ER Ca2+ leak channels, which may be strongly associated with insulin signaling. Long-term dantrolene feeding increases amyloid accumulation in the brains of AD model mice (10). Moreover, insulin is known to promote the release of amyloid β (33), and the knock-out of insulin receptor substrate, a direct downstream molecule of IR, down-regulates brain insulin signaling, followed by a decrease in amyloid β accumulation in AD model mice (34). In this study, we suggested that dantrolene treatment significantly increased the expression of IR (Fig. 5). Dantrolene might hyperactivate insulin signaling via the increase in IR expression, thereby exacerbating amyloid accumulation in the brain. Therefore, we assume that PS can modulate the pathology of AD through regulating the expression of several cell surface receptors in a γ-secretase-independent manner.
In addition to the pathology setting of AD, our results indicate that PS might decrease the level of IR, thereby modulating insulin signaling in adipose cells (Fig. 6). In adipose cells, insulin signaling contributes to glucose uptake, which controls the energy homeostasis (28). In addition, it is known to regulate adipogenesis (35). Our Oil Red O staining experiment showed that the red signals in −/− MEF cells were larger than those in WT cells at 4 days, which indicates the possibility that adipogenesis in −/− MEF cells might be faster than that in WT cells. This result might be caused by the up-regulation of insulin signaling in −/− MEF cells because up-regulation of insulin signaling is reported to positively regulate adipogenesis (36). On the other hand, the result might be caused by the inhibition of γ-secretase activity because recent literature has demonstrated that the γ-secretase inhibitor inhibits adipogenesis via inhibiting Notch signaling (37). We conclude that PS might play very important roles in both preadipocyte and mature adipose cells.
An increasing number of reports confirm that intracellular Ca2+ regulates the transcription of several target genes (38, 39). However, in this study we did not elucidate the mechanism by which intracellular Ca2+ regulates IR expression. Thus, this should be clarified in future studies. Moreover, not only FAD mutants but also modifications including phosphorylation or ubiquitination of PS were reported to modulate the physiological functions of PS/γ-secretase (27, 40). Therefore, whether FAD mutations and/or modifications of PS regulate the expression of cell surface receptors should be elucidated in future studies. Damage to these pathways may be the mechanistic basis for FAD and sporadic AD.
Supplementary Material
Acknowledgments
We thank Dr. B. DeStrooper for the gift of PS1/PS2 double knock-out MEFs (−/− MEFs), Dr. H. Itoh (Kyoto University, Kyoto, Japan) for the gift of Oil Red O, Dr. M. Kinoshita (Nagoya University, Nagoya, Japan) and Dr. A. Yanagida (Kyoto University) for technical assistance in real-time PCR assays, and Dr. K. Hosoda (Kyoto University) for advice.
The work was supported by Grant-in-aid 20300124 from the Ministry of Education, Culture, Sports, Science and Technology and a research grant from the Takeda Science Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
- PS
- presenilin
- AD
- Alzheimer disease
- FAD
- familial AD
- MEF
- mouse embryonic fibroblast
- ER
- endoplasmic reticulum
- IR
- insulin receptor
- APP
- amyloid precursor protein
- DAPT
- N-(N-(3,5-difluorophenacetyl)-l-alanyl)-S-phenylglycine t-butyl ester
- BAPTA-AM
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester.
REFERENCES
- 1. Yu G., Nishimura M., Arawaka S., Levitan D., Zhang L., Tandon A., Song Y. Q., Rogaeva E., Chen F., Kawarai T., Supala A., Levesque L., Yu H., Yang D. S., Holmes E., Milman P., Liang Y., Zhang D. M., Xu D. H., Sato C., Rogaev E., Smith M., Janus C., Zhang Y., Aebersold R., Farrer L. S., Sorbi S., Bruni A., Fraser P., St George-Hyslop P. (2000) Nature 407, 48–54 [DOI] [PubMed] [Google Scholar]
- 2. Francis R., McGrath G., Zhang J., Ruddy D. A., Sym M., Apfeld J., Nicoll M., Maxwell M., Hai B., Ellis M. C., Parks A. L., Xu W., Li J., Gurney M., Myers R. L., Himes C. S., Hiebsch R., Ruble C., Nye J. S., Curtis D. (2002) Dev. Cell 3, 85–97 [DOI] [PubMed] [Google Scholar]
- 3. Goutte C., Tsunozaki M., Hale V. A., Priess J. R. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 775–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Struhl G., Greenwald I. (1999) Nature 398, 522–525 [DOI] [PubMed] [Google Scholar]
- 5. Jarriault S., Brou C., Logeat F., Schroeter E. H., Kopan R., Israel A. (1995) Nature 377, 355–358 [DOI] [PubMed] [Google Scholar]
- 6. Yu G., Chen F., Levesque G., Nishimura M., Zhang D. M., Levesque L., Rogaeva E., Xu D., Liang Y., Duthie M., St George-Hyslop P. H., Fraser P. E. (1998) J. Biol. Chem. 273, 16470–16475 [DOI] [PubMed] [Google Scholar]
- 7. Meredith J. E., Jr., Wang Q., Mitchell T. J., Olson R. E., Zaczek R., Stern A. M., Seiffert D. (2002) Biochem. Biophys. Res. Commun. 299, 744–750 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Z., Hartmann H., Do V. M., Abramowski D., Sturchler-Pierrat C., Staufenbiel M., Sommer B., van de Wetering M., Clevers H., Saftig P., De Strooper B., He X., Yankner B. A. (1998) Nature 395, 698–702 [DOI] [PubMed] [Google Scholar]
- 9. Tu H., Nelson O., Bezprozvanny A., Wang Z., Lee S. F., Hao Y. H., Serneels L., De Strooper B., Yu G., Bezprozvanny I. (2006) Cell 126, 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang H., Sun S., Herreman A., De Strooper B., Bezprozvanny I. (2010) J. Neurosci. 30, 8566–8580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kang D. E., Yoon I. S., Repetto E., Busse T., Yermian N., Ie L., Koo E. H. (2005) J. Biol. Chem. 280, 31537–31547 [DOI] [PubMed] [Google Scholar]
- 12. Repetto E., Yoon I. S., Zheng H., Kang D. E. (2007) J. Biol. Chem. 282, 31504–31516 [DOI] [PubMed] [Google Scholar]
- 13. Knutson V. P. (1991) FASEB J. 5, 2130–2138 [DOI] [PubMed] [Google Scholar]
- 14. Saltiel A. R., Kahn C. R. (2001) Nature 414, 799–806 [DOI] [PubMed] [Google Scholar]
- 15. Kasuga K., Kaneko H., Nishizawa M., Onodera O., Ikeuchi T. (2007) Biochem. Biophys. Res. Commun. 360, 90–96 [DOI] [PubMed] [Google Scholar]
- 16. Marambaud P., Shioi J., Serban G., Georgakopoulos A., Sarner S., Nagy V., Baki L., Wen P., Efthimiopoulos S., Shao Z., Wisniewski T., Robakis N. K. (2002) EMBO J. 21, 1948–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uemura K., Kitagawa N., Kohno R., Kuzuya A., Kageyama T., Shibasaki H., Shimohama S. (2003) J. Neurosci. Res. 73, 166–175 [DOI] [PubMed] [Google Scholar]
- 18. Noguchi M., Hosoda K., Fujikura J., Fujimoto M., Iwakura H., Tomita T., Ishii T., Arai N., Hirata M., Ebihara K., Masuzaki H., Itoh H., Narumiya S., Nakao K. (2007) J. Biol. Chem. 282, 29574–29583 [DOI] [PubMed] [Google Scholar]
- 19. Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 20. Hass M. R., Sato C., Kopan R., Zhao G. (2009) Semin Cell Dev. Biol. 20, 201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gordon M. D., Nusse R. (2006) J. Biol. Chem. 281, 22429–22433 [DOI] [PubMed] [Google Scholar]
- 22. Raurell I., Codina M., Casagolda D., Del Valle B., Baulida J., de Herreros A. G., Duñach M. (2008) PLoS ONE 3, e4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aberle H., Bauer A., Stappert J., Kispert A., Kemler R. (1997) EMBO J. 16, 3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen F., Yang D. S., Petanceska S., Yang A., Tandon A., Yu G., Rozmahel R., Ghiso J., Nishimura M., Zhang D. M., Kawarai T., Levesque G., Mills J., Levesque L., Song Y. Q., Rogaeva E., Westaway D., Mount H., Gandy S., St George-Hyslop P., Fraser P. E. (2000) J. Biol. Chem. 275, 36794–36802 [DOI] [PubMed] [Google Scholar]
- 25. De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., Von Figura K., Van Leuven F. (1998) Nature 391, 387–390 [DOI] [PubMed] [Google Scholar]
- 26. De Strooper B., Annaert W. (2010) Annu. Rev. Cell Dev. Biol. 26, 235–260 [DOI] [PubMed] [Google Scholar]
- 27. Maesako M., Uemura K., Kubota M., Hiyoshi K., Ando K., Kuzuya A., Kihara T., Asada M., Akiyama H., Kinoshita A. (2011) Neuroscience 177, 298–307 [DOI] [PubMed] [Google Scholar]
- 28. White M. F. (2003) Science 302, 1710–1711 [DOI] [PubMed] [Google Scholar]
- 29. Zhao W. Q., Alkon D. L. (2001) Mol. Cell. Endocrinol. 177, 125–134 [DOI] [PubMed] [Google Scholar]
- 30. Maesako M., Uemura K., Kubota M., Ando K., Kuzuya A., Asada M., Kihara T., Kinoshita A. (2010) Neurosci. Lett. 483, 157–161 [DOI] [PubMed] [Google Scholar]
- 31. Schubert M., Gautam D., Surjo D., Ueki K., Baudler S., Schubert D., Kondo T., Alber J., Galldiks N., Küstermann E., Arndt S., Jacobs A. H., Krone W., Kahn C. R., Brüning J. C. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 3100–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang C., Wu B., Beglopoulos V., Wines-Samuelson M., Zhang D., Dragatsis I., Südhof T. C., Shen J. (2009) Nature 460, 632–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gasparini L., Gouras G. K., Wang R., Gross R. S., Beal M. F., Greengard P., Xu H. (2001) J. Neurosci. 21, 2561–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Freude S., Hettich M. M., Schumann C., Stöhr O., Koch L., Köhler C., Udelhoven M., Leeser U., Müller M., Kubota N., Kadowaki T., Krone W., Schröder H., Brüning J. C., Schubert M. (2009) FASEB J. 23, 3315–3324 [DOI] [PubMed] [Google Scholar]
- 35. Rosen E. D., MacDougald O. A. (2006) Nat. Rev. Mol. Cell Biol. 7, 885–896 [DOI] [PubMed] [Google Scholar]
- 36. Nakatsu Y., Sakoda H., Kushiyama A., Zhang J., Ono H., Fujishiro M., Kikuchi T., Fukushima T., Yoneda M., Ohno H., Horike N., Kanna M., Tsuchiya Y., Kamata H., Nishimura F., Isobe T., Ogihara T., Katagiri H., Oka Y., Takahashi S. I., Kurihara H., Uchida T., Asano T. (2011) J. Biol. Chem. 286, 20812–20822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang Y., Yang X., Wu Y., Jing W., Cai X., Tang W., Liu L., Liu Y., Grottkau B. E., Lin Y. (2010) Cell Prolif. 43, 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Crabtree G. R. (1999) Cell 96, 611–614 [DOI] [PubMed] [Google Scholar]
- 39. Aziz M. H., Manoharan H. T., Sand J. M., Verma A. K. (2007) Mol. Carcinog. 46, 646–653 [DOI] [PubMed] [Google Scholar]
- 40. Aoyagi N., Uemura K., Kuzuya A., Kihara T., Kawamata J., Shimohama S., Kinoshita A., Takahashi R. (2010) Biochem. Biophys. Res. Commun. 391, 1240–1245 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.