

Abstract
Cell based therapies for bone regeneration are an exciting emerging technology, but the availability of osteogenic cells is limited and an ideal cell source has not been identified. Amniotic fluid-derived stem (AFS) cells and bone-marrow derived mesenchymal stem cells (MSCs) were compared to determine their osteogenic differentiation capacity in both 2D and 3D environments. In 2D culture, the AFS cells produced more mineralized matrix but delayed peaks in osteogenic markers. Cells were also cultured on 3D scaffolds constructed of poly-ε-caprolactone for 15 weeks. MSCs differentiated more quickly than AFS cells on 3D scaffolds, but mineralized matrix production slowed considerably after 5 weeks. In contrast, the rate of AFS cell mineralization continued to increase out to 15 weeks, at which time AFS constructs contained 5-fold more mineralized matrix than MSC constructs. Therefore, cell source should be taken into consideration when used for cell therapy, as the MSCs would be a good choice for immediate matrix production, but the AFS cells would continue robust mineralization for an extended period of time. This study demonstrates that stem cell source can dramatically influence the magnitude and rate of osteogenic differentiation in vitro.
Key words or phrases: Mesenchymal stem cells, Fetal stem cells, Regenerative medicine, Tissue engineering, In vitro, Bone, Human
1. Introduction
Little is known about the optimal cell source for tissue engineering of cell-based therapies for musculoskeletal tissues. Bone graft substitutes composed of a biodegradable scaffold containing stem cells capable of osteogenic differentiation have shown promise as an alternative to bone grafting or delivery of osteoinductive proteins. This cellular augmentation is especially important in clinical cases where endogenous cellular supply is diminished, such as in older patients, smokers, or after chemotherapy or radiation therapy.
Mesenchymal stem cells (MSCs) are believed to be the source of osteoblastic cells during normal bone growth and remodeling and may be isolated from the bone marrow, among other tissues. Mesenchymal stem cells are a sub-population of bone marrow-derived cells characterized by their ability to differentiate to the mesenchymal lineage tissues of bone, fat and cartilage, as well as muscle[1-7]. Additionally, MSCs can be isolated from the intended recipient, thus reducing the probability of rejection[8]. Their relative ease of isolation and differentiation capability makes MSCs potentially useful for many applications, including bone deficits[9]. Osteogenic differentiation of MSCs has been well characterized. With the addition of just a few supplements to the basal growth media, often referred to as osteoinductive factors including dexamethasone, B-glycerophosphate and ascorbic acid, MSCs will mineralize the surface of a two-dimensional surface[2, 10, 11]. Furthermore, MSCs have been used in 3D constructs to mineralize bone graft substitutes[12-14].
Human amniotic fluid-derived stem (AFS) cells are isolated from amniotic fluid after routine amniocentesis[15-22]. The AFS cells express both embryonic and adult stem cell markers and are broadly multipotent; they can be induced to differentiate into cells representing all three embryonic lineages, such as cells of the osteogenic, adipogenic, chondrogenic, myogenic, endothelial, neuronal, and hepatic lineages[17, 23-29]. Unlike embryonic stem cells, AFS cells are not tumorigenic and can expand extensively without the use of feeder layers or expensive defined media[23]. The AFS cell lines have been shown to expand over 250 population doublings and retain telomere length and have a normal chromosomal karyotype[23]. Therefore, AFS cells have great potential for cellular tissue engineering. Previously, AFS cells have been shown to be capable of producing robust mineralized matrix in two-dimensional culture and throughout a three-dimensional medical grade poly-ε-caprolactone (PCL) scaffold and nanofiberous scaffolds[24, 30-32]. This study compares the ability of the adult and fetal stem cell sources to produce mineralized matrix in 2D and 3D culture.
Tissue engineering strategies using an extracellular matrix combined with stem cells capable of osteogenic differentiation may therefore be used to develop bone graft substitutes. The aim of this study was to compare two cell sources for potential application in the tissue engineering of bone graft substitutes. Comparison of human fetal-derived AFS cells and MSCs for the production of bone graft substitutes will help elucidate the most appropriate cell source for particular applications and may suggest appropriate delivery strategies. By culturing cells on 3D scaffolds, it was determined that the AFS cells had greater osteogenic potential overall, but they were observed to take longer to differentiate than the MSCs.
2 Results
2.1 2D differentiation
The morphology of the AFS cells was heterogeneous, but overall there appeared to be two general morphologies with one very small and compact and the other more spindle shaped (Supplemental Figure 1 A and B). The MSCs were also heterogeneous, but to a lesser extent than the AFS cells. The MSCs were spindle shaped and visibly larger than the AFS cells (Supplemental Figure 1C). The AFS cells and MSCs both differentiated into mineral producing cells when grown on tissue culture plastic. The mineralized matrix was visualized by the alizarin red S staining of calcium that covers the cells (Supplemental Figure 1 D-F).
2.2 Biochemical assay of 2D mineralized matrix
Although the staining appeared to be qualitatively similar between the cell types (Supplemental Figure 1 D-F), when the alizarin red was extracted and quantified, the AFS cells demonstrated significantly more mineral deposition/staining at 2 and 4 weeks (Figure 1A). The increased mineralized matrix was confirmed by calcium extraction and quantification by Arsenazo III reagent (Figure 1B). The AFS cell mineralized matrix contained significantly more Calcium than the matrix produced by the MSCs.
Figure 1.

A. To compare the amount of alizarin red staining of the mineralized matrix produced by the cells, the dye was extracted and the absorbance determined spectrophotometrically to determine the temporal changes in mineralized matrix. At 2 and 4 weeks the AFS cells had produced significantly more mineralized matrix than the MSCs. B. Calcium deposition by the AFS cells and MSCs was confirmed and quantified using the Arsenazo III reagent at 4 weeks. The AFS cells deposited significantly more calcium than the MSCs (p< 0.001). Both the AFS cells and MSCs deposited significantly more calcium than their control counterpart (n=6, p< 0.01). Real-time PCR quantification of Runx2 (C), Alkaline Phosphatase (D) and Osteocalcin (E) illustrate the temporal fluctuation of mRNA typical of osteoblasts. The MSCs had an early increase in transcript levels, which then decreased. The AFS cells required a longer induction time before the mRNA levels were increased. (n= 3, P<0.05)
2.3 Gene Expression
The expression levels of mRNA for Runx2, Alkaline Phosphatase and Osteocalcin were examined during osteogenic differentiation of the AFS cells and MSCs. Runx2 is a transcription factor involved in regulating osteogenic and chondrogenic differentiation and maintenance. Alkaline Phosphatase (ALP) is a phosphatase associated with mineralized matrix production. Osteocalcin is a vitamin D and K dependent protein produced by osteoblasts and is a common marker of bone formation.
In all three genes analyzed, the MSCs had a peak in gene expression between days 3 and 5 of differentiation (Figure 1, C-E). The AFS cells did not show this initial peak in gene expression but rather gradually increased expression over the 14 day culture. The ALP gene expression is consistent with previously reported findings that the MSCs have a higher ALP level at 1 week, at 2 weeks the levels are similar, and at 3 weeks the AFS cells have a higher ALP level[33]. The temporal differences in the gene expression support the hypothesis that the AFS cells are in a more primitive state and require additional time to differentiate into osteoblasts.
2.4 Osteogenic Differentiation in 3D PCL scaffolds
The AFS cells and MSCs were seeded onto PCL scaffolds with lyophilized collagen. The lyophilized collagen filled the scaffold pores to enhance cell retention and the composite scaffolds were placed in non-cell binding plates to encourage cell attachment. The seeding efficiencies of the cell types were not significantly different (Figure 4C). Both the AFS cells and MSCs had visually occluded the pores by 5 weeks.
Figure 4.
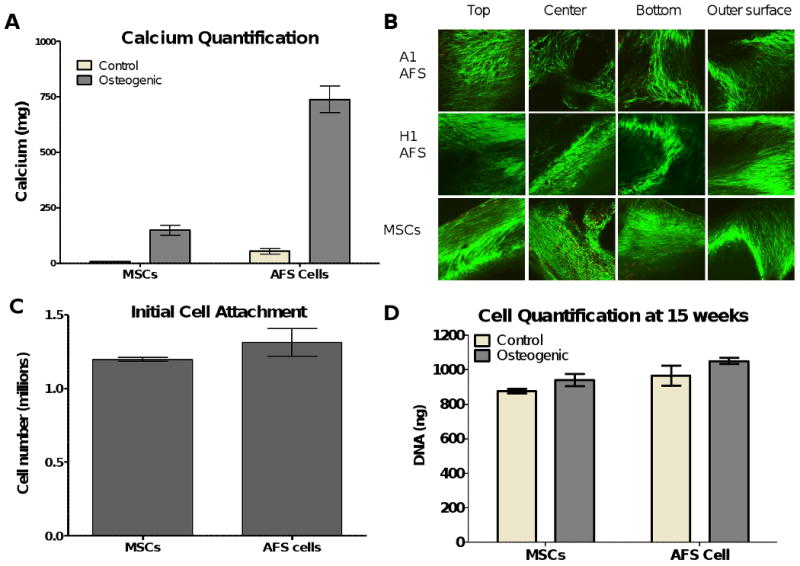
A. The calcium produced by the cells during the 15 weeks in culture was compared and quantified using the Arsenazo III reagent. 25% of the scaffold was used for this comparison. The AFS cells produced significantly more mineralized matrix than the MSCs when cultured in osteogenic media (p<0.001). Both cells produced significantly more mineralized matrix in osteogenic media than when cultured in control media (p<0.001 for AFS cells, p<0.01 for MSCs). B. Cell viability of the cells at 15 weeks in culture was determined by Calcien AM staining for live cells and ethidium homodimer-1 staining of the nuclei of dead cells. The exterior surfaces (top, bottom and outer surface) along with the middle of the scaffold (produced by cutting the scaffolds longitudinally) were visualized by confocal microscopy. Few dead cells were seen in the scaffolds and cells can be seen throughout the scaffolds, including spanning the pore space between the PCL struts. C. The cell attachment of the AFS cells and MSCs to the PCL scaffolds was determined by cell count after 3 days in culture. No differences were seen between the two groups. N=6. D. The cell proliferation and survival was similar after the 15 weeks in 3D culture, whether the scaffolds were seeded with MSCs or AFS cells and cultured in osteogenic or control media. N=6.
The mineralized matrix production by the cells was quantified by microCT analysis. The 3D renderings of the mineralized matrix are shown in Figure 2. The AFS cells had sparse mineral deposition at 3 and 5 weeks. By 10 weeks the AFS cells had produced substantial mineral throughout the PCL scaffold. Conversely, the MSCs showed more mineral production at the early time points, and the mineral content was not enhanced by the additional culture time. Additionally, the spatial distribution of the mineralized matrix appeared to be cell-source specific. The AFS cells routinely produced mineral throughout the entire scaffold, whereas the MSCs only produced mineral in the center of the scaffold, even though there were cells found throughout the scaffold (Figure 2 and Figure 5 E, I, and M). The mineralized matrix production by the AFS cells on 3D constructs is highly reproducible, as is illustrated by four representative microCT images of each cell line at 15 weeks, see Supplementary Data Figure 3.
Figure 2.
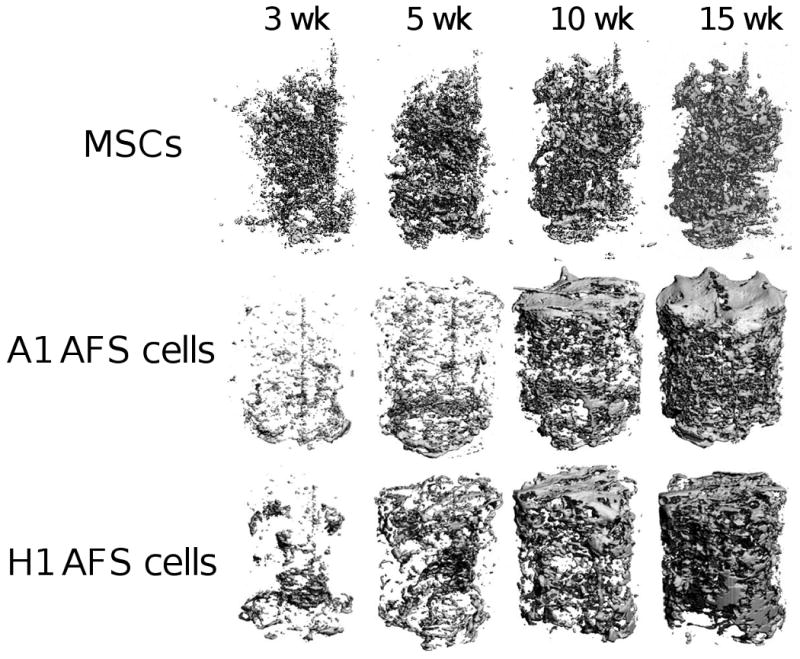
MSCs and AFS cells induced to produce mineralized matrix within the PCL scaffold. The scaffolds were aseptically scanned by microCT at weeks 3, 5, 10 and 15. The mineral volume of the same scaffold is shown for the A1 AFS cells, H1 AFS cells, and MSCs to illustrate the change in mineralized matrix over time. The mineralization of the PCL scaffold by the AFS cells is much more extensive, with the mineral distributed throughout the scaffold. The mineral produced by the MSCs is primarily found on the interior of the scaffold. N=12
Figure 5.
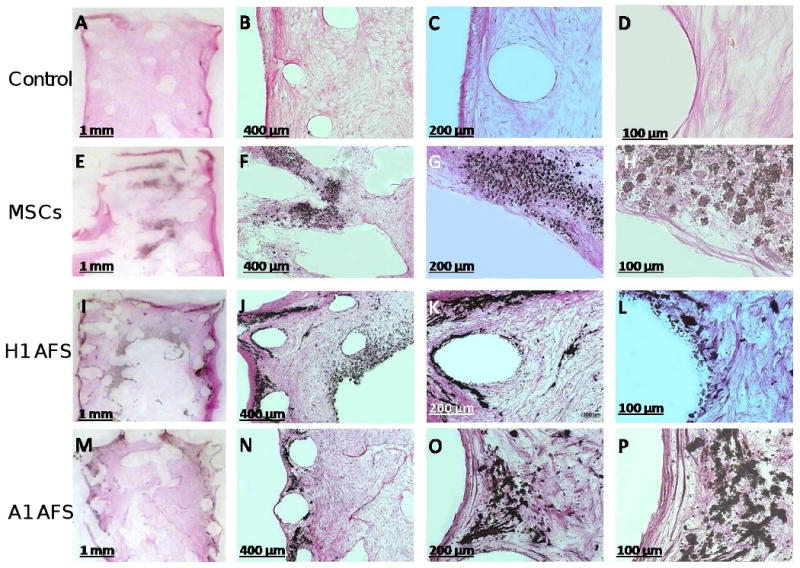
Histology revealed the onset of ectopic bone formation for osteoinduced MSC and AFS cells when seeded into 3D PCL scaffolds and cultured for 15 weeks. MSC control cell scaffold show no mineralisation, only fibrous tissue formation, which stained pink (A-D). Clear black mineral deposition can be observed for osteinduced MSCs (E-H) and AFS cells (I-P), within a fibrous network penetrating the scaffold pores.
2.5 Quantification of 3D Mineralized Matrix
The quantity of mineralized matrix was determined by the microCT scan, and the averages are shown in Figure 3A. The MSCs produced significantly more mineral at the 3 and 5 week time points, but their mineralization capacity was limited to the first few weeks in culture. Alternatively, the AFS cells did not produce substantial mineralized matrix until 5 weeks in culture, with mineralized matrix production increasing throughout the 15 weeks in culture. At 3 and 5 weeks, the MSCs had produced significantly more mineralized matrix than the AFS cells (p<0.001), but by 15 weeks the AFS cells had produced 3 times more mineralized matrix than the MSCs (p< 0.001). At all time points, cells grown in osteogenic media produced more mineralized matrix than those grown in control media (p<0.001).
Figure 3.

A. The volume of mineralized matrix produced by the osteogenic and control cells within the PCL scaffold was determined by microCT. At all time points there was significantly more mineral in the osteogenic scaffolds than the control scaffolds (p< 0.001). At 3 and 5 weeks, significantly more mineral was produced by the MSCs than the AFS cells (p< 0.001). At 10 and 15 weeks, both AFS cell lines produced significantly more mineral than the MSCs (p< 0.001). At 15 weeks, the H1 AFS line also produced significantly more mineral than the A1 cell line. N=6 for controls, n=12 for osteogenic. B. The rate of mineral deposition was calculated for each scaffold and then compiled with its cohort. The rate of mineral deposition was significantly higher in the osteogenic MSCs at 3 weeks (p< 0.001) when compared to osteogenic AFS cells, but at 5 weeks the rate of mineralization between the three osteogenic groups was not significantly different. At 10 and 15 weeks, the rate of mineralization of the osteogenic AFS cell groups was significantly greater than the osteogenic MSCs. At 15 weeks, the H1 mineralization rate was greater than the A1 AFS cell mineralization. At 10 and 15 weeks, the rate of mineralization of the MSCs was not significantly different from the control groups. N=6 for controls, n=12 for osteogenic. (p< 0.001). N=6 for controls, n=12 for osteogenic.
Two AFS cell lines were available at the time of this study and their differentiation potential was compared. Little variability was seen in the osteogenic differentiation of the A1 and H1 AFS cell lines. The mineralized matrix volume did not differ until 15 weeks in culture at which time the H1 AFS cells had produced more mineralized matrix than the A1 AFS cells. Both AFS cell lines produced approximately 5 times the mineralized matrix than the MSCs at 15 weeks. The AFS cell control media constructs also had more mineralized matrix than the MSC control constructs at 15 weeks (p<0.05), which may indicate greater spontaneous differentiation of the AFS cells in the absence of osteogenic cues.
2.6 Rate of 3D Mineralization
The scaffolds were scanned sterile and returned to culture. Therefore, the rate of mineralized matrix production was determined by quantifying the new mineral volume since the previous scan and dividing by the number of weeks between scans. The average rate of mineralized matrix production (Figure 3B) by the MSCs was greater at 3 weeks than the AFS cells (p<0.001). By 5 weeks, the rate of mineralization of the cell types was not significantly different. At 10 and 15 weeks, the AFS cells were producing mineralized matrix at a greater rate than the MSCs (p<0.001). Additionally, at 10 and 15 weeks the MSCs were producing mineralized matrix with a rate that is indistinguishable from the control cells.
2.7 Calcium Content and Cell Number
The AFS cells produced significantly more mineralized matrix, as analyzed by calcium concentration, than the MSCs (Figure 4A). Additionally, both the MSCs and AFS cells produced significantly more mineralized matrix when placed in osteogenic media than in control media (n=6, p<0.001). This increased mineralization was not due to an increased cell number, but to an increased matrix deposition as the AFS cells and MSCs attached to the scaffold in comparable numbers (Figure 4C). The seeding efficiency was comparable for both cell types on the PCL scaffold. After 15 weeks in culture, both the AFS cell and MSC seeded scaffolds had similar cell number whether cultured in control or osteogenic media, as determined by DNA content (Figure 4D). Therefore, human AFS cells demonstrated an increased long-term osteogenic differentiation and mineralized matrix production capacity compared to human MSCs.
2.8 Cell Viability
After 15 weeks in culture, cell viability was determined by a live-dead assay that relied on the intracellular esterase activity and plasma membrane integrity, two measures of cell viability, to discriminate between live and dead cells. Photomicrographs of the exterior surfaces (top, bottom and exterior circumference) are shown (Figure 4B). Additionally, the cells at the interior of the scaffolds were examined by cutting the scaffold longitudinally. It can be observed that even after 15 weeks in culture, the cell viability is very high in all the scaffolds.
2.9 Histology
Histological examination of the scaffolds after 15 weeks, using resin embedding and microtome sectioning to 5μm followed by von Kossa staining revealed no mineralisation for the control (MSCs in CCM) PCL scaffolds (Figure 5 A-D). Only pink stained fibrous tissue can be observed. Whereas the onset of bone formation was observed for osteoinduced MSC and AFS cells which were seeded into 3D PCL scaffolds. Mineral nodules containing calcium, stain black with the von Kossa staining by virtue of silver ions (positive charge) binding with the mineralised tissue (negative portion of the calcium salt) forming a silver salt which is black in colour. Clear black mineral deposition can be observed for osteinduced MSCs (Figure 5 E-H) and AFS cells (I-P), within a fibrous network penetrating the scaffold pores. Residual PCL scaffold was evident within all transplants as evidenced by voids in the tissue from longitudinal and transverse sectioning of the scaffold struts. It can be noted that the pattern of mineral deposition was different between each cell type and seems to favour a more central scaffold locality for MSC cells whereas the AFS cells seem to deposit throughout the entire scaffold both centrally and at the periphery.
3 Discussion
This study demonstrates the mineralization potential of a fetal and adult stem cell population. The fetal AFS cells demonstrated an increased duration of mineralized matrix production, but the MSCs were capable of robust differentiation at the earlier time points. After 15 weeks in 3D culture, the AFS cells had produced approximately 20 mm3 of mineralized matrix, whereas the MSCs had produced approximately 4 mm3 by 5 weeks, which did not increase thereafter. MicroCT analysis showed the mineralization for MSC scaffolds constructs was located mainly within the central core of the scaffold as opposed to the AFS cells which mineralized the entire scaffold including the outer edges.
In 2D culture, the AFS cells differentiate readily and using a standard time course it is not evident that there is a lag in differentiation by these cells. Quantitative PCR comparing osteogenic gene expression between the two cell sources in 2D revealed a lag in the AFS cells when compared to the MSCs. This supports the longer induction time necessary for the AFS cells to differentiate to osteoblasts. The sensitivity of the staining of the extracellular matrix in 2D culture was not capable of determining these early differences in the cell differentiation because by 14 days the gene expression by the AFS cells matched or exceeded the expression in the MSCs.
The 15 week in vitro culture protocol used in this study provided a very challenging culture system to examine long term potential of the cell sources. When grown in a large 6mm × 9mm PCL scaffold, the AFS cells had great potential but the cells required an extensive induction time prior to their osteogenic differentiation. Current studies often culture cells within 3D scaffolds for less than 6 weeks but this time period would not have shown the potential of the AFS cells. Early time points suggest the MSCs have a greater potential, but the AFS cells have a greater overall mineralized matrix production capacity when examined after an extensive culture period. This supports the hypothesis that the AFS cells have a longer induction period than the MSCs, which may be due to their fetal origins.
The mineral deposition distribution found by microCT was supported by histological examination which demonstrated that the pattern of black stained mineral deposition was different between each cell type and seems to favour a more central scaffold locality for MSC cells whereas the AFS cells seem to deposit throughout the entire scaffold both centrally and at the periphery. This could be due to the MSCs requiring local mineral presence in order to nucleate more mineral production which leads to a tendency to concentrate their cells actively capable of mineralisation within the same area (in the scaffold core in this case) whereas AFS cells might be more capable of spontaneous mineralisation without the requiring any prerequisite mineral existing in close proximity. Alternatively the mineral nucleation sites (such as those present on collagen or denatured collagen) [34] might have been distributed more centrally in the case of the MSC scaffold constructs compared to the AFS scaffold constructs. It certainly highlights the differences in mineralisation capacities of these different cells from a temporal, spatial and rate perspective.
Serial microCT scans of scaffolds were performed which allowed the determination of the change in matrix within each scaffold over the 15 weeks in culture. The rate of mineralized matrix production was higher for the MSCs at the beginning of the culture but the cells did not produce detectible mineralized matrix after the 5 week scan. The AFS cells had a low rate of mineralization for the first 3 weeks, but the rate increased thereafter and continued to increase over the 15 weeks of culture. This may be due to the more primitive state of the fetal AFS cells needing additional time or cell density before they will differentiate to osteoblasts.
Although the rate of MSC mineralized matrix production decreased after 5 weeks, the cells maintained high viability throughout the culture. At 15 weeks, the cells were found throughout the scaffolds and there was minimal cell death in scaffolds seeded with either cell type without bias for the spatial localization within the scaffold. Additionally, the AFS cells and MSCs had similar cell numbers attach to the scaffold after 15 weeks in culture so the difference in matrix production was not due to cell proliferation or apoptosis. Maintaining the high viability achieved in vitro will be imperative to have similar results in vivo. The long induction time for the AFS cells may make this even more challenging. Co-implantation of both MSCs and AFS cells may prove beneficial as the MSCs could provide immediate mineralized matrix production and the AFS cells will provide long-term support.
Cell-based tissue engineering strategies represent a clinical alternative to bone grafting and the delivery of osteoinductive proteins[35]. However, cell sourcing is a critical issue for cell-based therapies which aim to regenerate musculoskeletal tissues, and this issue needs to be addressed[9, 36]. Tissue engineering approaches that combine biodegradable scaffolds with stem cells capable of osteogenesis have shown promise as an effective bone graft substitute[37]. Cell-based engineered bone grafts are an attractive alternative to allografts or autografts, particularly when the endogenous supply of stem cells is depleted through advanced age or concurrent therapy[12, 38, 39]. Several factors are critical for the choice of transplanted cells, such as: a) availability in sufficient numbers for therapeutic use, b) immune-tolerance, and c) the ability to promote bone formation on a therapeutic timescale.
Despite well known developmental differences in tissue regeneration and scar formation, very little is known about the differences in phenotype and regenerative capacity of stem cells isolated from different human developmental stages. Although there is evidence that allogeneic stem cells promote bone repair, many tissue-engineering studies have been limited by a lack of quantitative outcome measures to allow direct comparisons between different stem cell sources. Purified mesenchymal stem cells (MSCs) derived from bone marrow have been shown to enhance repair of critically-sized defects in preclinical animal studies[38, 40].
Many questions still remain pertaining to the use of stem cells for regenerative medicine. Although we have shown that the AFS cells have an increased osteogenic capacity than the MSCs in vitro, the comparison of this capacity in vivo is of greater clinical significance. Critical factors are still unknown for the optimal delivery strategy, such as: a) should the cells be undifferentiated or pre-differentiated in culture prior to implantation, b) are exogenous factors such as pro-angiogenic growth factors important for revascularization, c) what is the optimal time point for implantation post-trauma, d) how many cells should be delivered and should they be implanted in one site as a bolus or at multiple implantation sites or specific designated times? There is vast potential for stem cells in regenerative medicine, and determining the optimal cell source will certainly improve patient outcome.
4. Methods
4.1 Amniotic fluid stem cell culture
Human AFS cells were kindly provided by the Institute for Regenerative Medicine at Wake Forest University[23]. In this study, two cell lines were available and both were analyzed, A1 and H1 human AFS cells. The AFS cells were received at passage 14, and further expanded 2-3 times in α-MEM medium containing 15% FBS, 2mM L-glutamine, 100 units penicillin, 100 μg streptomycin (all Invitrogen, Carlsbad, Ca), supplemented with 18% Chang B and 2% Chang C (Irvine Scientific, Santa Ana, CA) at 37°C with 5% CO2 atmosphere. This media is referred to in this paper as modified Chang media. AFS cells were sub-cultured at a dilution of 1:10 and not permitted to expand beyond 70% confluence in the modified Chang media on Integrid plates (BD Falcon, San Jose, CA). Cells were frozen in the modified Chang media supplemented with 5% DMSO. For experimental use, 1×106 AFS cells were quickly thawed to 37°C and placed in 10, 150-mm Integrid culture plates with modified Chang media. After 5 days, the cells were harvested with 0.25% trypsin-EDTA (Invitrogen), counted and used experimentally.
4.2 Mesenchymal stem cell culture
Human MSCs were generously provided by the Texas A&M Health Science Center through the NIH-funded center for preparation and distribution of adult stem cells (2P40RR017447-08). MSCs from four adult donors were expanded by plating the cells at an initial density of 50 cells per cm2 and cultured in complete culture media (CCM) or also referred to as Control Media. CCM consists of alpha-modified minimal essential media (αMEM, Invitrogen) supplemented with 17% FBS (Atlanta Biologicals), 2mM L-glutamine, 100 units penicillin, 100 μg streptomycin (all Invitrogen). Cells were cultured on tissue culture dishes (Nuncleon Δ surface, Thermo Fisher, Rochester, NY). After 7-9 days, the cells were harvested with 0.25% trypsin-EDTA (Invitrogen), counted and used experimentally. Cells were not permitted to expand beyond 70% confluence and were frozen in αMEM supplemented with 30% serum and 5% DMSO. For experimental use, all four MSC donor cells were quickly thawed to 37°C and placed in four 10-mm Nuncleon culture plates with CCM to recover from freezing. The next day the cells were removed from the plates with trypsin-EDTA and counted. Equal quantities of each donor MSCs were combined and placed in Nuncleon culture plates at 50 cells per cm2. After 7 days, the cells were harvested with 0.25% trypsin-EDTA, counted and used experimentally.
4.3 Osteogenic Differentiation
AFS cells or MSCs were cultured at 20,000 cells per cm2 in 6 well plates (Nunc, Rochester, NY) in either modified Chang media (AFS cells) or CCM (MSCs), N = 6 for all conditions. After allowing the cells to attach to the culture dish for 24 hours, the media was changed to the osteogenic induction conditions, which comprised CCM media supplemented with1 μM Dexamethasone, 6 mM β-glycerol phosphate, 50 μg/ml Ascorbic acid 2-Phosphate, and 50 ng/ml Thyroxine (all Sigma, St. Louis, MO)[10]. Control samples of both MSCs and AFS cells were grown in CCM to exclude spontaneous differentiation. The media was changed 2 times per week. Cells were analyzed 2 and 4 weeks after the start of differentiation.
4.4 Alizarin Red S Staining for Calcium
The cells were washed with excess PBS (Mg2+ and Ca2+ free, Invitrogen) and fixed in 10% neutral buffered formalin (Sigma) for 15 minutes. The cells were washed 3× with water, then stained with 2 ml 0.4mM Alizarin red S (pH 4.2, Sigma) for 20 minutes with rocking. Calcium forms an alizarin red S-calcium complex in a chelation process, producing a dark red stain. The excess Alizarin red S stain was removed and using vigorous washing with excess water (4 times for 5 minutes each, with rocking). Stained monolayers were visualized by phase microscopy using an inverted microscope (Nikon, Melville, NY).
4.5 Gene Expression by real-time RT-PCR
For each cell type, 50,000 cells per cm2 were placed into 6 well dishes and cultured overnight in CCM. After allowing the cells to attach to the culture dish for 24 hours, the media was changed to osteogenic media and the media was changed every 3 days throughout the culture. Cells were harvested at days 0, 1, 2, 3, 5, 7, 10 and 14 using a cell scraper (Nunc) and the RNA was isolated using Qiagen RNEasy Plus kit according to the manufacturer's instructions. cDNA was produced from 1 μg total RNA using Superscript III First-strand Synthesis SuperMix according to manufacturer's instructions (Invitrogen). Real-time PCR was performed on a StepOnePlus Real-Time PCR System (Applied Biosystems) with Power Sybr Green PCR Master Mix (Applied Biosystems). The cycle threshold was normalized to GapDH and compared to a relative standard curve for each primer set. Primer sequences and concentrations are given in Supplimentary Table S1.
4.6 Scaffold preparation
3-dimensional sheets of PCL lattice comprised of 300 μm struts spaced 500 μm apart in a 0, 60, 120 degree repeating pattern with 85% porosity and a height of 9mm were produced through fused deposition modeling as previously described[14, 30] with a height of 9 mm. Cylinders of a 6 mm diameter were cut from the sheet with a dermal biopsy punch (Miltex, York, PA). The cylinders were incubated in 5M NaOH for 2 hours at 37°C to clean and partially degrade the scaffold surface, thereby increasing surface roughness. The scaffolds were then washed 3× in excess sterile water and sterilized through 70% ethanol evaporation.
To produce a collagen network throughout the pores of the PCL lattice, a collagen gel was produced with type 1 rat tail collagen (Vitrogen, Fremont, CA). Briefly, 100 parts collagen (1.4 mg/ml in 0.05% acetic acid) was combined with 9 parts sodium bicarbonate, and 250 μl was placed in a custom mold. The PCL cylinder was then placed in the mold/collagen and the collagen allowed to gel for 30 minutes at room temperature, and then cooled to -80°C for 2 hours. The PCL/collagen was then placed in a lyophilizer overnight (Labconco, Kansas City, MO). After lyophilization, the scaffolds were removed from the mold and placed in a 12 well tissue culture dish (Nunc, low cell binding). To maintain scaffold orientation during culture, the cylinders were placed in a holder consisting of a sterile 3/4″ Teflon disk with 4 stainless steel pins surrounding the cylinder.
4.7 Cell seeding onto PCL/collagen scaffolds
AFS cells and MSCs were expanded as described above. Six million cells were resuspended in 150 μl of modified Chang media or CCM and slowly placed drop-wise on the top of the scaffold. The media were readily absorbed by the collagen mesh, with minimal pooling at the bottom. The cell/scaffold was placed in the 37°C incubator for 1 hour to promote cell attachment, after which time 4 ml of modified Chang media or CCM was carefully added to each well. The scaffolds were cultured under static conditions for 3 days to allow the cells to attach to the scaffold.
After 3 days, the media was carefully aspirated and osteogenic media was added to each osteogenic sample and control samples were cultured in CCM. At this time, the scaffolds were placed on a rocker plate to increase media perfusion through the scaffold (Belly Button® orbital shaker, 7.5 rpm, minimal pitch, Stovall, Greensboro, NC). The media was changed every 2-3 days for 15 weeks.
The seeding efficiency was determined at day 3, prior to the placement in osteogenic media. The scaffolds were washed 2× with PBS to remove unattached cells. The scaffolds were then immersed in 1ml of Trypsin-EDTA for 5 minutes at 37°C. The trypsin was inactivated with 5ml complete culture media and the cells pelleted by centrifugation at 450×g for 10 minutes. The cell pellets were resuspended in 3 ml CCM, stained with trypan blue and counted in duplicate on a hemocytometer. N=4.
4.8 MicroCT scan
At 3, 5, 10, and 15 weeks, the scaffolds were removed from the Teflon holders and placed in a sterile polysulfone sample holder. Mineralization of the scaffolds was quantified using the VivaCT (Scanco Medical, Switzerland) at a 21.5 voxel resolution. Samples were evaluated at a threshold of 80, a filter width of 1.2, and filter support of 1. For each scaffold, a measurement of the volume filled with hydroxyapatite was determined. The samples were then removed aseptically from the sample holder and returned to the Teflon holder for further culture. N=12.
4.9 Calcium Assay
At 15 weeks in either osteogenic or control media, the scaffold was dissected into 4 sections by slicing in a custom matrix so that each scaffold was cut vertically across the diameter of the cylinder and horizontally to produce 4 sections of equal size. ¼ of the scaffold, ½ of the top half of each scaffold, was used to determine the calcium within the matrix. Each scaffold was placed in 500μl of 1M acetic acid and placed on a vortex overnight at 4°C to extract the calcium from the mineralized matrix.
In a 96 well clear polycarbonate plate, 25μl of cell extract were mixed with 300μl of calcium reagent (arsenazo III, Diagnostic Chemicals Limited) and the absorbance determined at 615-nm with a spectrophotometer. N=6.
4.10 DNA Assay
The PicoGreen DNA quantification kit (Invitrogen) was used to following the protocol recommended by the manufacturer to determine at 15 weeks the relative amount of DNA within scaffolds from the different experimental groups. Lambda DNA standards were produced from 1μg to 1ng. The cell lysates were diluted 1:10 in Tris-EDTA buffer. 100 μl of the PicoGreen working solution and 100μl of each sample were placed in triplicate, in black 96-well plates. After a 5-minute incubation, the fluorescence was determined at an excitation of 485-nm and an emission of 535-nm (Perkin-Elmer HTX 7000 fluorescent plate reader, Waltham, MA). N=6.
4.11 Cell Viability Staining
Cell viability at 15 weeks was determined using the LIVE/DEAD Viability/Cytotoxicity Kit for mammalian cells according to manufacturer's instructions (Molecular Probes, Invitrogen). Briefly, scaffolds were cut longitudinally and washed 3 times in excess PBS and then incubated for 45 minutes in 4 μM Calcein AM and 4 μM Ethidium homodimer-1 with gentle shaking. The scaffolds were then washed 3 times with PBS and analyzed using a confocal microscope (LSM 510 UV, Carl Zeiss, Thornwood, NY). Micrographs were taken on the exterior surfaces: top, bottom, and outside surfaces as well as the cut surface to check the viability at the core of the scaffold.
4.12 Histology
For histological examination, scaffolds were removed from culture at 15 weeks and fixed in 4% paraformaldehyde. An ethanol gradient (30 minutes in 70%, 1 hour in 90%, 95% and 100% ethanol) was used to dehydrate the samples. They were next processed three times through xylenes for 40 minutes each, infiltrated with methylmathacrylate (MMA) for 3h and embedded in MMA containing 3% PEG softener. Five micrometre sections were cut with an osteomicrotome (SM2500; Leica Microsystems, Wetzlar, Germany), stretched flat with 70% ethanol onto a polylysine coated microscope slide (Lomb Scientific), overlayed with a plastic film and slides were clamped together before being dried overnight at 50°C. Sections were then stained using combined von Kossa and van Giesen stains to visualise the mineralised bone and connective tissue respectively [41].
4.13 Data Analysis
Data are reported as mean +/- SE and statistical analyses was performed using Graphpad Prism 5 software using a general linear model (ANOVA) and Tukey's post-hoc analysis with Bonferroni adjustment or pair wise comparisons; with p< 0.05 considered significant.
Supplementary Material
Supplementary Figure 1: A-C. Low density photomicrographs show the morphology of the AFS cells and the MSCs. Both the AFS cells and MSCs show heterogeneity, but the AFS cells (A and B) have a distinct small-sized cell population not seen in MSC culture (C). Osteogenic capability of the MSCs and AFS cells can be seen in D-F. Alizarin red S staining shows dense calcium deposits covering the cell layers.
Supplementary Figure 2: The mineralized matrix production by the MSCs and AFS cells on 3D constructs is highly reproducible. Representative scaffolds of the osteogenic AFS cells and MSCs at 15 weeks are shown to illustrate the distribution of mineralized matrix through the PCL scaffold. Scaffolds 1-4 (of 12) of each cell type are shown for comparison.
Supplementary Figure 3: The extracellular matrix accumulation can be easily visualized at 15 weeks in vitro in all scaffolds seeded with MSCs or AFS cells in both control and osteogenic media. The highly porous nature of the scaffold can be seen in the no cell control. The arrow points to the white calcified area of the MSC scaffold which is also seen on the microCT images. Additionally, the osteogenic AFS cells produced a white calcified shell around the scaffold.
Acknowledgments
This work was supported by NIH grant R01-AR056694, 2K12GM000680 and the Georgia Tech/Emory Center for the Engineering of Living Tissues (GTEC) NSF grant EEC-9731643.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Owen M, Friedenstein AJ. Stromal stem cells: marrow-derived osteogenic precursors. Ciba Found Symp. 1988;136:42–60. doi: 10.1002/9780470513637.ch4. [DOI] [PubMed] [Google Scholar]
- 2.Owen ME, Cave J, Joyner CJ. Clonal analysis in vitro of osteogenic differentiation of marrow CFU-F. J Cell Sci. 1987;87(Pt 5):731–738. doi: 10.1242/jcs.87.5.731. [DOI] [PubMed] [Google Scholar]
- 3.Bruder SP, Jaiswal N, Ricalton NS, Mosca JD, Kraus KH, Kadiyala S. Mesenchymal stem cells in osteobiology and applied bone regeneration. Clin Orthop Relat Res. 1998:S247–256. doi: 10.1097/00003086-199810001-00025. [DOI] [PubMed] [Google Scholar]
- 4.Tsutsumi S, Shimazu A, Miyazaki K, Pan H, Koike C, Yoshida E, Takagishi K, Kato Y. Retention of multilineage differentiation potential of mesenchymal cells during proliferation in response to FGF. Biochem Biophys Res Commun. 2001;288:413–419. doi: 10.1006/bbrc.2001.5777. [DOI] [PubMed] [Google Scholar]
- 5.Sekiya I, Vuoristo JT, Larson BL, Prockop DJ. In vitro cartilage formation by human adult stem cells from bone marrow stroma defines the sequence of cellular and molecular events during chondrogenesis. Proc Natl Acad Sci U S A. 2002;99:4397–4402. doi: 10.1073/pnas.052716199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnstone B, Hering TM, Caplan AI, Goldberg VM, Yoo JU. In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells. Exp Cell Res. 1998;238:265–272. doi: 10.1006/excr.1997.3858. [DOI] [PubMed] [Google Scholar]
- 7.Wakitani S, Saito T, Caplan AI. Myogenic cells derived from rat bone marrow mesenchymal stem cells exposed to 5-azacytidine. Muscle Nerve. 1995;18:1417–1426. doi: 10.1002/mus.880181212. [DOI] [PubMed] [Google Scholar]
- 8.Eliopoulos N, Stagg J, Lejeune L, Pommey S, Galipeau J. Allogeneic marrow stromal cells are immune rejected by MHC class I- and class II-mismatched recipient mice. Blood. 2005;106:4057–4065. doi: 10.1182/blood-2005-03-1004. [DOI] [PubMed] [Google Scholar]
- 9.Caplan AI. Review: mesenchymal stem cells: cell-based reconstructive therapy in orthopedics. Tissue Eng. 2005;11:1198–1211. doi: 10.1089/ten.2005.11.1198. [DOI] [PubMed] [Google Scholar]
- 10.Peister A, Mellad JA, Larson BL, Hall BM, Gibson LF, Prockop DJ. Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood. 2004;103:1662–1668. doi: 10.1182/blood-2003-09-3070. [DOI] [PubMed] [Google Scholar]
- 11.Marie PJ, Fromigue O. Osteogenic differentiation of human marrow-derived mesenchymal stem cells. Regen Med. 2006;1:539–548. doi: 10.2217/17460751.1.4.539. [DOI] [PubMed] [Google Scholar]
- 12.Hutmacher DW, Schantz T, Zein I, Ng KW, Teoh SH, Tan KC. Mechanical properties and cell cultural response of polycaprolactone scaffolds designed and fabricated via fused deposition modeling. J Biomed Mater Res. 2001;55:203–216. doi: 10.1002/1097-4636(200105)55:2<203::aid-jbm1007>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Cartmell S, Huynh K, Lin A, Nagaraja S, Guldberg R. Quantitative microcomputed tomography analysis of mineralization within three-dimensional scaffolds in vitro. J Biomed Mater Res A. 2004;69:97–104. doi: 10.1002/jbm.a.20118. [DOI] [PubMed] [Google Scholar]
- 14.Porter BD, Lin AS, Peister A, Hutmacher D, Guldberg RE. Noninvasive image analysis of 3D construct mineralization in a perfusion bioreactor. Biomaterials. 2007;28:2525–2533. doi: 10.1016/j.biomaterials.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Choi SA, Lee JH, Kim KJ, Kim EY, Park KS, Park YB, Li X, Ha YN, Park JY, Kim MK. 291 Isolation and Characterization of Mesenchymal Stem Cells Derived from Human Amniotic Fluid. Reprod Fertil Dev. 2011;23:243. [Google Scholar]
- 16.De Coppi P, Callegari A, Chiavegato A, Gasparotto L, Piccoli M, Taiani J, Pozzobon M, Boldrin L, Okabe M, Cozzi E, Atala A, Gamba P, Sartore S. Amniotic fluid and bone marrow derived mesenchymal stem cells can be converted to smooth muscle cells in the cryo-injured rat bladder and prevent compensatory hypertrophy of surviving smooth muscle cells. J Urol. 2007;177:369–376. doi: 10.1016/j.juro.2006.09.103. [DOI] [PubMed] [Google Scholar]
- 17.Delo DM, De Coppi P, Bartsch G, Jr, Atala A. Amniotic fluid and placental stem cells. Methods Enzymol. 2006;419:426–438. doi: 10.1016/S0076-6879(06)19017-5. [DOI] [PubMed] [Google Scholar]
- 18.Cananzi M, Atala A, De Coppi P. Stem cells derived from amniotic fluid: new potentials in regenerative medicine. Reprod Biomed Online. 2009;18 1:17–27. doi: 10.1016/s1472-6483(10)60111-3. [DOI] [PubMed] [Google Scholar]
- 19.In't Anker PS, Scherjon SA, Kleijburg-van der Keur C, Noort WA, Claas FH, Willemze R, Fibbe WE, Kanhai HH. Amniotic fluid as a novel source of mesenchymal stem cells for therapeutic transplantation. Blood. 2003;102:1548–1549. doi: 10.1182/blood-2003-04-1291. [DOI] [PubMed] [Google Scholar]
- 20.Prusa AR, Marton E, Rosner M, Bernaschek G, Hengstschlager M. Oct-4-expressing cells in human amniotic fluid: a new source for stem cell research? Hum Reprod. 2003;18:1489–1493. doi: 10.1093/humrep/deg279. [DOI] [PubMed] [Google Scholar]
- 21.Tsai MS, Lee JL, Chang YJ, Hwang SM. Isolation of human multipotent mesenchymal stem cells from second-trimester amniotic fluid using a novel two-stage culture protocol. Hum Reprod. 2004;19:1450–1456. doi: 10.1093/humrep/deh279. [DOI] [PubMed] [Google Scholar]
- 22.Roubelakis MG, Pappa KI, Bitsika V, Zagoura D, Vlahou A, Papadaki HA, Antsaklis A, Anagnou NP. Molecular and proteomic characterization of human mesenchymal stem cells derived from amniotic fluid: comparison to bone marrow mesenchymal stem cells. Stem Cells Dev. 2007;16:931–952. doi: 10.1089/scd.2007.0036. [DOI] [PubMed] [Google Scholar]
- 23.De Coppi P, Bartsch G, Jr, Siddiqui MM, Xu T, Santos CC, Perin L, Mostoslavsky G, Serre AC, Snyder EY, Yoo JJ, Furth ME, Soker S, Atala A. Isolation of amniotic stem cell lines with potential for therapy. Nat Biotechnol. 2007;25:100–106. doi: 10.1038/nbt1274. [DOI] [PubMed] [Google Scholar]
- 24.Kolambkar YM, Peister A, Soker S, Atala A, Guldberg RE. Chondrogenic differentiation of amniotic fluid-derived stem cells. J Mol Histol. 2007;38:405–413. doi: 10.1007/s10735-007-9118-1. [DOI] [PubMed] [Google Scholar]
- 25.Ditadi A, de Coppi P, Picone O, Gautreau L, Smati R, Six E, Bonhomme D, Ezine S, Frydman R, Cavazzana-Calvo M, Andre-Schmutz I. Human and murine amniotic fluid c-Kit+Lin-cells display hematopoietic activity. Blood. 2009;113:3953–3960. doi: 10.1182/blood-2008-10-182105. [DOI] [PubMed] [Google Scholar]
- 26.Zheng YM, Zhao HY, Zhao XE, Quan FS, Hua S, He XY, Liu J, He XN, Lin H. Development of cloned embryos from porcine neural stem cells and amniotic fluid-derived stem cells transfected with enhanced green fluorescence protein gene. Reproduction. 2009;137:793–801. doi: 10.1530/REP-08-0469. [DOI] [PubMed] [Google Scholar]
- 27.Cipriani S, Bonini D, Marchina E, Balgkouranidou I, Caimi L, Grassi Zucconi G, Barlati S. Mesenchymal cells from human amniotic fluid survive and migrate after transplantation into adult rat brain. Cell Biol Int. 2007;31:845–850. doi: 10.1016/j.cellbi.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 28.Carraro G, Perin L, Sedrakyan S, Giuliani S, Tiozzo C, Lee J, Turcatel G, De Langhe SP, Driscoll B, Bellusci S, Minoo P, Atala A, De Filippo RE, Warburton D. Human amniotic fluid stem cells can integrate and differentiate into epithelial lung lineages. Stem Cells. 2008;26:2902–2911. doi: 10.1634/stemcells.2008-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hauser PV, De Fazio R, Bruno S, Sdei S, Grange C, Bussolati B, Benedetto C, Camussi G. Stem cells derived from human amniotic fluid contribute to acute kidney injury recovery. Am J Pathol. 2010;177:2011–2021. doi: 10.2353/ajpath.2010.091245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peister A, Deutsch ER, Kolambkar Y, Hutmacher DW, Guldberg RE. Amniotic fluid stem cells produce robust mineral deposits on biodegradable scaffolds. Tissue Eng Part A. 2009;15:3129–3138. doi: 10.1089/ten.tea.2008.0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun H, Feng K, Hu J, Soker S, Atala A, Ma PX. Osteogenic differentiation of human amniotic fluid-derived stem cells induced by bone morphogenetic protein-7 and enhanced by nanofibrous scaffolds. Biomaterials. 2010;31:1133–1139. doi: 10.1016/j.biomaterials.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peister A, Porter BD, Kolambkar YM, Hutmacher DW, Guldberg RE. Osteogenic differentiation of amniotic fluid stem cells. Biomed Mater Eng. 2008;18:241–246. [PubMed] [Google Scholar]
- 33.Kolambkar YM, Peister A, Ekaputra AK, Hutmacher DW, Guldberg R. Colonization and osteogenic differentiation of different stem cell sources on electrospun nanofiber meshes. Tissue Eng Part A. 2010 doi: 10.1089/ten.tea.2010.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taubenberger AV, Woodruff MA, Bai H, Muller DJ, Hutmacher DW. The effect of unlocking RGD-motifs in collagen I on pre-osteoblast adhesion and differentiation. Biomaterials. 2010;31:2827–2835. doi: 10.1016/j.biomaterials.2009.12.051. [DOI] [PubMed] [Google Scholar]
- 35.Kimelman N, Pelled G, Helm GA, Huard J, Schwarz EM, Gazit D. Review: gene- and stem cell-based therapeutics for bone regeneration and repair. Tissue Eng. 2007;13:1135–1150. doi: 10.1089/ten.2007.0096. [DOI] [PubMed] [Google Scholar]
- 36.Barrilleaux B, Phinney DG, Prockop DJ, O'Connor KC. Review: ex vivo engineering of living tissues with adult stem cells. Tissue Eng. 2006;12:3007–3019. doi: 10.1089/ten.2006.12.3007. [DOI] [PubMed] [Google Scholar]
- 37.Meinel L, Karageorgiou V, Fajardo R, Snyder B, Shinde-Patil V, Zichner L, Kaplan D, Langer R, Vunjak-Novakovic G. Bone tissue engineering using human mesenchymal stem cells: effects of scaffold material and medium flow. Ann Biomed Eng. 2004;32:112–122. doi: 10.1023/b:abme.0000007796.48329.b4. [DOI] [PubMed] [Google Scholar]
- 38.Bruder SP, Fox BS. Tissue engineering of bone. Cell based strategies. Clin Orthop Relat Res. 1999:S68–83. doi: 10.1097/00003086-199910001-00008. [DOI] [PubMed] [Google Scholar]
- 39.Salgado AJ, Coutinho OP, Reis RL. Bone tissue engineering: state of the art and future trends. Macromol Biosci. 2004;4:743–765. doi: 10.1002/mabi.200400026. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez-Guijo FM, Blanco JF, Cruz G, Muntion S, Gomez M, Carrancio S, Lopez-Villar O, Barbado MV, Sanchez-Abarca LI, Blanco B, Brinon JG, del Canizo MC. Multiparametric comparison of mesenchymal stromal cells obtained from trabecular bone by using a novel isolation method with those obtained by iliac crest aspiration from the same subjects. Cell Tissue Res. 2009;336:501–507. doi: 10.1007/s00441-009-0778-x. [DOI] [PubMed] [Google Scholar]
- 41.Reichert JC, Woodruff MA, Friis T, Quent VM, Gronthos S, Duda GN, Schutz MA, Hutmacher DW. Ovine bone- and marrow-derived progenitor cells and their potential for scaffold-based bone tissue engineering applications in vitro and in vivo. J Tissue Eng Regen Med. 2010;4:565–576. doi: 10.1002/term.276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: A-C. Low density photomicrographs show the morphology of the AFS cells and the MSCs. Both the AFS cells and MSCs show heterogeneity, but the AFS cells (A and B) have a distinct small-sized cell population not seen in MSC culture (C). Osteogenic capability of the MSCs and AFS cells can be seen in D-F. Alizarin red S staining shows dense calcium deposits covering the cell layers.
Supplementary Figure 2: The mineralized matrix production by the MSCs and AFS cells on 3D constructs is highly reproducible. Representative scaffolds of the osteogenic AFS cells and MSCs at 15 weeks are shown to illustrate the distribution of mineralized matrix through the PCL scaffold. Scaffolds 1-4 (of 12) of each cell type are shown for comparison.
Supplementary Figure 3: The extracellular matrix accumulation can be easily visualized at 15 weeks in vitro in all scaffolds seeded with MSCs or AFS cells in both control and osteogenic media. The highly porous nature of the scaffold can be seen in the no cell control. The arrow points to the white calcified area of the MSC scaffold which is also seen on the microCT images. Additionally, the osteogenic AFS cells produced a white calcified shell around the scaffold.