

Abstract
Motivation: High-throughput techniques facilitate the simultaneous measurement of DNA copy number at hundreds of thousands of sites on a genome. Older techniques allow measurement only of total copy number, the sum of the copy number contributions from the two parental chromosomes. Newer single nucleotide polymorphism (SNP) techniques can in addition enable quantifying parent-specific copy number (PSCN). The raw data from such experiments are two-dimensional, but are unphased. Consequently, inference based on them necessitates development of new analytic methods.
Methods: We have adapted and enhanced the circular binary segmentation (CBS) algorithm for this purpose with focus on paired test and reference samples. The essence of paired parent-specific CBS (Paired PSCBS) is to utilize the original CBS algorithm to identify regions of equal total copy number and then to further segment these regions where there have been changes in PSCN. For the final set of regions, calls are made of equal parental copy number and loss of heterozygosity (LOH). PSCN estimates are computed both before and after calling.
Results: The methodology is evaluated by simulation and on glioblastoma data. In the simulation, PSCBS compares favorably to established methods. On the glioblastoma data, PSCBS identifies interesting genomic regions, such as copy-neutral LOH.
Availability: The Paired PSCBS method is implemented in an open-source R package named PSCBS, available on CRAN (http://cran.r-project.org/).
Contact: olshena@biostat.ucsf.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Changes in the number of copies of genomic DNA is an important step in the progression of cancer. Comparative genomic hybridization (CGH) was developed to identify these changes at a resolution of 10–20 Mb (Kallioniemi et al., 1992). Platforms for copy number (CN) analysis that employ microarray technology and that achieve high resolution include array CGH (Pinkel et al., 1998), ROMA (Lucito et al., 2003) and SNP arrays (Hardenbol et al., 2005; Peiffer et al., 2006; Zhao et al., 2004). Current technology has improved the resolution to as low as 1 kb. With custom arrays also available, the resolution in particular neighborhoods can be even higher.
Heretofore, CN analysis has consisted primarily of examining total copy number (TCN). TCN is the sum of the CNs from the two parental chromosomes. For normal human cells, total CN is two, one from each parental chromosome. SNP arrays allow separate estimates of CN from the parental chromosomes. This is parent-specific copy number (PSCN).
PSCN may be interesting for two major reasons. First, there may be alleles that differentially undergo CN change (Nagase et al., 2003). Estimating PSCN would help elucidate this situation. Second, when the total CN is C, the PSCNs may be more complicated than (1,C−1). For instance, diploid (C=2) CN is maintained when one parental copy is lost and the other is doubled. This type of alteration is called copy-neutral loss-of-heterozygosity (CN-LOH), and it occurs often in many cancers including glioblastoma (Kuga et al., 2008) and hematologic malignancies (O'Keefe et al., 2010). Such a region would be assumed normal if there was analysis only of total CN.
Direct estimates of PSCN can be made only for SNPs at which a subject is heterozygous. Homozygous SNPs are not directly informative because all the CN signal is in one allele. For example, if both parents contributed G, then there would only be a G CN signal, and this would result in no information additional to that contained in total CN. For heterozygous SNPs, however, there are two components to the CN information. If the subject was GT at a SNP, then there would be a CN estimate corresponding to G and one corresponding to T. One of the CNs would be expected to have come from one parent and the other to have come from the other parent. Additionally, the data are unphased; it is not directly known which measurement is associated with which parental chromosome.
CN alterations apply to contiguous regions, and the data on CN derived from microarrays can be noisy. Therefore, methods have been developed to analyze CN data that rely on the underlying spatial correlation. The idea is to split the genome into regions of equal total CN. Methods for this have included direct segmentation (Olshen et al., 2004; Picard et al., 2005; Venkatraman and Olshen, 2007), hidden Markov models (HMMs) (Fridlyand et al., 2004; Guha et al., 2008; Lai et al., 2008) and smoothing (Hsu et al., 2005; Tibshirani and Wang, 2008). When the earlier methods were compared (Lai et al., 2005; Willenbrock and Fridlyand, 2005), direct segmentation methods performed best.
The purpose of the present article is to extend segmentation to allele-specific data. We cannot simply perform two separate segmentations, one for each parental chromosome, because the data are unphased. Therefore, other techniques are needed. As part of our algorithm, we use the circular binary segmentation (CBS) method (Olshen et al., 2004; Venkatraman and Olshen, 2007), although any good segmentation method could replace CBS in our overall procedure. We call our method Paired Parent-Specific CBS (‘Paired PSCBS’ or just ‘PSCBS’), while acknowledging that due to the lack of phase information, we cannot assign segmentation-based estimates to the paternal or maternal chromosomes.
Other approaches to PSCN segmentation bear some resemblance to PSCBS. Here, we focus on the BAF segmentation method of Staaf et al. (2008), especially since their study provides a comparison to existing methods. It is similar in that it relies on CBS and it adapts to datasets consisting of paired tumor and normal samples. It essentially segments the mirrored B-allele frequency (mirrored BAF), which is the ratio of the higher parental copy number to the total copy number, after removing all homozygotes identified in the normal samples. It differs from PSCBS, as discussed in Section 2, in that it segments only heterozygous SNPs, whereas Paired PSCBS has an advantage in that it utilizes all SNPs as well as any non-polymorphic loci. Another advantage of Paired PSCBS over BAF segmentation is that it uses the normal sample to more accurately quantify the tumor data (Bengtsson et al., 2010).
Another paired method of which we are aware is a hidden Markov method that segments jointly on TCN and mirrored BAF (Lamy et al., 2007). But since it is specific to Affymetrix arrays, and we are interested only in general methods, we did not evaluate it. During the review of this article, Van Loo et al. (2010) published a paired joint segmentation method that was not studied here.
Other methodologies exist that are not based on paired samples. LaFramboise et al. (2005) used CBS to segment total CN data, and then estimated parental CN within segments. By not segmenting the allele-specific data, certain events may be missed. Li et al. (2008) developed a similar procedure using an HMM. They referred to the mirrored BAF as the major copy proportion, so their method is called MCP. SOMATICs (Assié et al., 2008) uses the BAF, which is the ratio of the B-allele to the total CN, to identify CN abnormalities that are then confirmed by the total CN. QuantiSNP (Colella et al., 2007) and PennCNV (Wang et al., 2007) are two HMM methods that rely on the same six-state model. Sun et al. (2009) is a ‘2d’ HMM method in the same vein as PennCNV and QuantiSNP, but that has been adapted to cancer studies. Recently, GAP (Popova et al., 2009) segments total CN and allelic ratio independently and then considers the segments defined by the union of the two sets of change-points. Chen et al. (2011) extended their HMM methodology (Lai et al., 2008) to allele-specific data. An advantage of Chen's HMM method (PSCN) is that there is no limit on the number of states. In addition, Greenman et al. (2010) developed the PICNIC method, which is also based on an HMM and assigns integer CN states.
In the present article, Section 2 covers our methods. Section 3 contains simulations that show the effectiveness of our procedure, as well as an example drawn from glioblastoma data. Finally, Section 4 has discussion.
2 METHODS
The paired parent-specific CBS (Paired PSCBS) algorithm leverages the CBS method (Olshen et al., 2004; Venkatraman and Olshen, 2007) for segmenting total CN data to the 2D unphased data arising from SNP arrays. The algorithm depends on paired test (tumor) and reference (normal) samples that are hybridized to separate arrays.
2.1 Parental-specific data at the locus level
In this subsection, we introduce the locus-level components that go into the segmentation and calling.
2.1.1 Total CNs and allele B fractions
SNP arrays quantify both total and allele-specific signals at the loci of a large number of SNPs. Some platforms also provide total CN estimates (TCNs) at a large number of non-polymorphic loci. For a locus i=1,2,…,m of either type, on a chromosome, chromosome arm or other region under consideration, let Xi denote the observed total CN ratio for a test sample relative to a reference (here a matched normal sample), where the ratio is multiplied by two for a diploid genome. If the locus is a SNP, we also have allele-specific CNs, which we denote (Ai,Bi), where the TCN is Xi=Ai+Bi (Fig. 1a). If the subject is homozygous at SNP i, then the minimum of Ai and Bi should be zero plus noise, and thus all of the true CN signal is in one of the alleles. If the subject is heterozygous at SNP i, then there should be significantly non-zero CN for both Ai and Bi, and in the case there is balanced heterozygosity, then Ai and Bi should be approximately equal.
Fig. 1.

Total CN (a), raw allele B fractions (b), TumorBoost normalized BAFs (c) and DH (d) of chromosome 7 of TCGA sample TCGA-02-0007. Normalized BAFs are less noisy than raw BAFs. As TCN quantifies the difference in total CN between tumor and normal, DH does the same for allelic ratios. From (a) and (d), we conclude that the p-arm (0–60 Mb) has approximately balanced CN between the two parental chromosomes, while the q-arm (60–160 Mb) has extreme allelic imbalance, indicating LOH.
A convenient representation for SNPs is (Xi,βi), where βi=Bi/Xi, which is the ratio of the B-allele CN to the total CN in the test sample (Fig. 1b). This quantity is known as the allele B fraction (BAF) (Bengtsson et al., 2010). It has also been called the B-allele frequency (Staaf et al., 2008), which may be misleading because it is not a frequency in the strict statistical sense in that it does not involve a count. We note that for a homozygous SNP i, βi is near zero or one, e.g. for SNPs that are AA and BB as well as AAA and BBB. For a balanced heterozygous SNP, βi is near one half, e.g. for SNPs that are AB as well as AABB. Note that by using this representation, we have a total CN signal Xi for any locus, regardless of whether it is a SNP.
2.1.2 Allelic imbalances
The total CN signals do not contain information on allelic imbalances. In addition, homozygous SNPs do not carry information on allelic imbalances, since the two parental components cannot be separated by the array. It is only SNPs that are heterozygous in the germline that provide this information (Assié et al., 2008; Bengtsson et al., 2010; Peiffer et al., 2006; Staaf et al., 2008). Because of this, all information on allelic imbalances is preserved in what we call decrease in heterozygosity (DH) (Bengtsson et al., 2010). DH is defined for heterozygous SNPs as
| (1) |
It provides a measure of how much the allelic composition of the tumor has diverged from the normal (germline) state. It is similar to the mirrored BAF (Staaf et al., 2008).
When there is a parental CN change, true TCN, true DH or both can change. We specify cases where only one of them changes. For instance, in a tumor without normal contamination where PSCNs shift from (0,1) to (0,2), the change can in principal only be observed in TCN. Conversely, when there is a shift from (1,1) to (0,2), the change can only be observed in DH.
2.1.3 Identifying heterozygous SNPs
In order to calculate the DHs, SNPs that are heterozygous in the germline must be identified. Genotypes can be called from the allele-specific signals (A′i,B′i), or equivalently from (X′i,β′i) of the normal sample. By default, Paired PSCBS uses the naive genotyping algorithm proposed by Bengtsson et al. (2010), which calls the genotypes by thresholding on the observed density function of the normal BAFs (β′i). One may substitute these in PSCBS with more sophisticated genotype calls. However, as Bengtsson et al. (2010) showed, a naive genotype caller will do nearly as well for the purpose of PSCN segmentation.
2.1.4 Normalization of DH signals
Another advantage of having a normal sample is that systematic SNP effects can be estimated from the matched normal and be removed from the tumor signals. More specifically, Bengtsson et al. (2010) suggested adjusting the BAFs to eliminate SNP-specific effects using a procedure called TumorBoost (Fig. 1c). For heterozygous SNPs, which are the only ones that need to be normalized for PSCBS, the TumorBoost adjustment is
| (2) |
where βi′ corresponds to allele B fractions of the matched normal sample. Note that the TumorBoost correction is performed independently of chromosomal events. In Bengtsson et al. (2010), it was shown that utilizing TumorBoost significantly improves the power to detect PSCN aberrations; in some cases, the power increases dramatically. The corresponding TumorBoost-normalized DH, ρTBi=2|βTBi−0.5|, follows immediately (Fig. 1d). For convenience, we drop the TB superscript and assume the DH has been TumorBoost-normalized unless stated otherwise.
2.2 Segmentation of parent-specific CNs
It was shown in the previous section that the data carrying information on parent-specific chromosomal aberrations are contained in (Xi,ρi) for loci i=1,2,…,m, where total CNs (Xi) are defined for all loci and DHs (ρi) are defined only for SNPs that are heterozygous in the germline. We next identify segments of constant parent-specific CN using a two-step segmentation in which an initial set of change-points is identified from total CN signals (Xi), which is then updated with additional change-points from the normalized decrease-of-heterozygosity signals (ρi).
The two-step PSCBS approach is capable of detecting a PSCN change while TCN remains constant. As mentioned in Section 2.1.2, it can detect CNs changed from (1,1) to (0,2) in the case of no normal contamination, which would not be recognized by TCN segmentation alone. O'Keefe et al. (2010) provide detailed descriptions on how copy-neutral changes, where a loss in one chromosome is counter-balanced by a perfectly overlapping gain in the other, may occur. Note also that such a copy-neutral event may also be observed instead of a sequence of change-points in regions where the coverage is low, e.g. in the centromere. Likewise, a change from CN-LOH (0,2) to a deletion (0,1) would not be detectable from the DHs alone (except when there is normal contamination).
Even in cases where a chromosomal change-point is reflected in both the true TCN and DH, the power to detect a particular change-point differs between TCN and DH as a function of the type of aberration. This is explained in great detail and argued for both theoretically and empirically by Bengtsson et al. (2010). In this context, it means that a true change-point may be missed in the initial round of TCN segmentation, but later be identified by a segmentation of DHs. This also emphasizes an advantage that PSCBS has over, for instance, Staaf's BAF segmentation and PICNIC, both of which segment based on only one of the two signals available; PSCBS utilizes the signal available in both TCN and DH to detect change-points.
2.2.1 Identification of change-points in total CNs
CBS is applied to total CNs ratios Xi. We do not log these ratios because absolute values and their variances increase without bound as the total CN decreases without bound. Note that in Olshen et al. (2004) and Venkatraman and Olshen (2007), log-ratios were indeed used. However, the main reason then was that two-color DNA microarrays were used at the time and log-ratios work well in this context.
CBS identifies change-points using T=max1≤i<j<m|Tij|, where Tij is the two sample t-statistic that compares the mean of the observations with index from i+1 to j, to the mean of the rest of the observations. That is
| (3) |
where 
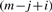 and sij2 is the corresponding sample variance. If the P-value corresponding to T is less than some predetermined threshold αTCN, we estimate the change−points as i and j for which Tij=T and repeat the procedure on the resulting segments. This process is repeated recursively and continues until no further change-points can be found. See Venkatraman and Olshen (2007) for a discussion of how to estimate P-values quickly in this context.
and sij2 is the corresponding sample variance. If the P-value corresponding to T is less than some predetermined threshold αTCN, we estimate the change−points as i and j for which Tij=T and repeat the procedure on the resulting segments. This process is repeated recursively and continues until no further change-points can be found. See Venkatraman and Olshen (2007) for a discussion of how to estimate P-values quickly in this context.
2.2.2 Identification of additional change-points using DH
In the second round of segmentation, the segments defined by CBS in the first round are split further based on the heterozygous SNPs. We identify additional change-points from DH (ρi) using CBS on each such subsegment. Change-points are not pruned; that is, those already identified in the TCN segmentation remain after the DH segmentation. New potential change-points are kept if their associated P-values are less than αDH. We specify αTCN and αDH so that, due to the Bonferroni inequality, our overall α≤αTCN+αDH is at a desired level. We choose αTCN≥αDH because most changes should be found in the first round of segmentation and total CN is available for every locus. The default for PSCBS is αTCN=0.009 and αDH=0.001 so that α≤0.01.
2.2.3 Defining boundaries of segments
The genomic position of a change-point is formally (Page, 1954) the locus after which the distribution of the data changes. But the actual change in CN can be anywhere in between the change-point locus and the next locus (conditional on having identified the change-point correctly). This was not a major problem when segmenting on TCN, but it is when segmenting on DH, since there are TCN loci that do not fall into either DH segment. As a convention, for DH segmentation we average the genomic positions between the change-point and the next heterozygous (DH) locus, and fix the change-point at the TCN locus immediately before this average.
2.2.4 Parent-specific CNs at the (non-called) region level
For each segment s=1,…,S defined by the change-points, the region-level TCN and DH,  , are estimated as:
, are estimated as:
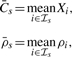 |
(4) |
where 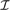 s is the set of loci that are located within segment s, that is, in region (xs−1,xs] and where non-defined DHs are excluded when calculating
s is the set of loci that are located within segment s, that is, in region (xs−1,xs] and where non-defined DHs are excluded when calculating 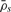 . The corresponding minor and major CNs,
. The corresponding minor and major CNs, 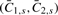 , are
, are
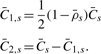 |
(5) |
so that  as well as
as well as  holds.
holds.
2.2.5 Bootstrapping
We utilize simple percentile bootstrap techniques to estimate standard errors and confidence intervals for the above estimates of CN. Later, we use these estimates in the calling described in Section 2.3. We resample the loci, that is (Xi,ρi), per segment with replacement such that the number of SNPs and the number of non-polymorphic loci, as well as the number of homozygous and heterozygous SNPs per segment, are preserved in each bootstrap sample. By default, PSCBS draws B=1000 bootstrap samples.
2.3 Calling parent-specific CN
In this subsection, we detail how to distinguish for every segment among the cases discussed in Section 2.1.1, i.e. equal PSCN, unequal PSCN with both parental CNs positive and LOH. Once calls are made, the PSCN estimates are updated. The segmentation and bootstrapping procedures rely on a minimum of prespecified parameters. Due to technical artifacts, normal contamination and lack of clonality in the tumor, some assumptions or tuning parameters are needed here. Note that calls cannot be made for very small segments since CN estimates are unstable, and the bootstrap estimates break down. In what follows, we will for clarity of notation drop segment index s.
2.3.1 Calling allelic balance
We start by distinguishing the case of equal PSCN from the case of unequal PSCN and both parental CNs positive. The former is a case of allelic balance, and the latter is a case of allelic imbalance. Formally, our null hypothesis for allelic balance is C1=C2, or equivalently DH=0.
As discussed in Section 2.2.5, we estimate confidence intervals for DH, and our tests are based on them. For every region, we take B bootstrap samples and estimate the region-level DH  as in the original data. We reject the null hypothesis if
as in the original data. We reject the null hypothesis if
| (6) |
where 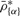 is the α:th percentile of
is the α:th percentile of  and ΔAB is a bias-correction term. This rejection region corresponds to the one-sided (1−αAB):th confidence interval not containing zero. The main reason for ΔAB is that
and ΔAB is a bias-correction term. This rejection region corresponds to the one-sided (1−αAB):th confidence interval not containing zero. The main reason for ΔAB is that  will always be a biased estimate of the true DH (when near zero), because DH is by definition always non-negative, cf. Equations (1) and (4), or equivalently because minor CN is by definition always less than or equal to major CN. This bias increases with the noise level. Therefore, we estimate ΔAB from the data in such a way that it adapts to the noise level, as further described in the Supplementary Materials. The procedure for choosing ΔAB assumes that at least some of the genome is in balance. This is a safe assumption for most samples. Nevertheless, there should be some safeguard in case it is not true. We recommend further examination if the resulting ΔAB is suspiciously large, e.g. ΔAB>0.20. In such cases, a predefined choice of ΔAB may be used. Our default value for αAB is 0.05.
will always be a biased estimate of the true DH (when near zero), because DH is by definition always non-negative, cf. Equations (1) and (4), or equivalently because minor CN is by definition always less than or equal to major CN. This bias increases with the noise level. Therefore, we estimate ΔAB from the data in such a way that it adapts to the noise level, as further described in the Supplementary Materials. The procedure for choosing ΔAB assumes that at least some of the genome is in balance. This is a safe assumption for most samples. Nevertheless, there should be some safeguard in case it is not true. We recommend further examination if the resulting ΔAB is suspiciously large, e.g. ΔAB>0.20. In such cases, a predefined choice of ΔAB may be used. Our default value for αAB is 0.05.
2.3.2 Calling LOH
Analogous to the above, we use a test to call LOH, where the null hypothesis is that a segment is not in LOH, or equivalently, the minor CN is ‘non-zero’ (The exact meaning of ‘non-zero’ will be explained below.) Segments already called to be in allelic balance will not be considered. Formally, we reject the null hypothesis if
| (7) |
where  is the α:th percentile of B bootstrapped
is the α:th percentile of B bootstrapped  mean estimates and ΔLOH≥0 is a parameter that is derived from data. Choosing ΔLOH, and, more generally, calling LOH requires strong assumptions. Even with these assumptions, there are difficulties.
mean estimates and ΔLOH≥0 is a parameter that is derived from data. Choosing ΔLOH, and, more generally, calling LOH requires strong assumptions. Even with these assumptions, there are difficulties.
Definition of LOH: LOH is not obviously defined when considering a cell population from a tumor study. The measured CN signals represent the average of a large number of often non-homogeneous cells, so that it is not clear what ‘zero’ minor CN is. This is because the tumor tissue extract will likely consist of some normal cells and possibly a mixture of different tumor cells. Thus, the term LOH can refer to the ‘zero’ minor CN state either of the mixed tumor and normal cells, or of just the (possibly) heterogeneous tumor cells. In either case, we cannot expect all cells to have truly zero minor CNs in segments that we wish to call LOH even when most cells do. Therefore, one option is to consider a segment to be in LOH when the fraction of the cells that has lost the contribution from one parent is ν, where 0≤ν≤1. Exactly which definition of LOH and which value of ν to use depends on the underlying biological question and needs to be chosen by the investigator.
On background signals including normal contamination: In theory, and as proposed by several (Assié et al., 2008; Lamy et al., 2007; Popova et al., 2009; Staaf et al., 2008; Sun et al., 2009; Van Loo et al., 2010; Yamamoto et al., 2007), it should be possible to estimate the amount of normal contamination from data. Unfortunately, its impacts on the PSCN estimates is confounded by additional sources of background signal, which makes it difficult in practice.
In the Supplementary Materials, we suggest a procedure for setting ΔLOH that reflects both the amount of background signal (including normal contamination) and the desired fraction (ν) of tumor cells to be in LOH. We note that the former component is data driven, while the latter is a predetermined data-independent tuning parameter. Stricter calling of LOH would be accomplished by decreasing ν. Unfortunately, it is not unusual to have a tumor with no LOH. In such a case, the above procedure fails to provide a useful ΔLOH. For this reason, as a rough guide, values of ΔLOH>0.75 should be evaluated further and possibly be replaced by a fixed value. By default, we use ν=0.50 and αLOH=0.05.
2.3.3 Estimating CN from called regions
If LOH has been found in the tumor, then the called PSCNs are estimated to be 0 and the total CN, that is  . Otherwise, if the two parental CNs are called equal, then the PSCNs are both estimated to be the total CN divided by 2, that is
. Otherwise, if the two parental CNs are called equal, then the PSCNs are both estimated to be the total CN divided by 2, that is  . If they are unequal, the called PSCN estimates are the same as the non-called PSCN estimates, that is,
. If they are unequal, the called PSCN estimates are the same as the non-called PSCN estimates, that is, 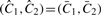 . Note that in the first two cases DH is not part of the CN estimates. Also note that for all segments it holds that
. Note that in the first two cases DH is not part of the CN estimates. Also note that for all segments it holds that 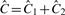 and
and  , cf. Section 2.2.4.
, cf. Section 2.2.4.
2.4 Algorithm and implementation
The paired parent-specific CBS and calling method, referred to as Paired PSCBS, is available in the PSCBS package, which is an open-access and open-source implementation in R. It is available on CRAN (http://cran.r-project.org/).
The method is designed and implemented to work with data from any generic SNP microarray technology, e.g. Affymetrix and Illumina. Great efforts have been made to make the implementation robust and straightforward to use. The low-level application programming interface (API) uses standard R data types, making it easy to incorporate PSCBS elsewhere. A high-level API that plugs into the Aroma Project framework (Bengtsson et al., 2008) is planned.
Since PSCBS is a single-pair method, it can be used to process any number of samples in bounded memory. The computational complexity to segment a sample with PSCBS is only slightly more than that for CBS. The reason for this is that the DH segmentation is significantly faster than TCN segmentation, because the regions being segmented are smaller and because homozygotes and non-polymorphic loci are not included.
3 RESULTS
Here, we assess the performance and correctness of Paired PSCBS. First, we assess the performance of PSCBS relative to extant methods on previously simulated data. Second, we show that PSCBS finds interesting genomic regions on glioblastoma data and that its results are similar for Affymetrix and Illumina arrays.
3.1 Simulation results
We compared PSCBS to other known methods using simulated data from Staaf et al. (2008). Specifically, they simulated a normal contamination series for a tumor based on HapMap sample NA06991 hybridized to the Illumina HumanHap550 array. To model a tumor, they added to the original data four regions of loss, three regions of gain and three regions of CN-LOH, as listed in Table 1. The alterations were reflected in the (log base 2) total CN and BAF. The percentage of normal cell contamination ranged from 0% (Supplementary Fig. S1) to 100% in increments of 5%; 10% normal contamination meant 10% normal cells and 90% tumor cells. The 100% normal contamination data was the same as the original sample, except as discussed in what follows. The other methods were ‘Paired BAF’ and ‘Unpaired BAF’ segmentation (Staaf et al., 2008), QuantiSNP (Colella et al., 2007), PennCNV (Wang et al., 2007) and SOMATICs (Assié et al., 2008). The results for all methods but PSCBS were taken from the Staaf analysis. Moreover, in agreement with the original authors, we have identified and corrected for a mistake in the simulated dataset causing the simulated total CNs to be slightly incorrect, cf. Supplementary Materials. The correction was not designed to give us an advantage in this assessment.
Table 1.
The regions of copy number alteration added to the HapMap sample NA06991 in the simulation
| Region type | Chrom. | Start | End | # Loci | # Het. |
|---|---|---|---|---|---|
| 1 CN-LOH | 5 | 1 | 47 700 000 | 9397 | 2756 |
| 2 Loss | 5 | 111 789 971 | 112 521 346 | 156 | 79 |
| 3 Gain | 8 | 1 | 45 200 000 | 12 564 | 3830 |
| 4 Gain | 8 | 128 432 670 | 129 207 869 | 218 | 91 |
| 5 Loss | 9 | 1 | 50 600 000 | 11 201 | 3889 |
| 6 Loss | 10 | 84 504 379 | 94 825 178 | 1988 | 648 |
| 7 Gain | 12 | 1 | 132 449 811 | 27 131 | 8818 |
| 8 Loss | 13 | 31 766 569 | 31 892 852 | 37 | 10 |
| 9 CN-LOH | 17 | 7 431 864 | 11 747 138 | 1150 | 308 |
| 10 CN-LOH | 17 | 22 800 000 | 78 774 742 | 9660 | 3191 |
Here ‘CN-LOH’ stands for copy-neutral loss of heterozygosity. This simulation was originally proposed by (Staaf et al., 2008).
3.1.1 Genotyping and PSCBS
We used the 100% normal contamination data as the reference sample for the purpose of genotyping. For segmentation and calling, we treated the test sample as if it already had been TumorBoost adjusted (Bengtsson et al., 2010). The reason is that because the tumor sample derived directly from the normal sample, we would be eliminating all noise in the tumor sample if we adjusted. Other than not doing TumorBoost, we ran PSCBS using all the default parameters.
3.1.2 Calling gains and losses
PSCBS is a segmentation and parent-specific calling algorithm, but it does not call gains and losses; we leave those decisions to the user. For purposes of the simulation assessment, we needed to make these other types of calls. We devised a simple calling algorithm. Regions were called gains or losses based on a sample-specific global threshold. That threshold was an estimated total CN from PSCBS that was more than 0.25 SDs from the median estimated total CN across all SNPs. The SDs were estimated using the residuals between the observed total CNs and the estimated total CNs from the PSCBS segments. Regions were called CN-LOH if they were not called gained or lost and if the parent-specific estimates were unequal. More explanation for this calling of CN-LOH can be found in Section 4.
3.1.3 Assessment
Following Staaf et al. (2008), the sensitivity for each altered region was the fraction of SNPs that were called altered, and the specificity was the fraction of SNPs outside an altered region that were called altered. Overall, as shown in Figure 2, PSCBS was typically sensitive until the contamination reached 90%. It also had an average specificity of 0.9996. We compared sensitivities by averaging the results across levels of contamination. Among all the methods, PSCBS, SOMATICs and Paired BAF segmentation were the most sensitive. Among the 10 comparisons, PSCBS was the most sensitive eight times, Paired BAF segmentation was most sensitive once (loss in Region 8), and SOMATICs was the most sensitive once (CN-LOH in Region 10). The sensitivity of SOMATICs was compromised by its low specificity, as can been seen in Figure 3; it is the only method without nearly perfect specificity.
Fig. 2.

Sensitivities for PSCBS and five other methods (Unpaired BAF, Paired BAF, QuantiSNP, PennCNV and SOMATICs) as a function of percentage normal contamination for 10 chromosomal aberrations. The performances were quantified using the Staaf et al. (2008) simulated dataset, in which copy-neutral LOH, single-copy gain, single-copy loss (hemizygous loss) and single-copy gain (including whole-chromosome trisomy) have been added to the HapMap sample NA06991 by adjusting the CN mean levels, cf. Table 1. The PSCBS results have been added to those obtained by Staaf et al. (2008).
Fig. 3.

Specificities for PSCBS and five other methods (Unpaired BAF, Paired BAF, QuantiSNP, PennCNV and SOMATICs) as a function of normal contamination. The same simulated dataset and annotations as in Figure 2 are used.
PSCBS did relatively least well with the small regions of alteration. This is counter-intuitive because PSCBS, since it incorporates all data rather than just heterozygotes like Paired BAF segmentation, should be particularly sensitive to small abnormalities. However, PSCBS is for the purpose of the assessment using a calling algorithm for gains and losses based solely on total CN. In this simulation, alterations are more strongly reflected in the allele B fraction than in the total CN, so it does not highlight one of the advantages of PSCBS. Overall, PSCBS compares favorably with other methods. Results for sensitivity can be found in Supplementary Table S1.
3.2 Glioblastoma data results
We examined the performance of the PSCBS algorithm on glioblastoma data from the Cancer Genome Atlas (TCGA). TCGA is a comprehensive effort to improve the understanding of cancer through application of genomic analysis (The Cancer Genome Atlas (TGCA) research Network, 2008). We evaluated the first batch of glioblastoma samples that were part of the TCGA for which there were 23 tumor/normal pairs. We analyzed data from both the Affymetrix GenomeWideSNP_6 array and the Illumina HumanHap550 array. These arrays contained approximately 900k and 550k SNPs, respectively. The Affymetrix array also contained about 900k non-SNP loci. We examined only the autosomes. The Affymetrix data was processed by us using an allele-specific version of the CRMAv2 method (Bengtsson et al., 2009a), while the Illumina data was processed by the TCGA consortium.
PSCBS was run at default parameter values, except that for both array types, we eliminated loci that were extreme outliers in total CN; these were more than 20 SDs from any of the 10 nearest loci. We also did not call regions with fewer than 10 heterozygotes.
We focused here on sample TCGA-02-0007 because it had an interesting pattern of alteration. For this particular sample, the methods for choosing the thresholds for calling segments suggest ΔAB=0.12 and ΔLOH=0.60 for the Affymetrix data, and ΔAB=0.077 and ΔLOH=0.59 for the Illumina data.
Segmentation results can be found in Figure 4 and in Supplementary Table S2. Large regions of LOH were found on both platforms for chromosomes 6, 7, 10, 11, 13, 14, 19 and 22. Large regions of gain were found on both for chromosomes 7 and 9. While we do not know the true CNs, it is encouraging that PSCBS when applied to both technologies gave similar results, which is consistent with previous studies comparing replicated CN data originating from different sources (Bengtsson et al., 2009b). Figure 5 has a closer look at chromosomes 7, 11 and 19 from the Affymetrix array. In addition to the alterations already mentioned, CN-LOH was found on chromosome 7q. All of 7p and part of 7q near the centromere showed gain in both parental chromosomes.
Fig. 4.

Whole-genome (chromosomes 1-22) PSCBS analysis of TCGA sample TCGA-02-0007. The top is from hybridization to the Affymetrix GenomeWideSNP_6 chip type (1 759 189 loci and 871 166 SNPs of which 234 058 are heterozygous in this sample) and the bottom is from hybridization to the Illumina HumanHap550 chip type (561 466 SNPs of which 175 585 are heterozygous). The black points represent total CN for all loci, and the gray points represent minimum CN for SNPs called heterozygous. The upper (purple) lines are PSCBS estimates of total CN, and the lower (blue) lines are the same for minor CN. Regions called LOH and allelic balance are highlighted at the horizontal axis as black and gray lines, respectively. The Affymetrix and the Illumina technologies show great similarity in their global segmentation patterns, such as finding all the same large regions of LOH.
Fig. 5.

Three chromosomes from the Affymetrix technology shown in Figure 4. The array identifies gain (chromosome 7), LOH (all three chromosomes) and CN-LOH (chromosome 7q). The same annotations were used as in Figure 4.
We show the segmentation of a second sample in Supplementary Figure S2. Note that, as shown in Figure 4, the analysis of the Affymetrix data was more complicated than that of the Illumina data because of the greater variability in the minimum CN. PSCBS, however, worked similarly for either type of data. Finally, to further assert that PSCBS produces valid results, we applied Paired BAF segmentation (Staaf et al., 2008) to the same sample and confirmed that the two methods agree on the major aberrant regions (Supplementary Table S3).
4 DISCUSSION
We developed an extension of CBS to estimate parent-specific CN from SNP data. In a simulation it identified gains and losses accurately and performed favorably compared with some of its competitors. A matched Affymetrix and Illumina real data example showed consistent and visually appealing segmentation results.
PSCBS consists of a concatenation of several tests and estimates. In an ideal world we would compute an exact probability that it estimates allele-specific CN correctly. Anyone who sees explicit computation as a target will conclude that at any step of the process, an accurate calculation would be conditional on the outcomes of all previous steps. Such a computation is impossible, no matter the order in which it is attempted. Despite our inability to provide a precise probability, we trust that readers will agree that there are persuasive arguments for use of the algorithm. They include demonstration that it is successful in achieving goals for which it is intended.
As mentioned in Section 3.1, regions in the simulation were called CN-LOH if they were not called gained or lost and if the parent-specific estimates were unequal. The first should be uncontroversial, but the second needs explanation. The simulation consisted of a mixture of tumor and normal cells. In a typical analysis, if there was a high degree of normal contamination, we would not want to call the region LOH. However, if the CN was normal but the parental CNs were imbalanced, we would conclude that there is some degree of LOH. So here we are equating copy-neutral LOH to some degree of copy-neutral LOH. Alternatively, we could have adjusted ΔLOH to get the same results, but this seems artificial.
An aspect of our algorithm is that it does not identify CN alterations that are already in the germline. For example, if there was a region with uniparental disomy (Robinson, 2000), where a subject inherits two copies from one parent, Paired PSCBS would not find it because there would be no difference between the tumor and the normal. While some may consider this to be a flaw, we believe it is a feature. Our purpose is to identify changes that come about during tumorigenesis and tumor evolution. A later analysis could be undertaken to find germline abnormalities.
Furthermore, it is important to acknowledge that PSCBS does not generate calibrated PSCNs, which is illustrated by the fact that although Affymetrix and Illumina agree to a great extent on the change-point locations, they differ somewhat in the estimated CN levels (Fig. 4). This means, for instance, that it is not valid to interpret the estimated PSCNs as true integer CN levels. This further stresses the importance of PSCN calibration, which is, to the best of our knowledge, still not investigated well enough; it is an important task from which all PSCN methods would gain and which we plan to undertake in a future study.
Contrary to popular belief, we wish to emphasize that for modern Affymetrix arrays, we can hereby produce high-quality PSCN segmentation from a single pair of tumor–normal samples without the need of external references; this brings many advantages (Bengtsson et al., 2010). The reason for this is that the CRMAv2 is a truly single-array method for estimating locus-level TCN and DH signals, and both TumorBoost as well as Paired PSCBS require only a single tumor–normal pair.
On the other hand, the Paired PSCBS algorithm does indeed require paired data. The germline reference sample is used to identify heterozygotes and to improve allele B fraction estimates (Bengtsson et al., 2010). While the latter is a luxury, the former is crucial, even if the genotypes are estimated using an external method. Thus, without germline genotypes it is not possible to perform DH segmentation. In Staaf et al. (2008), a heuristic is proposed for inferring which are the heterozygous SNPs based on the allele B fractions of the tumor. This is doable in segments where the homozygous and heterozygous BAFs are well separated, which may be the case when the DH level is not too large and the noise level of the BAFs is low. In case of pure LOH this would not work, although, as noted by several (Bengtsson et al., 2010; Staaf et al., 2008), normal contamination would to some extent play in our favor. Regardless, this heuristic would break down eventually, and there would be no possibility to distinguish homozygous and heterozygous SNPs from tumor alone. Instead, we are developing an alternative strategy for segmenting the allelic ratios in the setup of unpaired data, which will be the subject of a future publication.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Nancy Zhang and Ronglai Shen for helpful comments, Johan Staaf for extensive help with the simulation, and the reviewers for constructive discussions. The results published here are in whole or part based upon data generated by The Cancer Genome Atlas (TCGA) project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions who constitute the TCGA Research Network can be found at http://cancergenome.nih.gov/.
Funding: National Institutes of Health (1U24 CA143840-01 to A.B.O., 4R37EB002784-35 to R.A.O., 1U24 CA126551 to H.B., P.N. and P.S., 1U24 CA143799 to P.S., P30 CA82103 to H.B., and CA124504 to V.E.S.).
Conflict of Interest: none declared.
REFERENCES
- Assié G., et al. SNP arrays in heterogeneous tissue: highly accurate collection of both germline and somatic genetic information from unpaired single tumor samples. Am. J. Hum. Genet. 2008;82:903–915. doi: 10.1016/j.ajhg.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson H., et al. Technical Report 745. Berkeley: Department of Statistics, University of California; 2008. aroma.affymetrix: A generic framework in R for analyzing small to very large Affymetrix data sets in bounded memory. [Google Scholar]
- Bengtsson H., et al. A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix arrays including GenomeWideSNP 5 & 6. Bioinformatics. 2009a;27:2149–2156. doi: 10.1093/bioinformatics/btp371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson H., et al. A single-sample method for normalizing and combining full-resolution copy numbers from multiple platforms, labs and analysis methods. Bioinformatics. 2009b;25:861–867. doi: 10.1093/bioinformatics/btp074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson H., et al. TumorBoost: normalization of allele-specific tumor copy numbers from a single pair of tumor-normal genotyping microarrays. BMC Bioinformatics. 2010;11:245. doi: 10.1186/1471-2105-11-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., et al. Estimation of parent specific DNA copy number in tumors using high-density genotyping arrays. PLoS Comput. Biol. 2011;7:e1001060. doi: 10.1371/journal.pcbi.1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella S., et al. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlyand J., et al. Application of Hidden Markov Models to the analysis of the array CGH data. J. Multivar. Anal. 2004;90:132–153. [Google Scholar]
- Greenman C.D., et al. PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostat. 2010;11:164–175. doi: 10.1093/biostatistics/kxp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha S., et al. Bayesian hidden Markov modeling of array CGH data. J. Am. Stat. Assoc. 2008;103:485–497. doi: 10.1198/016214507000000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardenbol P., et al. Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res. 2005;15:269–275. doi: 10.1101/gr.3185605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu L., et al. Denoising array-based comparative genomic hybridization data using wavelets. Biostatistics. 2005;6:211–26. doi: 10.1093/biostatistics/kxi004. [DOI] [PubMed] [Google Scholar]
- Kallioniemi A., et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- Kuga D., et al. Prevalence of copy-number neutral LOH in glioblastomas revealed by genomewide analysis of laser-microdissected tissues. Neuro-oncology. 2008;10:995–1003. doi: 10.1215/15228517-2008-064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFramboise T., et al. Allele-specific amplification in cancer revealed by SNP array analysis. PLoS Comput. Biol. 2005;1:e65. doi: 10.1371/journal.pcbi.0010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai W.R., et al. Comparative analysis of algorithms for identifying amplifications and deletions in array-CGH data. Bioinformatics. 2005;21:3763–3770. doi: 10.1093/bioinformatics/bti611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai T.L., et al. Stochastic segmentation models for array-based comparative genomic hybridization data analysis. Biostat. 2008;9:290–307. doi: 10.1093/biostatistics/kxm031. [DOI] [PubMed] [Google Scholar]
- Lamy P., et al. A hidden markov model to estimate population mixture and allelic copy-numbers in cancers using affymetrix snp arrays. BMC Bioinformatics. 2007;8:434. doi: 10.1186/1471-2105-8-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., et al. Major copy proportion analysis of tumor samples using SNP arrays. BMC Bioinformatics. 2008;9:204. doi: 10.1186/1471-2105-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucito R., et al. Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation. Genome Res. 2003;13:2291–2305. doi: 10.1101/gr.1349003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H., et al. Allele-specific Hras mutations and genetic alterations at tumor susceptibility loci in skin carcinomas from interspecific hybrid mice. Cancer Res. 2003;63:4849–4853. [PubMed] [Google Scholar]
- O'Keefe C., et al. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood. 2010;115:2731–2739. doi: 10.1182/blood-2009-10-201848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen A.B., et al. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Page E.S. Continuous inspection schemes. Biometrika. 1954;41:100–115. [Google Scholar]
- Peiffer D.A., et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F., et al. A statistical approach for array CGH data analysis. BMC Bioinformatics. 2005;6:27. doi: 10.1186/1471-2105-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkel D., et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- Popova T., et al. Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome Biol. 2009;10:R128. doi: 10.1186/gb-2009-10-11-r128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson W.P. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays. 2000;22:452–459. doi: 10.1002/(SICI)1521-1878(200005)22:5<452::AID-BIES7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Staaf J., et al. Segmentation-based detection of allelic imbalance and loss-of-heterozygosity in cancer cells using whole genome SNP arrays. Genome Biol. 2008;9:R136. doi: 10.1186/gb-2008-9-9-r136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W., et al. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic Acids Res. 2009;37:5365–5377. doi: 10.1093/nar/gkp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas (TGCA) research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R., Wang P. Spatial smoothing and hot spot detection for CGH data using the fused lasso. Biostatistics. 2008;9:18–29. doi: 10.1093/biostatistics/kxm013. [DOI] [PubMed] [Google Scholar]
- Van Loo P., et al. Allele-specific copy number analysis of tumors. Proc. Natl Acad. Sci. 2010;107:16910. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman E.S., Olshen A.B. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- Wang K., et al. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenbrock H., Fridlyand J. A comparison study: applying segmentation to array-CGH data for downstream analyses. Bioinformatics. 2005;21:4084–4091. doi: 10.1093/bioinformatics/bti677. [DOI] [PubMed] [Google Scholar]
- Yamamoto G., et al. Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of Affymetrix single-nucleotide-polymorphism genotyping microarrays. Am. J. Hum. Genet. 2007;81:114–126. doi: 10.1086/518809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004;64:3060–3071. doi: 10.1158/0008-5472.can-03-3308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.