

Abstract
Motivation: In recent years, development of a single-method fold-recognition server lags behind consensus and multiple template techniques. However, a good consensus prediction relies on the accuracy of individual methods. This article reports our efforts to further improve a single-method fold recognition technique called SPARKS by changing the alignment scoring function and incorporating the SPINE-X techniques that make improved prediction of secondary structure, backbone torsion angle and solvent accessible surface area.
Results: The new method called SPARKS-X was tested with the SALIGN benchmark for alignment accuracy, Lindahl and SCOP benchmarks for fold recognition, and CASP 9 blind test for structure prediction. The method is compared to several state-of-the-art techniques such as HHPRED and BoostThreader. Results show that SPARKS-X is one of the best single-method fold recognition techniques. We further note that incorporating multiple templates and refinement in model building will likely further improve SPARKS-X.
Availability: The method is available as a SPARKS-X server at http://sparks.informatics.iupui.edu/
Contact: yqzhou@iupui.edu
1 INTRODUCTION
Given a query protein sequence with unknown structure, the most reliable structure prediction technique is to recognize its matching structural folds from existing known structures with or without significant sequence similarity (called homology modeling and fold recognition, respectively). This approach is also known as template-based modeling. Template-based modeling becomes increasingly powerful because most popular structure folds (adopted by multiple sequences) are known (Dai and Zhou, 2011; Kihara and Skolnick, 2003; Zhang et al., 2006).
However, recognizing structurally similar folds in the absence of sequence similarity (fold recognition) is challenging, as revealed from the critical assessment of structure prediction (CASP) techniques. CASP experiments highlighted the importance of post-treatment of models predicted by individual fold recognition methods through the use of consensus predictions [For example, ROBETTA (Chivian et al., 2003), Pmodeller6 (Wallner et al., 2007), Fams-ace (Terashi et al., 2007), Phyre (Bennett-Lovsey et al., 2008)] and/or constrained template–fragment recombination and refinement [For example, Chunk-TASSER (Zhou et al., 2007a), I-TASSER (Zhang, 2007)]. The experiments also indicated a convergence of techniques that can be broadly characterized as mixing and matching of multiple fragments and templates (Bujnicki, 2006; Zhou et al., 2010). Examples of recently developed new methods include the combined use of fragment and template comparison (Zhou and Skolnick, 2010), non-linear scoring function from conditional random field model (Peng and Xu, 2009) and profile entropy (Peng and Xu, 2010), employment of predicted torsion angles (Wu and Zhang, 2008; Zhang et al., 2008) and a combined use of profile–profile alignment and pairwise and solvation potentials (Lobley et al., 2009).
We have developed a series of single fold recognition methods (SPARKS, SP2, SP3, SP4 and SP5) that are based on weighted matching of multiple profiles that include sequence profiles generated from multiple sequence alignment (Altschul et al., 1997), predicted versus actual secondary structures (Rost et al., 1997; Zhou and Zhou, 2004, 2005a), knowledge-based profile (single-body) score function (Zhou and Zhou, 2004), depth-dependent sequence profiles derived from template structures (Zhou and Zhou, 2005a), predicted versus actual solvent accessible surface area (Liu et al., 2007) and predicted versus actual dihedral angles (Zhang et al., 2008). Statistically significant improvement is observed for the accuracy and sensitivity of fold recognition as the number of matching profiles increases from 3 to 5 (Liu et al., 2007; Zhang et al., 2008; Zhou and Zhou, 2004, 2005a). In particular, SPARKS, SP3 and SP4 were ranked among the top performers for automatic servers in CASP 6 (Tress et al., 2005; Zhou and Zhou, 2005b) and CASP 7 (Battey et al., 2007; Liu et al., 2007) experiments.
One issue in the methods developed above is that matching predicted 1D profiles of query sequence with actual profiles of templates is based on simple difference matrices. It does not account for the probability of errors in predicted 1D structural properties such as secondary structure, backbone torsion angles and solvent accessible surface area. In this article, we introduce energy terms based on the estimated probability of a match between predicted and actual 1D structural properties, a technique commonly used in fold recognition based on hidden Markov models (Hargbo and Elofsson, 1999). In addition, we take advantage of recently improved accuracy in predicted secondary structure [Q3=81−82% by SPINE X (E.Faraggi et al., submitted for publication)], torsion angles [SPINE X (Faraggi et al., 2009b), mean absolute error = 33° for ψ and 22° for ϕ] and solvent accessibility (ASA) [correlation coefficient of 0.74 between predicted and actual values, Real-SPINE 3.0 (Faraggi et al., 2009a)]. The above proposed algorithm leads to the new method called SPARKS-X in order to distinguish from previous SP series methods.
We tested SPARKS-X alignment accuracy, fold recognition and structure prediction by using several benchmarks, compared it to several state-of-the-art techniques and participated in the automatic server part of CASP (CASP 9). All results indicate that SPARKS-X is one of the best single-method fold recognition servers. The performance of the method can likely be further improved significantly by incorporating the techniques of multiple templates and refinement in model building that are employed in many other automatic servers.
2 METHODS
2.1 Alignment score
The alignment score of SP5 for aligning query position i with the template position j is (Zhang et al., 2008)
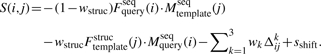 |
(1) |
with weight parameters (wstruc, wk) and a constant shift sshift. The first term in Equation (1) is the profile–profile comparison between the sequence profile from the query sequence and that from the template sequence. Fseqquery(i) is the sequence-derived frequency profile of the query sequence, Mseqtemplate(j) and Mseqquery(i) are the sequence-derived log odd profile of the template sequence and that of query sequence, respectively. These sequence profiles are constructed by three iterations of PSIBLAST (Altschul et al., 1997) searching (E value cutoff of 0.001) against non-redundant (NR) sequence database, which was filtered to remove low-complexity regions, transmembrane regions and coiled-coil segments (Jones, 1999). The second term in Equation (1) compares the sequence profile from the query sequence and that derived from the template structure (sequence profiles that would ‘fit’ to the structure). Fstructemplate(j) is a depth-dependent sequence profile generated from the sequences of those structural fragments that are similar to 9-residue segment structures of the template (Zhou and Zhou, 2005). The third term in Equation (1) measures the difference Δkij between the predicted 1D structural properties of the query sequence and the actual properties of the template (three-state secondary structure, real-value solvent accessibility and real value torsion angles).
By comparison, the alignment score developed in this article for SPARKS-X is as follows:
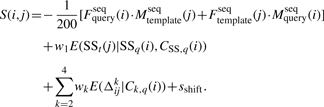 |
(2) |
There are two major changes from Equation (1) in SP5 to Equation (2) in SPARKS-X: the removal of sequence profile derived from template structure (designed sequences for templates) and replacement of simple difference Δkij by energy terms dependent on the predicted confidence—E(SSt(i)|SSq(j),CSS,q(j)) and E(Δkij|Ck,q(j)). Here, torsion angles ϕ and ψ are treated separately so that the maximum value of k is 4. We dropped the structure-derived sequence profile (the main novel feature in SP3) because we found that including this term no longer improves our results in our new formulation.
The first energy term for secondary structure (k=1) is calculated based on:
| (3) |
where P(SSt|SSq,CSS,q) is the probability of predicted secondary structure SSq by SPINE-X with confidence score CSS,q (Faraggi et al., 2009b) for a native secondary structure SSt, and the reference probability P(SSt) is the probability of secondary structure SSt in native proteins. For obtaining the probabilities, secondary structures were predicted by SPINE-X (E.Faraggi, submitted for publication) with three states for templates defined according to DSSP (Kabsch and Sander, 1983). CSS,q is evenly divided into eight discrete states.
The second energy term is based on
| (4) |
where P(Δk|Ck,q) is the probability of the predicted property having difference of Δk to corresponding native values with confidence score Ck,q; and the reference probability P0(Δk|Ck,q) is obtained by comparing the predicted one to all native values in the dataset. There are a total of three terms with k=2 for real valued ϕ, k=3 for real valued ψ, and k=4 for the real value solvent accessibility. Real value torsion angles (ϕ and ψ) are predicted by SPINE-X (Faraggi et al., 2009b). The difference for ϕ and ψ are evenly divided into 18 bins, Ck,q are evenly discretized into eight states. Real value solvent accessibility is predicted by Real-SPINE 3 (Faraggi et al., 2009a). The difference values are divided into 20 states, and C4,q is employing 20 amino acids to represent the prediction confidence. All energy terms were obtained from a NR dataset of 2479 proteins with length <500 amino acids from the original SPINE database [25% sequence identity cutoff, X-ray resolution lower than 3 Å and no unknown structural regions (Dor and Zhou, 2007)].
2.2 Parameter training and template ranking
As in SP5, the Smith–Waterman alignment algorithm (Smith and Waterman, 1981) is used to optimize the score that matches the query profiles with template profiles. To reduce the number of parameters, we set w2=w3 (equal weights for two torsion angles). All weight parameters and two gap penalty parameters (gap opening go and gap extension ge) were trained on the Prosup structural alignment benchmark (Domingues et al., 2000). The parameters were trained using the Powell method by many repeats from different random seeds (Press et al., 1992). The final parameters used are w1=1.04, w2=w3=0.23, w4=3.21, go=10.2, ge=0.69 and sshift=−1.52.
The templates are ranked by the greater one of two Z-scores, which is calculated based on the raw alignment score normalized by Lα or lα with L, the full alignment length, l, the non-end gap alignment length and α, a free parameter. The fractional exponent is introduced to mimic the fractional exponent employed in calculating domain–domain interactions (Zhou et al., 2007b). We find that α=3/4 yields a slightly improved (0.4% in TMscore of built model for the SCOP_20 dataset, see below) ranking. This ranking method is the same as used in SP3, SP4 or SP5 except that α=1 was used previously.
2.3 CASP 9 template library and model building
An automatically updated template library is used for the threading. When a new protein is input to the library, it is first divided into domains according to the ‘Author’ parameters in DDOMAIN (Zhou et al., 2007b). The divided domains are compared to existing domains in the library. If the sequence identity is < 40%, or the TM score [by TM align (Zhang and Skolnick, 2005)] between them is smaller than 0.5, the new domains and its chain will be included in the library. The automatically updated library had 31 750 templates on July 15, 2010 at the completion of server predictions in CASP 9.
The model is built by modeller9v7 (Sali et al., 1995) using the alignment generated by SPARKS-X. When there are gaps of > 30 residues in the termini, the program will be recalled to build a model for the missing parts in the region. After that, these different models are linked and steric clashes are removed by using the DFIRE potential functions (Yang and Zhou, 2008; Zhou and Zhou, 2002).
3 RESULTS
3.1 Alignment accuracy
As in SP3 and SP4, SPARKS-X was optimized by using the Prosup benchmark (Domingues et al., 2000) and tested in SALIGN (Marti-Renom et al., 2004). The Prosup benchmark, prepared by Sippl's group, consists of 127 pairs of proteins with alignment by the structural alignment program Prosup (Domingues et al., 2000). The SALIGN benchmark (Marti-Renom et al., 2004) contains 200 selected pairs with an average pair sharing 20% sequence identity or less and 65% (or more) of structurally equivalent Cα atoms superposed with an rmsd of 3.5 Å (Marti-Renom et al., 2004). Reference alignment is obtained from the structural alignment obtained from the TMalign program (Zhang and Skolnick, 2005) [i.e. TM overlap].
Table 1 shows the alignment accuracy of different methods given by different benchmarks. There is a consistent gradual improvement (1–2%) from SP3, SP4 to SP5 but a much larger improvement from SP5 to SPARKS-X (4–6%). This accuracy is comparable with the recently developed BoostThreader (Peng and Xu, 2009) or the new version of Raptor (Peng and Xu, 2010).
Table 1.
The alignment accuracy for Prosup and SALIGN benchmarks
| SP3 (%) | SP4 (%) | SP5a (%) | SP−Xb (%) | BTa (%) | PXa (%) | |
|---|---|---|---|---|---|---|
| Prosupc | 65.3 | 66.8 | 68.7 | 72.7 | 74.1 | 76.1 |
| d | 82.2 | 83.8 | – | 90.1 | 88.9 | – |
| SALIGNe | 56.3 | 57.3 | 59.7 | 65.9 | 63.6 | 64.4 |
aProsup was a test set for SP5, BT (BoostThreader) (Peng and Xu, 2009) and PX (Peng-Xu) (Peng and Xu, 2010) but training set for others.
bSP-X: SPARKS-X, this work.
cOne-to-one match given by the method and Prosup.
dWithin four residues by the method and Prosup.
eOne-to-one match given by the method and TMalign.
It is of interest to know the contribution to the overall accuracy of SPARKS-X made by individual terms in Equation (2). Table 2 compares the accuracy made by individual scoring terms by either adding to sequence profile [Position Specific Scoring Matrix (PSSM)] or removing from SPARKS-X. The results are obtained by training with the Prosup benchmark and testing with the SALIGN benchmark. It is clear that all three terms (secondary structure, torsion angles and ASA) contributed to the accuracy of alignment. Adding them to the PSSM increases the alignment accuracy while removing them from SPARKS-X decreases the accuracy. The contribution from ASA is the largest (5% adding to PSSM in SALIGN or 4% removing from SPARKS-X in SALIGN). Smaller but significant contributions are observed for secondary structure or torsion angles (3–4% for adding to PSSM and 0.4–1% for removing from SPARKS-X). The results from training and testing are consistent with each other.
Table 2.
The contribution of individual terms to the alignment accuracy for Prosup and SALIGN benchmarks
| Prosup (%) |
SALIGN | Prosup (%) |
SALIGN | ||||
|---|---|---|---|---|---|---|---|
| Method | 1–1a | ≤4b | 1–1c (%) | Method | 1–1a | ≤4b | 1–1c (%) |
| PSSMd | 63.4 | 80.5 | 59.1 | SP−Xe | 72.7 | 90.1 | 65.9 |
| +SSf | 68.4 | 87.1 | 62.9 | −SSg | 72.4 | 88.7 | 64.7 |
| +ϕ/ψf | 68.7 | 85.5 | 61.7 | −ϕ/ψg | 72.3 | 90.2 | 65.5 |
| +ASAf | 70.2 | 86.7 | 63.9 | −ASAg | 69.3 | 86.3 | 62.1 |
aOne-to-one match given by the method and Prosup.
bWithin four residues by the method and Prosup.
cOne-to-one match given by the method and TMalign.
dUsing PSSM matrix from PSIBLAST only.
eSP-X: SPARKS-X.
fUsing PSSM plus secondary structure, or ϕ/ψ, or ASA as noted.
gExcluding secondary structure, or ϕ/ψ, or ASA as noted.
3.2 Testing fold recognition with Lindahl benchmark
The purpose of improving alignment is to increase the ability of recognizing the correct structural fold of a query sequence from a template library. We employed the Lindahl Benchmark for comparing SPARKS-X with different methods. The benchmark is a large data set of 976 proteins, with 555,434, and 321 pairs of proteins in the same family, superfamily and fold, respectively (Lindahl and Elofsson, 2000). Here, the fold recognition sensitivity of each method is tested by aligning each protein with the rest 966 proteins, and checking whether or not the method can recognize the member of same family, superfamily or fold as the first ranked or within top five ranked templates. Thus, the benchmark tests both the modeling accuracy and the ranking methods for fold recognition.
Table 3 shows the fraction of correctly recognized matches for proteins in the same family, superfamily, fold as the first ranked or within top five ranked templates given by various methods. Although many published methods have been applied to this benchmark (Kim et al., 2003; Shi et al., 2001; Xu et al., 2003; Zhou and Zhou, 2004), we only list the most recent ones (Cheng and Baldi, 2006; Liu et al., 2007; Peng and Xu, 2009; Zhou and Zhou, 2004, 2005a). This is because of the time-dependent nature of sequence databases for sequence profiles.
Table 3.
Success rate for recognizing proteins within the same family, superfamily or fold in the Lindahl benchmark
| Family (%) |
Superfamily (%) |
Fold (%) |
||||
|---|---|---|---|---|---|---|
| Methods | Top 1 | Top 5 | Top 1 | Top 5 | Top 1 | Top 5 |
| SPARKSa | 81.6b | 88.1 | 52.5 | 69.1 | 24.3 | 47.7 |
| FOLDproc | 85.0 | 89.9 | 55.5 | 70.0 | 26.5 | 48.3 |
| SP3d | 81.6 | 86.8 | 55.3 | 67.7 | 28.7 | 47.4 |
| SP4e | 80.9 | 86.3 | 57.8 | 68.9 | 30.8 | 53.6 |
| SP5f | 82.4 | 87.6 | 59.8 | 70.0 | 37.9 | 58.7 |
| SPARKS-X | 84.1 | 90.3 | 59.0 | 76.3 | 45.2 | 67.0 |
| BoostThreaderg | 86.5 | 90.5 | 66.1 | 76.4 | 42.6 | 57.4 |
aFrom (Zhou and Zhou, 2004).
bThe percentage in each cell is the fraction of correctly recognized match of proteins in the same fold, super family, family as the first ranked or within top 5 ranked templates.
cFrom Ref. (Cheng and Baldi, 2006).
dFrom Ref. (Zhou and Zhou, 2005).
eFrom Ref. (Liu et al., 2007).
fFrom Ref. (Zhang et al., 2008).
gFrom Ref. (Peng and Xu, 2009).
Table 3 indicates that the improvement over SP3, SP4, SP5 in success rate of fold recognition by SPARKS-X exists in all three levels (family, superfamily and fold) except the Top 1 ranked model in superfamily where the success rate is similar between SP5 (59.8%) and SPARKS−X (59.0%). The largest improvement over SP5 is observed in fold level (7% absolute increase in Top 1 and 8% absolute increase for the best in Top 5). This is somewhat expected because the method was trained for remote homolog recognition (structurally similar protein with < 30% sequence identity in the Prosup benchmark). Comparing to BoostThreader, SPARKS-X is less successful in homology detection (family and superfamily in Top 1) but more successful in fold recognition (2% improvement in Top 1 and 10% improvement in Top 5) as trained.
The above success rates of matching sequences within the same SCOP classification are based on somewhat subjective SCOP definition of family, superfamily and fold (Murzin et al., 1995). A more direct measurement of accuracy is to calculate the accuracy of the first-ranked model built from the fold recognition alignment. First, the model is built by transferring the Cα coordinates of the template structures to the aligned residues in the query sequence. Then, the constructed model is assessed by using the MaxSub score between the model and the known native structure. MaxSub score (Siew et al., 2000) between two structures is a measure of similarity between them with 0.0 indicating no similarity and 1.0 a perfect match. The value is calculated by searching for the largest subset of well-superimposed residues (≤3.5 Å). Table 4 reports the MaxSub scores for the models built by SP3, SP4, SP5 and SPARKS-X methods averaged over the number of proteins. Again SPARKS-X improves over SP5, SP4 and SP3 in all levels. The relative improvement of SPARKS-X over SP5 in MaxSub score is 12, 22 and 13% in family, superfamily and fold levels, respectively.
Table 4.
The model quality of top-1 ranked models in Lindahl benchmark per protein
| Totala | Familyb | Superfamilyc | Foldd | |
|---|---|---|---|---|
| SP3 | 0.358e | 0.529 | 0.232 | 0.107 |
| SP4 | 0.361 | 0.532 | 0.251 | 0.116 |
| SP5 | 0.374 | 0.538 | 0.257 | 0.153 |
| SPARKS-X | 0.422 | 0.601 | 0.314 | 0.173 |
aAll 976 proteins.
bFamily only.
cSuperfamily only.
dFold only.
eThe average MaxSub score for the first-ranked models.
3.3 Testing fold recognition with SCOP-20 dataset
We built a SCOP-20 dataset by using domains of sequence identity <20% and chain lengths > 60 from SCOP 1.75. After removing domains with Cα atoms only, we obtained 6367 domains. We also compared our results with HHPRED (Soding et al., 2005) (version 1.5.1) and PRC (Madera, 2008) (version 1.5.6) because these two programs could be downloaded and installed on our local machine. The profiles of the domains for HHPRED are directly downloaded from HHPRED's web page (http://toolkit.tuebingen.mpg.de/hhpred). The profiles for PRC are using profiles generated from three iterations of PSIBLAST. For both these two predictors, default parameters were used. We would like to emphasize that we have only assessed PRC with the sequence profiles generated from PSIBLAST. Its performance may be different if other profiles are employed.
First, we tested the ability of HHPRED and SPARKS-X to recognize a match in the same family, same superfamily (after removing family members from the templates) and same fold (after further removing superfamily members) according to the SCOP definition within top-N templates. Note that on a given search we removed the query protein from the template library. Figure 1 shows the success rates of recognizing at least one template within same family, superfamily or fold as a function of the number (N) of top predicted matching templates. At the family and superfamily level, HHPRED has a higher success rate than SPARKS-X based on top 1–12 templates but a lower success rate afterwards. At the fold level, SPARKS-X has a consistent higher success rate than HHPRED and the difference becomes greater as more top templates are included. Similar results are observed in the ROC curve when the true positive rate is plotted as a function of the false positive rate (Fig. 2) for all pairs of the SCOP-20 dataset. Here, true positives denote the detection of the templates within the same classification (family, superfamily or fold). The performance of SPARKS-X is consistently better than that of HHPRED at the fold level while HHPRED has a higher true positive rate only at low false positive rate at the family and superfamily levels.
Fig. 1.

The success rate of having at least one correct prediction of templates within the same family, superfamily and fold in top N templates predicted for the SCOP-20 dataset. The performance of SPARKS-X (solid line) is compared with that of HHPRED (dashed line).
Fig. 2.

As in Figure 1, but for the true positive rate versus the false positive rate of detecting matching templates within the same family, superfamily and fold, respectively, for all pairs in the SCOP-20 dataset.
To avoid somewhat subjective definition of family, superfamily and fold, another way to compare the ability of recognizing structural similarity is to directly calculate the structural similarity between the target structure and the structure recognized without actually building the model. Results of average TM score between query and Top 1 template are shown in Table 5 where structural similarity is measured by TM align (Zhang and Skolnick, 2005). The table shows that the average TM score given by SPARKS-X is about 3% higher than that given by HHPRED when all templates are employed. The difference between the TM scores given by SPARKS-X and that given by HHPRED is larger if easily recognized templates are removed. SPARKS-X's average TM scores are 5, 14 and 16% higher than that given by HHPRED when templates from same family, superfamily and fold are excluded from the templates library. This result indicates that SPARKS-X has a higher ability than HHPRED or PRC to recognize structurally similar proteins regardless if they are in the same family, superfamily or same fold. The results in Table 5 can be further illustrated by a ROC curve for all SCOP-20 templates (Fig. 3). The positives are defined by templates having TM score > 0.5 to query structures by TM align (i.e. to test the ability to recognize a similar structure). The figure shows that the performance of SPARKS-X is consistently better than that of HHPRED at detecting structurally similar templates from all templates, without the same family members, and without the same family and superfamily members. The difference between the two methods is small at very low false positive rates (see the insert of Fig. 3) but increases significantly at low false positive rates. The difference between Figure 3 and Figure 2 is because family and superfamily members in SCOP are defined according to sequence evolution origins, rather than structural similarity. Our results suggest that using structural similarity is more direct and accurate assessment of the performance of structure prediction techniques.
Table 5.
The average TM score between query and Top 1 template of SCOP_20 dataset
| PRC | HHPRED | SPARKS-X | |
|---|---|---|---|
| Alla | 0.569 | 0.583 | 0.596 |
| Familyb | 0.445 | 0.469 | 0.489 |
| Superfamilyb | 0.330 | 0.341 | 0.387 |
| Foldb | 0.313 | 0.315 | 0.361 |
aAll templates in the dataset.
bAll templates except those belonging to same family, or same superfamily, or same fold, as the query sequence.
Fig. 3.

As in Figure 2. However, the positives are defined as those predicted template structures with a TM score > 0.5 to the query structure by TM align. Three sets of results from top to bottom are shown: all pairs, excluding the templates within the same family, and excluding those within the same family and superfamily.
The results reported in Table 5 and Figure 3 are based on direct structural comparison between target and template structures. A more common comparison is to measure the accuracy of the model built based on sequence template alignment. We found that this will further improve the performance of SPARKS-X relative to that of HHPRED/PRC because SPARKS-X uses local-global alignment while HHPRED and PRC are based on local alignment. As a result, SPARKS-X typically gives a longer alignment than HHPRED and PRC. This leads to improved scores for models built. For example, the average TM score of Top 1 model from all templates for HHPRED and SPARKS-X are 0.476 and 0.517, respectively. This is 9%, rather than 3% improvement based on structural alignment of target and template structures (Table 5). We also tested HHPRED with the option of ‘-mact 0.05’ because this option leads to almost global alignment and better scoring models. Although it does not change the ability of recognizing structurally similar proteins (Table 5), this option indeed increases the average TM score of Top 1 model from 0.476 to 0.502, which is 3% rather than 9% behind SPARKS-X.
3.4 CASP9 blind prediction
SPARKS-X participated in CASP 9 blind test and ranked #21 within automatic servers in SUM-Zscore, and #12 within independent groups (after removing redundant servers). The majority of the methods ranked before SPARKS-X are consensus techniques except HHPRED and RAPTORX. If the total TM score of Top 5 models (http://zhanglab.ccmb.med.umich.edu/casp9/) are employed as a criterion, SPARKS-X is ranked #12 (#6 in groups). This comparison of Top 5 models is meaningful as all top servers except HHPRED series submitted five models. Moreover, if ranked by TM score plus hydrogen bond score of Top 1 model, SPARKS-X is ranked #6 (#5 in groups) behind QUARK/Zhang-Server, ROSETTA, Seok-Server and GWS only. This indicates that the models built by SPARKS-X have better hydrogen bonds than many servers. As a reference, our method can be compared to MUSTER (Wu and Zhang, 2008), an extension to the SP4 server (Liu et al., 2007) by incorporating torsion angles and hydrophobicity. The summed TM score of SPARKS-X server is 5% higher than that of MUSTER.
4 DISCUSSION
We have reported a new fold recognition server called SPARKS-X that is significantly different from our previous versions in how the profile–profile matching score is obtained. Moreover, we also employed significantly improved secondary structure prediction, real value torsion angle prediction and solvent accessibility prediction. All these techniques made an improvement over our previous SP series possible. We found that predicted ASA contributes the most to the overall accuracy of SPARKS-X.
One interesting observation is that SPAKRS-X performs significantly better in recognizing structurally similar proteins (3%) and in building better models (3%) based on the large dataset of SCOP-20 and the latest version of HHPRED available on the web. On the other hand, limited CASP 9 blind prediction suggests the opposite. The official average GDT score for 147 domains given by HHPREDB is 59.5, compared with 57.7 given by SPARKS-X (http://predictioncenter.org/casp9). This 3% improvement of HHPRED over SPARKS-X is likely due to significantly more sophisticated model building techniques employed in the unreleased version of HHPRED by using distance restraints derived from multiple templates together with alignment confidence. Furthermore, SPARKS-X is only 8% behind the best automatic server in official average GDT score of Zhang server (62.2). This 8% is likely due to combined effect of consensus prediction from multiple fold recognition servers, the use of multiple templates and model refinement. This is an area of focus in our future work for further improving SPARKS.
ACKNOWLEDGEMENT
We would like to thank Johannes Soding for helpful comments and for making HHPRED available and Martin Madera for making PRC available.
Funding: National Institutes of Health (grants R01 GM 085003 and 067168).
Conflict of Interest: none declared.
REFERENCES
- Altschul S.F., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battey J.N.D., et al. Automated server predictions in CASP7. Proteins. 2007;69(Suppl. 8):68–82. doi: 10.1002/prot.21761. [DOI] [PubMed] [Google Scholar]
- Bennett-Lovsey R.M., et al. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins. 2008;70:611–625. doi: 10.1002/prot.21688. [DOI] [PubMed] [Google Scholar]
- Bujnicki J.M. Protein-structure prediction by recombination of fragments. Chembiochem. 2006;7:19–27. doi: 10.1002/cbic.200500235. [DOI] [PubMed] [Google Scholar]
- Cheng J., Baldi P. A machine learning information retrieval approach to protein fold recognition. Bioinformatics. 2006;22:1456–1463. doi: 10.1093/bioinformatics/btl102. [DOI] [PubMed] [Google Scholar]
- Chivian D., et al. Automated prediction of CASP-5 structures using the robetta server. Proteins. 2003;53(Suppl. 6):524–533. doi: 10.1002/prot.10529. [DOI] [PubMed] [Google Scholar]
- Dai L., Zhou Y. Characterizing the existing and potential structural space of proteins by large-scale multiple loop permutations. J. Mol. Biol. 2011;408:585–595. doi: 10.1016/j.jmb.2011.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingues F.S., et al. Structure-based evaluation of sequence comparison and fold recognition alignment accuracy. J. Mol. Biol. 2000;297:1003–1013. doi: 10.1006/jmbi.2000.3615. [DOI] [PubMed] [Google Scholar]
- Dor O., Zhou Y. Achieving 80% ten-fold cross-validated accuracy for secondary structure prediction by large-scale training. Proteins. 2007;66:838–845. doi: 10.1002/prot.21298. [DOI] [PubMed] [Google Scholar]
- Faraggi E., et al. Improving the accuracy of predicting real-value backbone torsion angles and residue solvent accessibility by guided learning through two-layer neural networks. Proteins. 2009a;74:847–856. doi: 10.1002/prot.22193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraggi E., et al. Predicting continuous local structure and the effect of its substitution for secondary structure in fragment-free protein structure prediction. Structure. 2009b;17:1515–1527. doi: 10.1016/j.str.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraggi E., et al. SPINE X: Going beyond 80% in accuracy of protein secondary structure prediction by multi-step learning coupled with prediction of solvent accessible surface area and backbone torsion angles. 2011 doi: 10.1002/jcc.21968. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargbo J., Elofsson A. Hidden markov models that use predicted secondary structures for fold recognition. Proteins. 1999;36:68–76. [PubMed] [Google Scholar]
- Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kihara D., Skolnick J. The PDB is a covering set of small protein structures. J. Mol. Biol. 2003;334:793–802. doi: 10.1016/j.jmb.2003.10.027. [DOI] [PubMed] [Google Scholar]
- Kim D., et al. PROSPECT II: protein structure prediction program for the genome-scale. Protein Eng. 2003;16:641–650. doi: 10.1093/protein/gzg081. [DOI] [PubMed] [Google Scholar]
- Lindahl E., Elofsson A. Identification of related proteins on family, superfamily and fold level. J. Mol. Biol. 2000;295:613–625. doi: 10.1006/jmbi.1999.3377. [DOI] [PubMed] [Google Scholar]
- Liu S., et al. Fold recognition by concurrent use of solvent accessibility and residue depth. Proteins. 2007;68:636–645. doi: 10.1002/prot.21459. [DOI] [PubMed] [Google Scholar]
- Lobley A., et al. pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics. 2009;25:1761–1767. doi: 10.1093/bioinformatics/btp302. [DOI] [PubMed] [Google Scholar]
- Madera M. Profile comparer (prc): a program for scoring and aligning profile hidden markov models. Bioinformatics. 2008;24:2630–2631. doi: 10.1093/bioinformatics/btn504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Renom M.A., et al. Alignment of protein sequences by their profiles. Protein Sci. 2004;13:1071–1087. doi: 10.1110/ps.03379804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin A.G., et al. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Peng J., Xu J. Research in Computational Molecular Biology. 2009. Boosting protein threading accuracy; pp. 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J., Xu J. Low-homology protein threading. Bioinformatics. 2010;26:i294–i300. doi: 10.1093/bioinformatics/btq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press W., et al. Numerical Recipes in C. 2nd. Cambridge, UK: Cambridge University Press; 1992. [Google Scholar]
- Rost B., et al. Protein fold recognition by prediction-based threading. J. Mol. Biol. 1997;270:471–480. doi: 10.1006/jmbi.1997.1101. [DOI] [PubMed] [Google Scholar]
- Sali A., et al. Evaluation of comparative protein modelling by MODELLER. Proteins. 1995;23:318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- Shi J., et al. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- Siew N., et al. Maxsub: an automated measure for the assessment of protein structure prediction quality. Bioinformatics. 2000;16:776–785. doi: 10.1093/bioinformatics/16.9.776. [DOI] [PubMed] [Google Scholar]
- Smith T.F., Waterman M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Soding J., et al. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashi G., et al. Fams-ace: a combined method to select the best model after remodeling all server models. Proteins. 2007;69(Suppl. 8):98–107. doi: 10.1002/prot.21785. [DOI] [PubMed] [Google Scholar]
- Tress M., et al. Assessment of predictions submitted for the CASP6 comparative modeling category. Proteins. 2005;61(Suppl. 7):27–45. doi: 10.1002/prot.20720. [DOI] [PubMed] [Google Scholar]
- Wallner B., et al. Pcons.net: protein structure prediction meta server. Nucleic Acids Res. 2007;35:W369–W374. doi: 10.1093/nar/gkm319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S., Zhang Y. MUSTER: improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 2008;72:547–556. doi: 10.1002/prot.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., et al. Protein structure prediction by linear programming. Pac. Symp. Biocomput. 2003;8:264–275. [PubMed] [Google Scholar]
- Yang Y., Zhou Y. Ab initio folding of terminal segments with secondary structures reveals the fine difference between two closely-related all-atom statistical energy functions. Protein Sci. 2008;17:1212–1219. doi: 10.1110/ps.033480.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins Suppl. 2007;69:108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., et al. On the origin and completeness of single domain structures. Proc. Natl Acad. Sci. 2006;103:2605–2610. doi: 10.1073/pnas.0509379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., et al. SP5: improving protein fold recognition by using predicted torsion angles and profile-based gap penalty. PLoS One. 2008;6:e2325. doi: 10.1371/journal.pone.0002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Skolnick J. Improving threading algorithms for remote homology modeling by combining fragment and template comparisons. Proteins. 2010;78:2041–2048. doi: 10.1002/prot.22717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Zhou Y. Single-body residue-level knowledge-based energy score combined with sequence-profile and secondary structure information for fold recognition. Proteins. 2004;55:1005–1013. doi: 10.1002/prot.20007. [DOI] [PubMed] [Google Scholar]
- Zhou H., Zhou Y. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins. 2005a;58:321–328. doi: 10.1002/prot.20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Zhou Y. SPARKS 2 and SP3servers in CASP 6. Proteins. 2005b;61(Suppl. 7):152–156. doi: 10.1002/prot.20732. [DOI] [PubMed] [Google Scholar]
- Zhou H., et al. Analysis of TASSER-based CASP7 protein structure prediction results. Proteins. 2007a;69(Suppl. 8):90–97. doi: 10.1002/prot.21649. [DOI] [PubMed] [Google Scholar]
- Zhou H., et al. DDOMAIN: dividing structures into domains using a normalized domain-domain interaction profile. Protein Sci. 2007b;16:947–955. doi: 10.1110/ps.062597307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., et al. Trends in template/fragment-free protein structure prediction. Theor. Chem. Acc. 2010;128:3–16. doi: 10.1007/s00214-010-0799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]