

Abstract
Unnatural amino acids (Uaas) can be translationally incorporated into proteins in vivo using evolved tRNA/aminoacyl-tRNA synthetase (RS) pairs, affording chemistries inaccessible when restricted to the 20 natural amino acids. To date, most evolved RSs aminoacylate Uaas chemically similar to the native substrate of the wild-type RS; these conservative changes limit the scope of Uaa applications. Here, we adapt Methanosarcina mazei PylRS to charge a noticeably disparate Uaa, O-methyl-l-tyrosine (Ome). In addition, the 1.75 Å X-ray crystal structure of the evolved PylRS complexed with Ome and a non-hydrolyzable ATP analogue reveals the stereochemical determinants for substrate selection. Catalytically synergistic active site mutations remodel the substrate-binding cavity, providing a shortened but wider active site. In particular, mutation of Asn346, a residue critical for specific selection and turnover of the Pyl chemical core, accommodates different side chains while the central role of Asn346 in aminoacylation is rescued through compensatory hydrogen bonding provided by A302T. This multifaceted analysis provides a new starting point for engineering PylRS to aminoacylate a significantly more diverse selection of Uaas than previously anticipated.
Incorporation of Uaas into proteins using a host’s endogenous translation machinery opens the door to addressing questions with chemical precision that is unattainable using naturally occurring amino acids. This expanded toolset allows one to pose and answer more in-depth molecular questions without the limitations imposed by the 20 natural amino acids used in traditional mutagenic analyses.1,2 Aminoacyl-tRNA synthetases (RSs) obtained by structure-based engineering and directed evolution efficiently recognize and activate Uaas through ATP-dependent adenylation and subsequently catalyze transfer to their cognate tRNA. To date, more than 70 Uaas are now amenable to translational insertion into proteins in bacteria, yeast, or mammalian cells using these artificially evolved tRNA/RS pairs.(3) By choosing particular matching sets of tRNA/RSs from diverse organisms, the pairs can function in vivo in an orthogonal manner. In other words, there is limited if any crosstalk between the expression host’s native tRNA/RS pairs and the orthogonal tRNA/RS pair; however, the orthogonal pair is still able to functionally couple with the host’s protein translational machinery. The tRNATyr/TyrRS pair derived from the archaeon Methanocaldococcus jannaschii was the first successful pair to direct the genetically encoded incorporation of the Uaa O-methyl-l-tyrosine (Ome) into translated proteins in vivo.4,5 This initial result then spawned the incorporation of the majority of Tyr-based Uaas possessing distinct functional groups into recombinant proteins. Recently, another matching set, namely, the tRNAPyl/PylRS pair, was used to incorporate Uaas bearing functional groups similar to Pyl.6−12
In most cases regarding tRNATyr/TyrRS systems, sterically innocuous modifications of the phenyl ring, typically focused on the para position, are employed as unnatural substrates. Similarly, most Uaas incorporated by the tRNAPyl/PylRS pair all contain slight variations of the extended Pyl side chain, while the core Lys moiety and the Nε-carbonyl group of Pyl remain unchanged.(3) In short, most evolved orthogonal tRNA/RS pairs demonstrate specificity for Uaas that maintain the chemical core of the native amino acid substrate, thus limiting the stereochemical diversity of Uaas accessible to the researcher. Since the RS component of the pair plays a key role in amino acid substrate selection, it is of tremendous importance to probe the extent to which it can be engineered not only to activate a chemically and structurally divergent amino acid but also its ability to transfer this new amino acid to its cognate tRNA. Given that the degree of substrate specificity exhibited by an RS is functionally coupled to the accuracy of protein translation, the RS is restrained by tremendous selective pressure; therefore, integration of structural approaches with directed evolution of RSs can expand upon our understanding of the evolutionary tenets governing substrate specificity and selection.13−15 This unified approach applied in the form of multiple rounds of structure-based design and directed evolution can often produce RSs with altered specificity for Uaa substrates in relatively rapid fashion.
Moreover, the inability to efficiently employ mammalian cells and multicellular organisms for evolving tRNA/RS pairs further limits the scope of unnatural substrates available for probing in vivo protein function in organisms more complex than microbial hosts. Currently, directed approaches for evolving a Uaa-specific RS involve the generation of an initial RS mutant library minimally containing 108 to 109 members.1,4 These numbers, coupled with the need for efficient selection procedures, limit applicability to organisms possessing high transformation efficiency and favorable growth characteristics.(1) The approach was first developed in E. coli(4,16) and later expanded to include another unicellular organism, Saccharomyces cerevisiae.17,18 Nevertheless, maintenance of large mutant libraries and selection methods are difficult to implement in more complex systems such as mammalian cells and multicellular organisms.
One solution is to evolve an RS that possess characteristics favorable to deployment in the intended mammalian hosts in recombinant organisms such as E. coli or yeast. Assuming the appropriate RS is chosen, the evolved RSs should then be readily integrated into mammalian cells or a multicellular organism for further optimization once the large collection of mutants is narrowed substantially. This divide and conquer approach thus shortcuts some of the anticipated problems faced during the initial rounds of selection.(19) This kind of strategy was applied to the tRNATyr/TyrRS pair from E. coli, wherein the TyrRS was first evolved in yeast and then transferred to mammalian cell hosts.19,20 Nonetheless, most reported mutant RSs evolved in yeast generally lack the in vivo efficiency of the original M. jannaschii TyrRS evolved and exploited in E. coli. Moreover, M. jannaschii TyrRS does not operate in an orthogonal manner in mammalian cells, often cross-charging mammalian tRNAs.
With these limitations in mind, tRNAPyl/PylRS is an attractive alternative because of the demonstrated orthogonal behavior of this particular pair in E. coli and mammalian cells.9,10,21,22 Mutating the active site does not impact orthogonality so the evolved pairs are highly unlikely to cross-react with the endogenous tRNA/RS pairs of their mammalian hosts. One remaining issue, however, concerns the extent to which the current assortment of orthogonal pairs can be evolved to specifically adenylate and then charge their cognate tRNAs with substrates substantially different from their natural substrates. Strikingly, to date, Uaas incorporated by mutant RSs derived from PylRS bear close chemical and steric resemblance to the natural Pyl substrate.(3)
High fidelity Uaa incorporation is essential for generating chemically homogeneous proteins for investigation. An optimally evolved RS should function similarly to a wild-type RS, which achieves high incorporation fidelity under physiological growth conditions and maintains fidelity when cells are grown in nutrient-rich media. For Uaa incorporation in E. coli, nutrient-rich media is necessary to ensure high in vivo incorporation efficiency of the evolved RS; however, when an evolved RS possesses non-optimal specificity for the Uaa, minimal media lacking certain or all natural amino acids must be used to limit the mis-incorporation of any natural amino acids into the recombinant protein.(23) Unfortunately, minimal media cannot be applied to traditional fermentations, mammalian cell cultures, and whole organism engineering; this drawback prevents the transfer and usage of non-optimally evolved RSs in these situations. Moreover, the active site of a non-optimal RS must lack the complementary stereochemical features for selection of the given Uaa substrate. Structural analyses then become critical for providing architectural guides to explain and further optimize the substrate selection and turnover. Recently, Wang et al. reported the incorporation of two Phe-based Uaas into proteins via evolved PylRSs, albeit in E. coli grown solely in minimal media.(24) To date, efficient evolution of PylRS to specifically incorporate a Uaa significantly deviating from the native Pyl in rich media followed by transfer to mammalian hosts has not been demonstrated. Moreover, the structural transformations that occur in the active site of such a highly specific PylRS mutant necessary for accommodating a dramatic substrate change remain unclear.
Here we show that the Methanosarcina mazei PylRS (MmPylRS) can be evolved to efficiently charge a Uaa with a short aromatic side chain, in contrast to the long aliphatic side chain of the native substrate Pyl. The evolved RS incorporates the Uaa into proteins with high fidelity in both E. coli grown in rich media and mammalian cells. Additionally, we solved and refined the X-ray crystal structure of the evolved PylRS complexed with the new Uaa and an ATP analogue to a nominal resolution of 1.75 Å. The three-dimensional structure and active site architecture in the trapped substrate-bound form confirms that the mutations obtained contribute directly to the high stereochemical selectivity of our mutant PylRS for turnover of Tyr-based Uaas. Furthermore, this combined structure–function investigation provides the experimental support necessary to continue diversifying and optimizing PylRS variants in E. coli to further expand the variety of smaller Uaas for later use in mammalian cells.
Results and Discussion
Laboratory Evolution of the Methanosarcina mazei tRNACUAPyl/PylRS pair
To expand the amino acid repertoire available to the orthogonal tRNACUAPyl/PylRS pair, we employed MmPylRS as a model for rationally engineering high specificity toward Tyr-based Uaa analogues. On the basis of the X-ray crystal structure of wild-type MmPylRS,(25) six residues (Ala302, Leu309, Asn346, Cys348, Val401, and Trp417) lining the active site cavity were chosen for mutational expansion to create a genetically diverse PylRS library (Figure 1b). The Y384F mutation of PylRS, previously shown to increase the aminoacylation rate,(26) was used as the starting PylRS sequence and remained fixed in our population of mutants. In general, RS engineering and directed evolution has subtly reshaped RS amino acid specificity by limiting mutagenic exploration to amino acid positions that encompass a portion of the extended amino acid side chain. Accordingly, mutant RS libraries generated from TyrRS and PylRS have restricted genetic diversity to deep within the amino acid binding pocket.1,3,4,10 To accommodate the dramatic substrate switch from the long aliphatic Pyl tail to a short aromatic moiety, we mutated active site residues lining the entire surface of the side chain binding pocket in the Y384F PylRS mutant library. In particular, Asn346, proposed to serve as a “gate keeper” for Pyl binding,25,27 has until now been retained and not subjected to mutational expansion in most reported PylRS libraries. We, by contrast, specifically targeted this residue for mutational expansion to drive substrate selectivity substantially away from the natural Pyl substrate.
Figure 1.
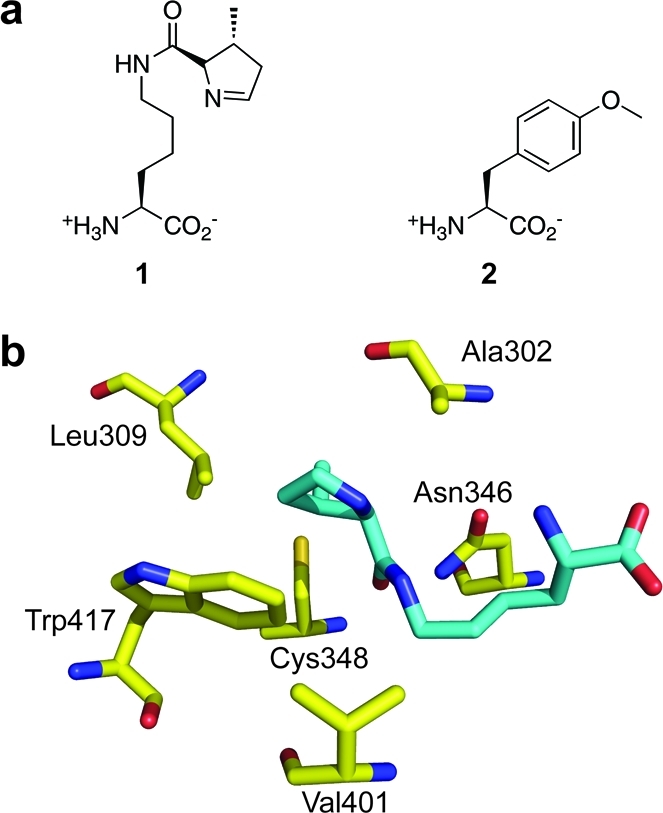
Evolving M. mazei PylRS to aminoacylate Uaas bearing aromatic side chains. (a) Structures of pyrrolsyine 1 and O-methyl-l-tyrosine (Ome, 2). (b) Structure of the active site of M. mazei PylRS (PDB ID: 2ZCE). Pyl is shown in cyan. The codons of the six residues highlighted in yellow were diversified with NNK oligonucleotides by PCR to generate a mutant library. MmOmeRS was selected from this library using Uaa Ome.
Our mutant library contains approximately 109 mutational variants, assessed by counting colony-forming units after efficient E. coli transformation. Cells were then subjected to positive selection in the presence of 1 mM of Ome (Figure 1a). Survival relies on the suppression of an amber stop codon introduced at a permissive site in the chloramphenicol acetyltransferase gene upon growth in the presence of 30 μg mL–1 chloramphenicol.4,16 Y384F PylRS mutant variants able to charge the orthogonal tRNACUAPyl with either natural or the added Uaa should then suppress the amber codon, lead to chloramphenicol acetyltransferase expression, and afford chloramphenicol resistance to the host E. coli strain.
A total of 48 colonies survived on agar plates containing 30 μg mL–1 chloramphenicol and 1 mM Ome. A secondary screen to further narrow mutant selection employed a quantitative assessment of the growth of the original 48 colonies on 1 mM Ome using a matrix of chloramphenicol concentrations. One mutant, named MmOmeRS, afforded an IC50 of 15 μg mL–1 chloramphenicol in the absence of Ome and 80 μg mL–1 chloramphenicol in the presence of Ome. Sequencing revealed that the MmOmeRS gene contained the fixed mutation Y384F and four additional changes, A302T, N346V, C348W, and V401L.
MmOmeRS Specifically Incorporates the Selected Uaa in E. coli
To measure the in vivo translation efficiency and fidelity of MmOmeRS for incorporation of Ome into recombinant proteins in E. coli, the sperm whale myoglobin gene with an amber UAG codon at position 4 (Myo4TAGHis6) was used together with the tRNACUAPyl/MmOmeRS pair in E. coli strain DH10β under control of the isopropyl β-d-1-thiogalactopyranoside(IPTG)-inducible T5 promoter. As shown in Figure 2a, when the nutrient-rich growth media 2xYT was supplemented with Ome (1 mM), full-length myoglobin appeared. Recombinant His6-tagged myoglobin was purified by Ni2+ affinity chromatography followed by gel filtration chromatography to ∼98% homogeneity yielding 4.6 mg L–1 myoglobin. In the absence of the Uaa, small amounts (0.68 mg L–1) of full-length myoglobin appeared, most likely due to low level charging of natural amino acids to tRNACUA by MmOmeRS. For comparison, the Ome-specific tRNACUATyr/synthetase pair previously evolved from wild-type M. jannaschii tRNATyr/TyrRS(4) was used to incorporate Ome into myoglobin, yielding 3.2 mg L–1 of recombinant myoglobin containing Ome.
Figure 2.

MmOmeRS specifically incorporates the cognate Uaa Ome into proteins in E. coli. (a) Suppression of the amber codon in sperm whale myoglobin at position 4 by M. janaschii OmeRS-tRNACUATyr and M. mazei OmeRS-tRNACUA with and without Ome, respectively. Myoglobin proteins were purified using Ni-NTA resin, separated by SDS-PAGE and stained with Commassie. Samples were normalized for constant cell numbers for each lane. (b, c) High resolution FT ion trap MS of the intact myoglobin protein expressed in the presence of MmOmeRS-tRNACUAPyl supplemented with 1 mM Ome (b) and without Ome in the growth media (c). Average and monoisotopic (indicated by MI) masses are labeled. In the absence of Ome, MmOmeRS incorporates Phe and Trp at low efficiencies. In the presence of Ome, only the Uaa was found to be incorporated at the UAG-encoded position by MmOmeRS. A peak with monoisotopic mass of 18508.61 was detected corresponding to myoglobin containing Ome at position 4; oxidation and deamidation peaks for this protein were also detected and labeled. Another peak with monoisotopic mass of 18377.58 was detected corresponding to Ome-containing myoglobin lacking the initiating Met; an oxidation for this species was also detected. (d) MS analysis of GFP expressed in the presence of MmOmeRS-tRNACUA supplemented with 1 mM Ome. The spectrum shows the intact precursor ions (charge deconvoluted) for one tryptic fragment of GFP (+H3N-HNIEDGSVQLADHXQQNTPIGDGPVLLPDNHYLSTQ SALSK-CO2H, X representing the UAG encoded site). The labeled peak corresponds to the incorporation of Ome at the UAG encoded position. Tandem MS spectrum of this peptide is shown in Supplementary Figure 1, which unambiguously confirms the peptide sequence and Ome at position X.
To measure the fidelity of Uaa incorporation, the purified myoglobin was analyzed by electrospray ionization Fourier transform ion trap mass spectrometry (ESI-FTMS). For myoglobin obtained with the tRNACUAPyl/MmOmeRS strain, a peak with a monoisotopic mass of 18508.61 Da was observed (Figure 2b). This mass corresponds to intact myoglobin containing a single Ome residue at position 4 (expected [M + H]+ = 18,508.77 Da). A second peak measured corresponds to Ome-containing myoglobin lacking the initiating Met (expected [M – Met + H]+ = 18377.73 Da, measured 18377.58 Da). Notably, no peaks corresponding to proteins containing any other amino acids at the amber codon position were observed. The signal-to-noise ratio of >1,000 observed in the intact protein mass spectra translates to a fidelity of >99.9% for the incorporation of Ome with the tRNACUA/MmOmeRS pair. Recombinant myoglobin produced using the tRNACUAPyl/MmOmeRS pair in media lacking Ome was also purified and analyzed by mass spectrometry (Figure 2c). The monoisotopic masses obtained indicate that Phe or Trp were incorporated at the UAG sites (expected [M(Phe) + H]+ = 18477.75 Da, measured 18477.52 Da; expected [M(Phe) – Met + H]+ = 18346.71 Da, measured 18347.44 Da; expected [M(Trp) – Met + H]+ = 18385.72 Da, measured 18385.48 Da). While these proteins contain natural amino acids, it is notable that the incorporated amino acids are similar to the Ome Uaa and quite distinct from the cognate Pyl amino acid of the original wild-type PylRS.
To assess the generality of MmOmeRS, a GFP gene containing a UAG codon at position 182 was employed. In the absence and presence of Ome, 0.13 and 13.89 mg L–1 of GFP were obtained, respectively. Ome-containing GFP was digested with trypsin and analyzed by ESI-FTMS. Ions for the tryptic GFP peptides clearly indicate that Ome was incorporated at the UAG site (expected [M + H]+ = 4486.19 Da, measured 4486.18 Da), and no peaks corresponding to any other amino acid incorporated at the UAG site were detected (Figure 2d). The precursor ion [M + H]+ corresponding to the peptide +H3N-HNIEDGSVQLADHXQQNTPIGDGPVLLPDNHYLSTQSALSK-CO2− (where X represents the UAG site) was also analyzed by tandem MS, and ion masses unambiguously confirmed the peptide sequence and assigned Ome to the UAG site (Supplementary Figure 1). In summary, these experimental observations demonstrate that while MmOmeRS will accept the sterically similar Phe or Trp natural amino acids at low levels in the absence of Ome, the tRNACUAPyl/MmOmeRS pair specifically incorporates Ome with high fidelity and efficiency when its cognate amino acid Ome is present.
Use of MmOmeRS for Uaa Incorporation in Mammalian Cells
One historical drawback associated with tRNA/RS pairs evolved in bacteria is that they are generally not orthogonal in eukaryotic cells and thus cannot be transferred to more complex systems such as mammalian cells or whole organisms. A significant advancement is the ability to evolve pairs in easily manipulated and efficiently transformed bacterial cells and then transfer the optimized orthogonal pairs directly to mammalian cells while preserving efficient and high fidelity Uaa incorporation into expressed proteins.10,19,20 MmOmeRS was tested for its ability to incorporate Uaas in HEK293 and HeLa cells. The tRNACUAPyl was expressed under control of the polymerase (Pol) III H1 promoter with the 3′ flanking sequence of the human tRNATyr appended to the 3′ end of the tRNACUA gene for correct 3′ end processing.(19) MmOmeRS was conditionally expressed using the mammalian PGK promoter.(19) The GFP gene with a UAG stop codon introduced at a permissive site Tyr182 was cotransfected with the tRNACUAPyl/MmOmeRS into HEK293 cells. Cells were grown in media with or without the Uaa Ome. As shown in Figure 3a, green fluorescence appeared when cells were grown in the presence of Ome. In the absence of the Uaa, no bright green fluorescent cells were observed. To accurately quantify the Ome incorporation efficiency, a HeLa cell line with the GFP(182TAG) gene stably integrated into the chromosome(19) was used as a reporter. After transfecting the tRNACUA/MmOmeRS pair, flow cytometry was then used to measure cellular GFP fluorescence. Total fluorescence intensity was normalized to the quantified fluorescence of HeLa cells transfected with the E. coli tRNACUATyr/TyrRS pair (Figure 3b). Incorporation efficiency for the tRNACUA/MmOmeRS pairs was 4.54% with a background suppression of 0.09% detected in the absence of Ome.
Figure 3.

MmOmeRS incorporates the cognate Uaa Ome in mammalian cells. (a) Fluorescent imaging of HEK293 cells transfected with different genes as labeled. Cells transfected with the E. coli tRNACUATyr/TyrRS pair, which incorporates Tyr at the UAG codon, were used as a positive control. For cells transfected with the GFP(182TAG) reporter gene and the tRNACUA /MmOmeRS pair, green GFP fluorescence was detected in cells cultured in media containing 1 mM Ome, indicating that the Uaa is incorporated into GFP. No GFP fluorescence was detected in cells cultured in media lacking Ome. (b) Incorporation efficiency of MmOmeRS measured with flow cytometry. The stable HeLa-GFP(182TAG) reporter cell line was transfected with the tRNACUAPyl/MmOmeRS pair and grown in media with and without 1 mM Ome. Cellular GFP fluorescence intensities were measured by flow cytometry and normalized to cells transfected with the E. coli tRNACUA/TyrRS pair. Error bars represent standard error of mean (SEM, n = 3).
A remaining challenge for the incorporation of Uaas into proteins in mammalian cells is the efficient transcription of an orthogonal tRNA incapable of being charged by the host’s endogenous RSs. Previously, we developed a general method to transcribe bacterial tRNAs in mammalian cells under control of type-3 Pol III promoters.(19) These promoters, including the H1 and U6 promoters, enable the efficient transcribing of tRNAs that do not possess the consensus A- and B-boxes normally required for Pol III transcription in mammalian cells. The M. mazei tRNACUAPyl used in this current study comes from the domain Archaea, and unlike bacterial tRNAs, this archaeal tRNA contains a consensus eukaryotic B-box and an imperfect A-box sequences. The M. mazei tRNACUA can also be functionally expressed under transcriptional control by the H1 promoter in mammalian cells. The results described here support the deployment of type-3 Pol III promoters for expressing other archaeal tRNAs in mammalian cells.(9)
Three-Dimensional Architecture of MmOmeRS
Following structure-guided mutation and selection, MmOmeRS switched amino acid specificity from the native substrate Pyl to the structurally divergent substrate Ome. Notably, the new specificity obtained for Ome differs substantially from the wild-type specificity for the long aliphatic side chain of Pyl. To understand the molecular basis underlying this evolutionary change, we determined the high-resolution X-ray crystal structure of the catalytic domain of MmOmeRS complexed with Ome and the non-hydrolyzable ATP analogue AMP-PNP.
MmOmeRS, like its wild-type ancestor PylRS, purified as a homodimer and crystallized in space group P64 with unit cell dimensions of a = 104.81 Å, b = 104.81 Å, c = 71.67 Å and angles α = β = 90°, γ = 120° using the previously published crystallization conditions for wild-type MmPylRS.(25) MmOmeRS was also cocrystallized with Mg2+, the non-hydrolyzable ATP analogue AMP-PNP, and the Uaa Ome. The crystal structure of the ternary Ome-AMP-PNP-Mg2+ complex was solved by molecular replacement and refined to 1.75 Å with an Rfactor of 18.3% and an Rfree of 21.2% (see Table 1 for data processing and refinement statistics). The SIGMAA weighted 2Fo – Fc experimental electron density maps exhibited clear electron density for Ome, AMP-PNP, and two octahedrally coordinated Mg2+ ions. MmOmeRS crystals contained one ternary complex per asymmetric unit; two ternary complexes related by a 2-fold crystallographic symmetry axis form the likely physiological dimer of MmOmeRS.
Table 1. X-ray Diffraction Data Processing and Refinement Statistics.
| protein | MmOmeRS |
| ligands | Ome, AMP-PNP, Mg2+ |
| PDB ID | 3QTC |
| Data Collection and Processing | |
| space group | P64 |
| resolution (Å) | 1.75 |
| cell dimensions | |
| a, b, c (Å) | 104.81, 104.81, 71.67 |
| α, β, γ (DEG) | 90, 90, 120 |
| molecules in asymmetric unit | 1 |
| no. measured reflectionsa | 458839 (45558) |
| no. unique reflectionsa | 44965 (7161) |
| redundancy | 10.2 (6.4) |
| Rmerge (%)a,b | 4.0 (36.8) |
| completeness (%)a | 99.7 (98.9) |
| I/σ(I)a | 35.1 (4.3) |
| Refinement | |
| resolution range (Å) | 50.0–1.75 |
| no. reflections | |
| working set | 42693 |
| test set | 2268 |
| Rwork/Rfreec | 0.1834/0.2124 |
| no. atoms | |
| protein | 2157 |
| ligands | 306 |
| water | 235 |
| mean overall B-factor (Å2) | 29.93 |
| rmsd bond lengths (Å)d | 0.01 |
| rmsd bond angles (DEG°) | 1.30 |
| refinement program | Refmac |
Values in parentheses represent data from the highest resolution shell.
Rmerge = ΣhklΣi∣Ii(hkl) – <I(hkl)>∣/Σhkl ΣiIi(hkl)
Rfactor = Σ∣∣Fobs∣ – ∣Fcalc∣∣/Σ∣Fobs∣. Rwork is the Rfactor calculated using all diffraction data included in the refinement. Rfree is the Rfactor calculated using the randomly chosen 5% of diffraction data that were not included in the refinement.
rmsd = root-mean-square deviation
PylRS and the MmOmeRS mutant belong to the class-II division of aminoacyl-tRNA synthetases (PF00587).(25) This fold consists of a central antiparallel seven-stranded β-sheet decorated by several peripheral helices. The overall tertiary structure of MmOmeRS (Figure 4a) is consistent with all previously reported crystal structures for PylRSs, generating root-mean-square deviation (rmsd) values ranging from 0.16 to 0.23 Å upon superposition of C-α atoms with representative family members (PDB IDs 2ZCE, 2ZIN, and 2Q7G). Structural alignment of the MmOmeRS structure reported here with the previously reported PylRS structure complexed with AMP-PNP and Pyl (PDB ID: 2ZCE)(27) illustrates conserved AMP-PNP binding with the sterically and chemically divergent amino acid Ome occupying a similar active site location as the wild-type cognate amino acid Pyl.25−27
Figure 4.
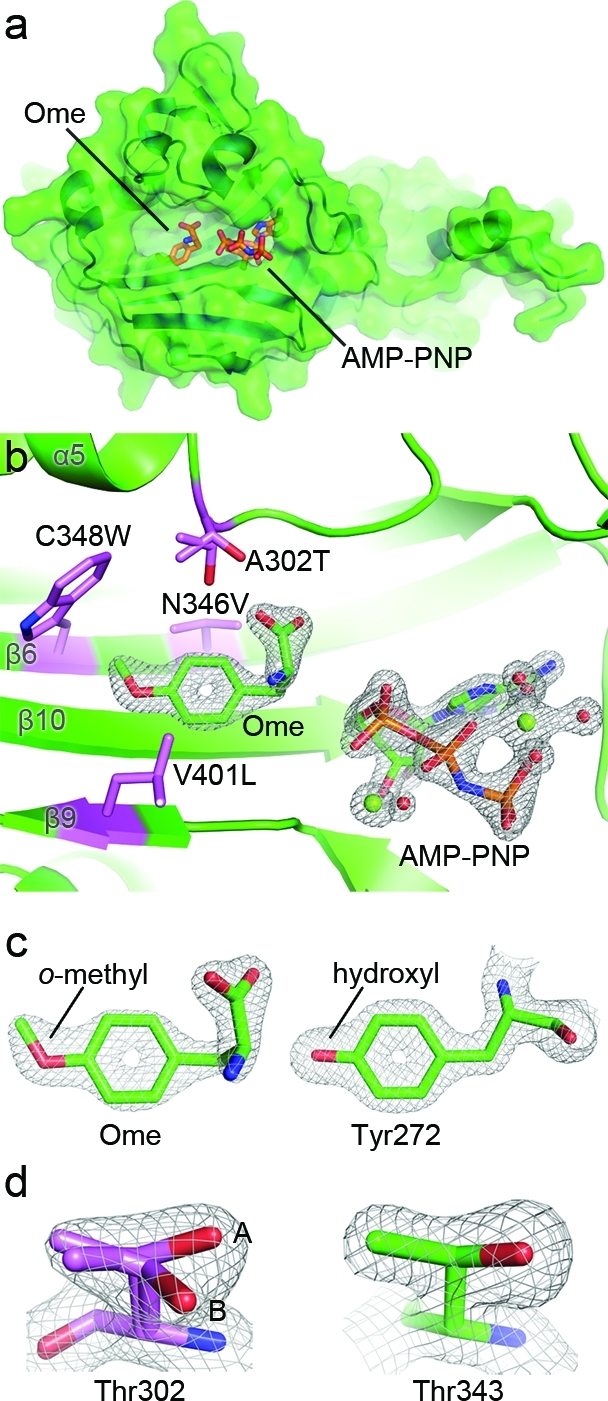
Crystal structure of MmOmeRS reveals the stereochemical basis for Ome selectivity. (a) Global view of the crystal structure of MmOmeRS expressed as a filled surface. (b) Depiction of the four mutations defining the transformation of PylRS to MmOmeRS in the crystal structure of MmOmeRS complexed with Ome, Mg2+, and AMP-PNP. Both the Uaa (Ome) and the non-hydrolyzable ATP-analogue (AMP-PNP) are shown together with the calculated SIGMAA-weighted 2Fo – Fc electron density contoured at 2σ and 2.5σ, respectively. (c) Comparison of the calculated SIGMAA-weighted 2Fo – Fc electron density contoured at 2σ for both Ome (left) and a structurally related amino acid Tyr272 (right) demonstrates that the quality of electron density is excellent for both protein residues and substrates. Moreover, the O-methyl moiety is unequivocally present in Ome. (d) Differences in the shapes of the electron density contoured at 1.75σ for both the active site Thr302 (left) and a second Thr343 (right) support the presence of two alternate conformers for Thr302 (labeled A and B).
One notable structural difference between the MmOmeRS crystal structure reported here and other published PylRS structures encompasses residues across the β7−β8 loop (Val177–Gly185). Previous crystal structures of wild-type PylRS establish several distinct conformations of this loop, including open, intermediate, and closed conformations that are seemingly independent of the bound Pyl amino acid ligand.(27) In the crystal structure of MmOmeRS, electron density for the β7−β8 loop is noticeably absent, signifying that the loop most likely contributes in a dynamic manner to active site closure and catalysis.
Active Site Substitutions Lead to an Unexpected Recognition of the Ome Side Chain Moiety
MmOmeRS contains a single fixed mutation, Y384F, and four additional mutations selected for during directed evolution: A302T (located on the N-terminal end of the α5 helix), N346V and C348W (abutting residues on the C-terminal end of the β6 strand), and V401L (located on the C-terminal end of the β9 strand) (Figure 4b). The methyl group of Ome is clearly visible on the tip of the bound ligand, distinguishing Ome from the structurally similar natural amino acid Tyr (Figure 4c). In all previous studies of PylRS, the wild-type residue Asn346 serves to (1) directly bind the secondary carbonyl (Nε-carbonyl) of Pyl or Pyl analogues through hydrogen bonding and (2) coordinate the α-amino group or α-carboxyl group, depending on the orientation of the main chain atoms in Pyl or Pyl analogues. This latter interaction occurs through a water-mediated hydrogen bond (Figure 5b,c).25−27 Evolved MmOmeRS possesses a non-hydrogen bond donor–acceptor at position 346 (Figure 4b). Curiously, previous results demonstrate that mutation of Asn346 to Ala significantly impairs aminoacylation activity in PylRS,(27) suggesting some form of compensatory changes in the selected MmOmeRS.
Figure 5.
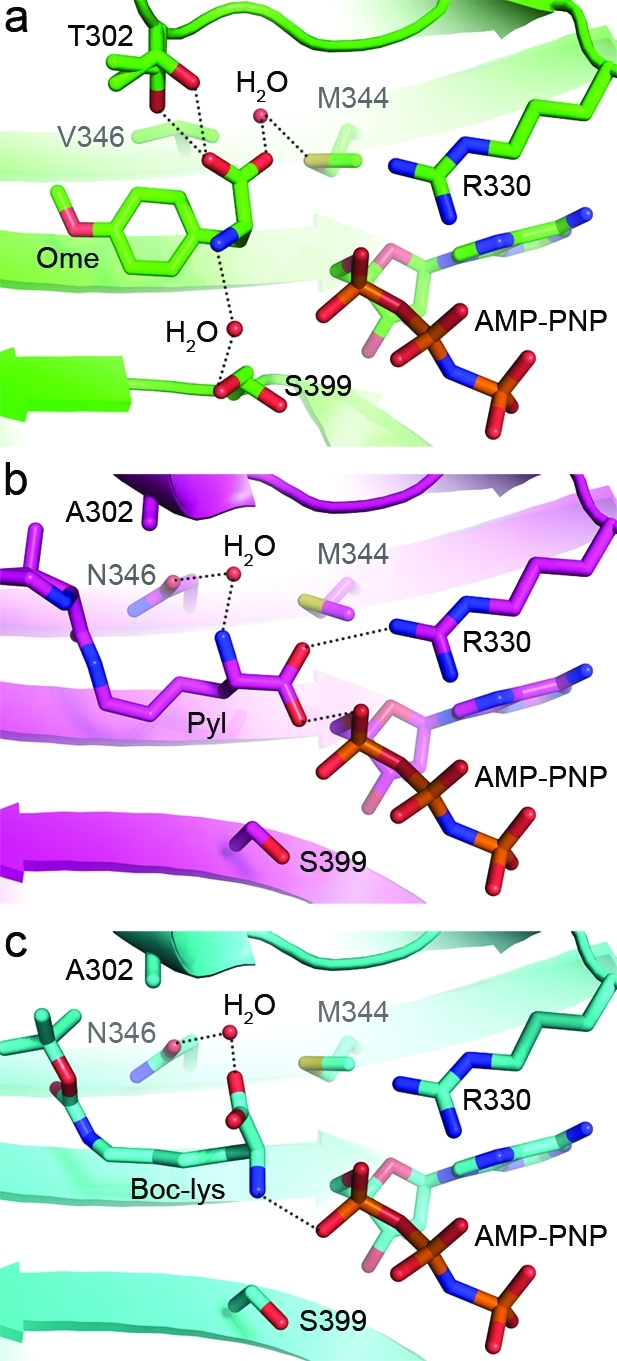
Structural depiction of interactions between the amino acid substrate and the surrounding active site residues within MmOmeRS and PlyRS. (a) Interactions between main chain atoms of the Ome substrate and surrounding protein residues in the crystal structure of MmOmeRS complexed with Ome, Mg2+, and AMP-PNP. Both Thr302 and Ser399 exist in two rotameric conformations. Dashed lines between atoms indicate that they are within hydrogen bonding distance. (b) Interactions between main chain atoms of Pyl and surrounding protein residues in the crystal structure of PylRS complexed with Pyl, Mg2+, and AMP-PNP (PDB ID: 2ZCE). (c) Interactions between main chain atoms of Boc-Lys and surrounding protein residues in the crystal structure of PylRS complexed with Boc-Lys and AMP-PNP (PDB ID: 2ZIN).
The X-ray crystal structure of the MmOmeRS-Ome-AMP-PNP-Mg2+ ternary complex illustrates how the A302T mutation positions the hydroxyl-bearing Thr side chain as a bump on the active site surface in proximity to the β-sheet defining the floor of the amino acid binding site. On this floor, the other critical change, N346V, projects up and into the active site (Figure 4b). The Thr hydroxyl moiety sterically and electronically compensates for the loss of the hydrogen bond contributed by Asn346 in wild-type PylRS. In particular, Thr302 in MmOmeRS assumes two distinct rotameric conformations (Figure 4d), both of which hydrogen bond directly to the α-carboxyl group of Ome without the intervention of a water molecule (Figure 5a). In addition, the N346V mutation abolishes the previously observed hydrogen-bonding interaction between Asn346 and the side chain carbonyl group of Pyl or Pyl analogues in PylRS.
In wild-type PylRS, Leu305, Tyr306, Cys348, and Trp417 form a deep hydrophobic pocket in the active site that accommodates the elongated methyl-pyrroline ring of Pyl. In MmOmeRS, the C348W mutation places the indole moiety of Trp348 directly into this elongated cavity, thereby shortening the depth of the cavity (Figure 4b). Consequently, the active site volume decreases from 2883 to 2174 Å3 when comparing PylRS (PDB ID: 2ZCE) to MmOmeRS, respectively. In PylRS, the mutation C348A does not exert a measurable effect on activity, suggesting that this residue does not contribute significantly to stabilization of the transition state accompanying amino acid charging.(27) In contrast, MmOmeRS uses Trp348 to not only facilitate recognition of Ome but also to stabilize the O-methyl group on Ome through a quadrupole-dipole interaction that is a direct result of the indole plane sitting perpendicular to the O-methyl moiety of Ome. Finally, the V401L mutation extends the wild-type Val side chain by a methylene group allowing the Leu side chain moiety to reach and better accommodate the shorter but wider Ome side chain (Figure 4b). In total, these selected mutations in MmOmeRS widen part of the amino acid binding cavity while condensing its depth, ultimately providing a complementary shape to accommodate the bulky phenyl ring of Ome.
Recognition of the Ome Main Chain and Coordination of the Adenyl-Donor AMP-PNP-Mg2+
In MmOmeRS, the α-amino and α-carboxyl groups of Ome rotate counterclockwise 103° along the Cα–Cβ bond relative to their positions in PylRS; this rotation effectively flips the positions of the α-amino and α-carboxyl groups in Ome-bound MmOmeRS compared to Pyl-bound PylRS. This interchange results in distinctly different stabilizing interactions (Figure 5a,b). For example, in wild-type PylRS, the α-amino group of Pyl interacts with the side chain of Asn346 via a water-mediated interaction. In MmOmeRS, the α-amino group of Ome hydrogen-bonds via a water-mediated interaction to one of two observed rotameric conformations of Ser399. Conspicuously, Ser399 resides distal to position 346 along the surface of the active site cavity. In PylRS, Arg330 interacts through electrostatic hydrogen bonds with the negatively charged α-carboxyl group of Pyl; this electrostatic role is replaced by a neutral hydrogen-bonding network in MmOmeRS triggered by the A302T mutation. Additionally, the second oxygen atom of the α-carboxyl group of Ome is further solvated by a water molecule that is unusually within hydrogen-bonding distance (2.9 Å) to the thioether sulfur atom of Met344; in contrast, for Pyl-bound PylRS (PDB ID: 2ZCE), this distance extends to 3.6 Å, which is more consistent with a van der Waals interaction rather than a more stabilizing hydrogen-bonding interaction. The α-amino-carboxyl group flip was also seen in the previously reported crystal structure of PylRS complexed with AMP-PNP and Boc-Lys (PDB ID: 2ZIN). In this latter case, the Asn346 side chain hydrogen bonds to the α-carboxyl moiety through a water molecule instead of with the α-amino group of Boc-Lys as seen in other wild-type PylRS crystal structures (Figure 5c).(26)
The main chain atoms of Pyl sit in a catalytically competent orientation consistent with the expected in-line nucleophilic attack of an oxygen atom from the α-carboxyl group on the α-phosphate atom of AMP-PNP (Figure 6b)25,27 By contrast, in MmOmeRS, the α-amino group of Ome sits proximal to the α-phosphate of AMP-PNP through an intervening network of water-mediated interactions (Figure 6a). The orientation of the main chain atoms in Ome is not uncommon among structures of PylRS complexed with various analogues; for example, Boc-Lys also has its α-amino group close to the α-phosphate of AMP-PNP (PDB ID: 2ZIN, Figure 6c).(26) These Uaas could readily rotate the α-carboxyl and α-amino groups along the Cα-Cβ bond to achieve a catalytically competent orientation.
Figure 6.
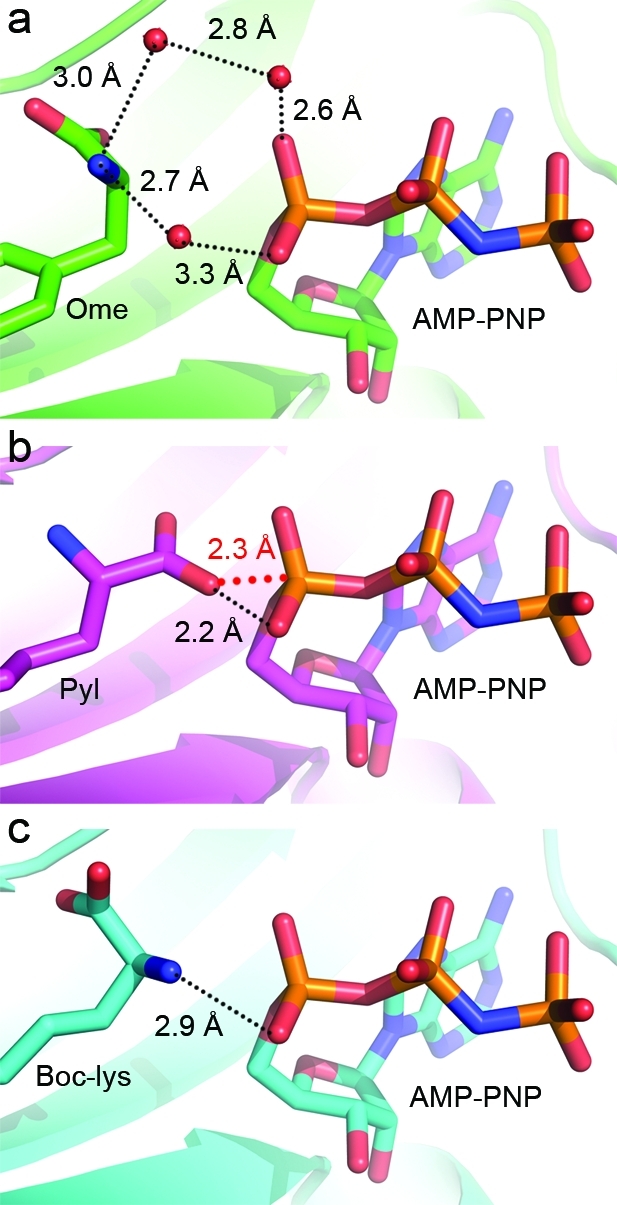
Differences in coordination geometry between an ATP analogue and various amino acid substrates in PylRS and MmOmeRS. (a) Indirect coordination of the Ome amino group to AMP-PNP is mediated by several waters in the crystal structure of MmOmeRS complexed with Ome, Mg2+, and AMP-PNP. The main chain carboxyl moiety sits in an orientation that is not directly in-line for attacking the α-phosphate of AMP-PNP (ATP). (b) Direct coordination of an oxygen atom from the carboxyl group of Pyl to the α-phosphate of AMP-PNP in the crystal structure of PylRS complexed with Pyl and AMP-PNP (PDB ID: 2ZCE). Unlike in the case of MmOmeRS, the main chain carboxyl group of Pyl resides in an orientation consistent with adenylation. The red dashed line represents the distance gap between the two atoms that undergo bond formation during the aminoacylation reaction. (c) Hydrogen-bonding network of the amino group of Boc-Lys and an oxygen atom of the α-phosphate of AMP-PNP in the crystal structure of PylRS complexed with Boc-Lys and AMP-PNP (PDB ID: 2ZIN). This Uaa sits in an orientation similar to that observed in the MmOmeRS-Ome-AMP-PNP-Mg2+ complex.
Conclusions
A challenge facing the deployment of tRNA/RS pairs in mammalian cells is the inability to streamline the directed evolution of such pairs to produce orthogonal and catalytically efficient tRNA/RS sets possessing high specificity for a particular Uaa. The current solution to this bottleneck consists of evolving E. coli RSs in yeast and transferring the resultant mutants to mammalian cells.19,20 The major drawbacks of using yeast include its lower transformation efficiency and its restriction to less efficient selection methods compared to those originally developed in E. coli.4,16 Unfortunately, M. jannaschii tRNATyr/TyrRS, which has been extensively evolved in E. coli, does not act in an orthogonal manner in mammalian cells. On the other hand, the more recently employed tRNACUAPyl/PylRS pair does have a currently unique property of being orthogonal in E. coli and mammalian cells, possibly due to its special role encoding the rarer amino acid Pyl that is not used by E. coli or mammalian cells. Thus, PylRS mutants evolved in E. coli can be directly transferred to mammalian cells for efficient Uaa incorporation.
To date, nearly all Uaas incorporated by evolved PylRS mutants contain a carbonyl group at the Nε position of Pyl. Our report represents a successful breakthrough regarding this limitation. The chosen amino acid for driving selection, namely, Ome, does not contain this carbonyl moiety of Pyl, and its side chain is significantly different from Pyl and reported Pyl analogues. Another striking switch in synthetase substrate specificity is the directed evolution of E. coli LeuRS to incorporate a fluorescent Uaa dansylalanine.(28) However, the E. coli LeuRS must be evolved in yeast as it is not orthogonal in E. coli. Although the previously reported evolution of PylRS to incorporate Phe-based Uaas into proteins in E. coli was somewhat promising, incorporation was achieved in E. coli grown in glycerol minimal media only.(24) Minimal media is used in published reports to greatly reduce the mis-incorporation of natural amino acids,(23) raising concerns with regard to the specificity of these evolved PylRSs when used in rich media, a condition necessary for fermentation, mammalian cell cultures, and whole organism engineering. Here, the evolved tRNA/MmOmeRS pair efficiently and specifically incorporated Ome into proteins both in E. coli using rich media and in mammalian cells, and we have been able to assign this high level of specificity to interactions revealed through structural analysis.
The crystal structure of MmOmeRS reported here illustrates the stereochemical features governing Uaa recognition at the molecular level. It was previously thought that the carbonyl group of the Pyl side chain played a critical role during the aminoacylation reaction catalyzed by PylRS;(26) Asn346 in wild type PylRS interacts simultaneously with this side chain carbonyl group and the α-amino group of Pyl or a Pyl analogue (PDB IDs: 2ZCE, 2ZIN, 2Q7G).25−27 In contrast, we chose to subject position 346 to mutational analysis. The resulting mutation selected at this position, N346V, creates an ideal hydrophobic pocket for the encapsulation of the Ome phenyl ring. Additionally, the A302T mutation complements the loss of the Asn346 hydrogen bond by forming a network of interactions with the main chain of the Ome substrate. These surprising observations indicate that bold yet progressive approaches toward library design allowed for access to regions of sequence space not typically explored due to deleterious effects of one mutation, suggesting that context dependence via compensatory mechanisms encouraged the successful evolution of MmOmeRS.
A similar interaction network is observed in several other class II amino acid RSs. For example, in the crystal structure of HisRS from E. coli (PDB ID: 2EL9), Thr85 (A302T in MmOmeRS) is within hydrogen-bonding distance of the α-amino group of the His substrate, while Gly129 (N346V in MmOmeRS) plays no apparent role in substrate binding. Interestingly, Thr85 is a conserved residue among all HisRSs, and Gly129 is partially conserved, indicating that a naturally evolved stabilization mechanism for some RSs is comparable to the laboratory evolved substrate binding of MmOmeRS reported here. This switch in the stereochemistry of substrate recognition in MmOmeRS due to the epistatic nature of two laboratory evolved mutations, N346V and A302T, has not been reported in any engineered RS to date. Like the fixed Y384F PylRS mutation that served as a starting point for this current study, the fixation of the N346V and A302T mutations may be a necessary step forward for the design and directed evolution of PylRS-based mutant synthetases that possess an even greater expansion of selectivity for Uaas.
Apart from these two critical positions, a third mutation is C348W. The X-ray crystal structure of the ternary complex again demonstrates the stereochemistry of this selected change. The bulky side chain indole moiety of Trp348 effectively shortens the elongated amino acid binding tunnel of wild type PylRS to accommodate the shorter Ome side chain.
In some synthetases, the tRNA binding event alters the conformation of the amino acid binding cleft and yields changes in amino acid substrate binding affinity.29,30 Although the crystal structure for the M. mazei tRNAPyl/PylRS complex has not been reported, the crystal structure for the Desulfitobacterium hafniense tRNAPyl/PylRS complex has previously been solved to a resolution of 3.1 Å.(31) A comparison of this tRNAPyl/PylRS complex to our M. mazei OmeRS structure reveals that the synthetase components of each structure are very similar; superposition of Cα atoms between the two structures yields an rmsd value of 1.188 Å. The only active site motif that subtly changes conformation is the β9 strand where the V401L mutation resides. However, this subtlety can be accounted for by differences in amino acid residues between the two proteins at these positions and is most likely not relevant to the tRNA binding event (Supplementary Figure 2). It is of note that the crystal structure of the tRNAPyl/PylRS complex models the β7-β8 loop in a solvent exposed orientation (away from the active site in an open position). This loop is absent from the MmOmeRS structure, and therefore its orientation in a complex with tRNA is unpredictable. Overall, the absence of the tRNA molecule in the crystal structure of MmOmeRS most likely does not impact library design in a directed evolution experiment. However, it is of note that structural differences between aaRS and tRNA/aaRS complex, if present, should be taken into consideration for mutant aaRS library design. In addition, rational design of amino acid specificity between natural amino acids in an aaRS has been accomplished and shown to require many substitutions throughout the catalytic domain.(38) Therefore, increase of mutant aaRS library size to target more residues for mutation may further expand the Uaa diversity and enhance incorporation efficiency.
In summary, this study presents the first stereochemical explanation for the laboratory evolved switch in PylRS substrate specificity from long aliphatic moieties to shortened aromatic side chains; this information provides both a molecular and a structural basis for engineering synthetases with expanded repertoires of Uaas that can be deployed in an orthogonal manner in complex hosts such as mammalian cells and multicellular organisms.
Methods
Plasmid Construction
Plasmids were constructed using standard cloning procedures and confirmed by DNA sequencing. The pTak-GFPtag plasmid contained the M. mazei tRNACUAPyl and the GFP(182TAG) gene tagged to a His6 polypeptide appended to the C-terminus. Transcription of the tRNACUA gene was driven by the lpp promoter and terminated by the rrnC terminator. GFP(182TAG) gene transcription was driven by the T5 promoter under regulation of the lac operator sequence and transcription was terminated with the λt0 terminator. Plasmid pTak-Myo4TAG was created by replacing the GFP(182TAG) gene in pTak-GFPtag with the sperm whale myoglobin gene containing an amber codon at position 4. Plasmid pLei-Myo4TAG is identical to pTak- Myo4TAG except the M. mazei tRNACUAPyl was replaced with the M. jannaschii tRNACUA, J17.(5) Plasmid pMPcua-OmeRS was derived from pEYcua-TyrRS(19) by replacing the E. coli tRNACUATyr with the M. mazei tRNACUA and the E. coli TyrRS with MmOmeRS. DNA sequences and primers are listed in Supporting Information.
Library Construction and in Vivo Selection
Three rounds of overlapping PCRs were performed on the M. mazei PylRS gene to randomize the codons for each selected active site residue using oligonucleotides containing NNK at each site destined for mutagenic diversification. The amplified full-length PCR product was ligated into the precut pBK5-MmPylRS plasmid to afford the mutant DNA library. The library was electroporated into DH10βT1 competent cells harboring pREP-PylT.(10) Selection was carried out on LB plates containing 12.5 μg mL–1 tetracycline, 50 μg mL–1 kanamycin, 30 μg mL–1 chloramphenicol and 1 mM Ome.
Protein Expression and Uaa Incorporation in E. coli
To translationally incorporate Uaas into myoglobin in E. coli, DH10β cells were transformed with pTak-Myo4TAG/pBK-MmOmeRS and pLei-Myo4TAG/pBK-MjOmeRS, respectively. For each sample, a colony was picked and grown overnight in 50 mL 2xYT supplemented with 30 μg mL–1 chloramphenicol and 50 μg mL–1 kanamycin at 37 °C. This culture was used to inoculate 1 L of 2xYT containing antibiotics and supplemented with or without 1 mM of Ome. When OD600 reached 0.5, cells were induced for protein expression by adding 0.5 μM IPTG. After 3 h, cells were pelleted and sonicated to lyse in 40 mL lysis buffer (50 mM TrisHCl, pH 8.0, 500 mM NaCl, 20 mM imidazole pH 8.0, 1% (v/v) Tween 20, 10% (v/v) glycerol, 10 mM 2-mercaptoethanol, and 0.5 mg mL–1 lysozyme). Lysed cells were centrifuged for 10 min at 6,000 g, and clarified supernatant was passed through a 0.5 mL column of Ni2+-NTA agarose resin (Qiagen). The column was washed with 10 column volumes of wash buffer (lysis buffer without Tween 20 and lysozyme). Protein was eluted with 1 mL of elution buffer (wash buffer containing 250 mM imidazole pH 8.0). Imidazole was removed from the eluent by buffer exchange using a Microcon Ultracel YM-10 spin column (Millipore). The sample was further purified by fast protein liquid chromatography using a HiLoad 26/60 Superdex 200 gel filtration column in 50 mM TrisHCl, pH 8.0 with 500 mM NaCl. To incorporate Ome into GFP, DH10β cells were transformed with pTak-GFPtag and pBK-MmOmeRS. GFP was purified using the same procedure described above.
Protein Expression and Uaa Incorporation in Mammalian Cells
Uaa incorporation was carried out as previously described.(32) Briefly, HEK293 cells were transfected with pCLHF-GFP(182TAG)(19) and pMPcua-OmeRS. The HeLa-GFP(182TAG) stable cells were transfected with pMPcua-OmeRS or pEYcua-TyrRS. Cells were grown in DMEM supplemented with 10% FBS; 1 mM of Ome was added to or withheld from the media for cells transfected with pMPcua-OmeRS. HEK293 cells were imaged 48 h after transfection with an Olympus IX8I microscope equipped with a Hamamatsu EM-CCD under same conditions for all samples (λex = 480 ± 20 nm, λem = 535 ± 40 nm). Transfected HeLa cells were trypsinized and washed with PBS twice. After resuspending in 1.0 mL PBS and 5 μL of propidium iodide, cells were analyzed with a FACScan (Becton & Dickinson, excitation 488 nm, emission 530 ± 30 nm). For each sample the total fluorescence intensity of 30,000 cells was recorded and was normalized to cells transfected with pEYcua-TyrRS, which expresses the E. coli tRNACUATyr/TyrRS.(19)
Mass Spectrometry
For intact protein analysis, myoglobin samples were analyzed by high resolution Fourier-transform (FT) mass spectrometry (MS) on a Thermo LTQ-Orbitrap XL mass spectrometer(33) (ThermoFisher). Samples were loaded onto a capillary column with integrated spray tip, which was filled with reversed phase material (Zorbax SB C-18, particle size 5 μm). Protein was eluted using 0.1% (v/v) formic acid and a gradient of increasing concentrations of acetonitrile at a flow rate of 300 nL min–1. The eluate was electrosprayed directly into the mass spectrometer. FT-MS spectra were recorded a resolution of 60,000 for a scan range of m/z = 400–1800. This was followed by a select ion scan of the most intense ion at resolution 60,000. Data were charge-deconvoluted using the Thermo Qualbrowser 2.0 Xtract program.
For GFP samples, ∼1 μg of protein was first desalted using Microcon Ultracel Y-10 spin columns, and then digested with trypsin (200 ng, Sigma, Proteomics grade) in 50 mM ammonium bicarbonate buffer for 16 h. An aliquot of the resulting digest (5–10%) were dried down and redissolved in 0.1% formic acid. High resolution FT-MS analysis was performed on a Thermo LTQ-Orbitrap XL. Samples were loaded onto the same capillary column described above, eluted and electrosprayed using the same procedures. FT-MS spectra were recorded at a resolution of 60,000 in the Orbitrap FT analyzer followed by MS/MS scans of the top 5 ions in the linear ion trap. MS/MS data were searched against a custom database consisting of the protein of interest using the Mascot algorithm (Matrixscience). For the search, a variable modification at Tyr residues with a mass of 14.01564 (corresponding to the mass difference between Ome and Tyr) was allowed.
Cloning, Expression, and Purification of MmOmeRS
The C-terminal catalytic domain (residues 185–454) of MmOmeRS was amplified from the pBK vector containing full length MmOmeRS by PCR with the following primers: 5′-TTC CCA TGG CGC AAG CGC CCC AGC TCT G-3′ and 5′-CGC AAG CTT TTA CAG GTT AGT AGA AAT ACC ATT GTA ATA GGA CTC GG-3′. The PCR product was cloned into an in-house pET28a(+) vector (Novagen) modified to contain an N-terminal His8-tag. Both the destination vector and insert were digested with Nco I and Hind III and ligated using T4 DNA ligase (New England Biolabs, Inc.). The insert containing plasmid was recovered after initial transformation in E. coli BP5α cells (BioPioneer), confirmed by DNA sequencing and transformed into E. coli BL21 (DE3) cells (Novagen). One colony was grown at 37 °C in TB media to an OD600 of 1.0 and then induced with 1 mM IPTG overnight at 22 °C. The cells were harvested, resuspended in lysis buffer (50 mM HEPES-Na+, pH 7.5, 500 mM NaCl, 20 mM imidazole, 1% (v/v) Tween-20, 10% (v/v) glycerol, 10 mM 2-mercaptoethanol) containing lysozyme (0.5 mg mL–1), and stirred for 45 min at 4 °C. The solution was then sonicated and centrifuged to clarify the supernatant, which was subsequently passed through a 1 mL column of Ni-NTA agarose resin (Qiagen). The column was washed with ten column volumes of lysis buffer and wash buffer (lysis buffer without Tween20) prior to elution of the His8-tagged MmOmeRS with elution buffer (wash buffer containing 250 mM imidazole). The eluent was dialyzed overnight in dialysis buffer (5 mM MgCl2, 300 mM NaCl and 10 mM HEPES-Na+, pH 7.5) and passed through a HiLoad 16/16 SuperdexTM 200 prep grade (GE Healthcare) gel filtration column. The purified MmOmeRS protein was concentrated to 10 mg mL–1 and stored at −80 °C.
Crystallization, Structure Solution, and Refinement
Co-crystallization of MmOmeRS with Ome and AMP-PNP was accomplished using a previously described method.(25) Briefly, 1 μL of MmOmeRS (10 mg mL–1) containing 5 mM Ome and 5 mM AMP-PNP was added to 1 μL of reservoir solution (100 mM TrisHCl, pH 8.0, 11% (w/v) PEG 2000 MME) at 25 °C. Crystals were cryostabilized in the reservoir solution supplemented with 30% (v/v) ethylene glycol and frozen in liquid nitrogen.
Diffraction data were collected on ALS beamline 8.2.2 (Lawrence Berkeley National Laboratory) using an ADSC Q315 CCD detector. Images were processed and scaled with XDS.(34) A model of MmOmeRS was constructed using PylRS as a reference structure, and molecular replacement followed by rounds of refinement were performed using the ccp4 programs Molrep and Refmac, respectively.(35) The program COOT was used for model visualization and building throughout the refinement process.(36) PROCHECK was used for quality assessment of the final MmOmeRS structure.(37) The data and refinement statistics are presented in Table 1.
Acknowledgments
We thank Dr. G. Louie for help with data processing of crystallographic data and Dr. W. Fisher and J. Read for help with mass spectrometry. N.D. acknowledges support of an HHMI predoctoral fellowship. This material is based in part upon work supported by the National Science Foundation under Award No. MCB-0645794 (J.P.N.) and by the Salk Institute Cancer Center Award No. P30CA014195 from the National Cancer Institute (J.P.N. and L.W.). J.P.N. is an investigator with the Howard Hughes Medical Institute. Portions of this research were conducted at the Advanced Light Source (ALS), a national user facility operated by Lawrence Berkeley National Laboratory, on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The Berkeley Center for Structural Biology is supported in part by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences. L.W. acknowledges support from the Ray Thomas Edwards Foundation, March of Dimes Foundation (#5-FY08-110), California Institute for Regenerative Medicine (RN1-00577-1), and National Institutes of Health (1DP2OD004744-01).
MmOmeRS: PDB ID 3QTC
Supporting Information Available
DNA sequences for described synthetases and other proteins, primers for plasmid construction, and supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ These authors contributed equally to this work.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wang L.; Schultz P. G. (2005) Expanding the genetic code. Angew. Chem., Int. Ed. 44, 34–66. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Parrish A. R.; Wang L. (2009) Expanding the genetic code for biological studies. Chem. Biol. 16, 323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. (2010) Adding new chemistries to the genetic code. Annu. Rev. Biochem. 79, 413–444. [DOI] [PubMed] [Google Scholar]
- Wang L.; Brock A.; Herberich B.; Schultz P. G. (2001) Expanding the genetic code of Escherichia coli. Science 292, 498–500. [DOI] [PubMed] [Google Scholar]
- Wang L.; Schultz P. G. (2001) A general approach for the generation of orthogonal tRNAs. Chem. Biol. 8, 883–890. [DOI] [PubMed] [Google Scholar]
- Srinivasan G.; James C. M.; Krzycki J. A. (2002) Pyrrolysine encoded by UAG in Archaea: charging of a UAG-decoding specialized tRNA. Science 296, 1459–1462. [DOI] [PubMed] [Google Scholar]
- Polycarpo C. R.; Herring S.; Berube A.; Wood J. L.; Soll D.; Ambrogelly A. (2006) Pyrrolysine analogues as substrates for pyrrolysyl-tRNA synthetase. FEBS Lett. 580, 6695–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namy O.; Zhou Y.; Gundllapalli S.; Polycarpo C. R.; Denise A.; Rousset J. P.; Soll D.; Ambrogelly A. (2007) Adding pyrrolysine to the Escherichia coli genetic code. FEBS Lett. 581, 5282–5288. [DOI] [PubMed] [Google Scholar]
- Mukai T.; Kobayashi T.; Hino N.; Yanagisawa T.; Sakamoto K.; Yokoyama S. (2008) Adding l-lysine derivatives to the genetic code of mammalian cells with engineered pyrrolysyl-tRNA synthetases. Biochem. Biophys. Res. Commun. 371, 818–822. [DOI] [PubMed] [Google Scholar]
- Chen P. R.; Groff D.; Guo J.; Ou W.; Cellitti S.; Geierstanger B. H.; Schultz P. G. (2009) A facile system for encoding unnatural amino acids in mammalian cells. Angew. Chem., Int. Ed. 48, 4052–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. T.; Mahapatra A.; Longstaff D. G.; Bechtel J.; Zhao G.; Kang P. T.; Chan M. K.; Krzycki J. A. (2009) Specificity of pyrrolysyl-tRNA synthetase for pyrrolysine and pyrrolysine analogs. J. Mol. Biol. 385, 1156–1164. [DOI] [PubMed] [Google Scholar]
- Wang Y. S.; Wu B.; Wang Z.; Huang Y.; Wan W.; Russell W. K.; Pai P. J.; Moe Y. N.; Russell D. H.; Liu W. R. (2010) A genetically encoded photocaged Nepsilon-methyl-L-lysine. Mol. Biosyst. 6, 1557–1560. [DOI] [PubMed] [Google Scholar]
- Turner J. M.; Graziano J.; Spraggon G.; Schultz P. G. (2006) Structural plasticity of an aminoacyl-tRNA synthetase active site. Proc. Natl. Acad. Sci. U.S.A. 103, 6483–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T.; Sakamoto K.; Takimura T.; Sekine R.; Kelly V. P.; Kamata K.; Nishimura S.; Yokoyama S. (2005) Structural basis of nonnatural amino acid recognition by an engineered aminoacyl-tRNA synthetase for genetic code expansion. Proc. Natl. Acad. Sci. U.S.A. 102, 1366–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddha M. R.; Crane B. R. (2005) Structure and activity of an aminoacyl-tRNA synthetase that charges tRNA with nitro-tryptophan. Nat. Struct. Mol. Biol. 12, 274–275. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zhang Z.; Brock A.; Schultz P. G. (2003) Addition of the keto functional group to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 100, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J. W.; Cropp T. A.; Anderson J. C.; Mukherji M.; Zhang Z.; Schultz P. G. (2003) An expanded eukaryotic genetic code. Science 301, 964–967. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Wang L. (2008) New methods enabling efficient incorporation of unnatural amino acids in yeast. J. Am. Chem. Soc. 130, 6066–6067. [DOI] [PubMed] [Google Scholar]
- Wang W.; Takimoto J. K.; Louie G. V.; Baiga T. J.; Noel J. P.; Lee K. F.; Slesinger P. A.; Wang L. (2007) Genetically encoding unnatural amino acids for cellular and neuronal studies. Nat. Neurosci. 10, 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Brock A.; Chen S.; Chen S.; Schultz P. G. (2007) Genetic incorporation of unnatural amino acids into proteins in mammalian cells. Nat. Methods 4, 239–244. [DOI] [PubMed] [Google Scholar]
- Blight S. K.; Larue R. C.; Mahapatra A.; Longstaff D. G.; Chang E.; Zhao G.; Kang P. T.; Green-Church K. B.; Chan M. K.; Krzycki J. A. (2004) Direct charging of tRNA(CUA) with pyrrolysine in vitro and in vivo. Nature 431, 333–335. [DOI] [PubMed] [Google Scholar]
- Ambrogelly A.; Gundllapalli S.; Herring S.; Polycarpo C.; Frauer C.; Soll D. (2007) Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc. Natl. Acad. Sci. U.S.A. 104, 3141–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T. S.; Ahmad I.; Yin J. A.; Schultz P. G. (2010) An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374. [DOI] [PubMed] [Google Scholar]
- Wang Y. S.; Russell W. K.; Wang Z.; Wan W.; Dodd L. E.; Pai P. J.; Russell D. H.; Liu W. R. (2011) The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of l-phenylalanine and its derivatives. Mol. Biosyst. 7, 714–717. [DOI] [PubMed] [Google Scholar]
- Kavran J. M.; Gundllapalli S.; O’Donoghue P.; Englert M.; Soll D.; Steitz T. A. (2007) Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc. Natl. Acad. Sci. U.S.A. 104, 11268–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa T.; Ishii R.; Fukunaga R.; Kobayashi T.; Sakamoto K.; Yokoyama S. (2008) Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(epsilon)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 15, 1187–1197. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T.; Ishii R.; Fukunaga R.; Kobayashi T.; Sakamoto K.; Yokoyama S. (2008) Crystallographic studies on multiple conformational states of active-site loops in pyrrolysyl-tRNA synthetase. J. Mol. Biol. 378, 634–652. [DOI] [PubMed] [Google Scholar]
- Summerer D.; Chen S.; Wu N.; Deiters A.; Chin J. W.; Schultz P. G. (2006) A genetically encoded fluorescent amino acid. Proc. Natl. Acad. Sci. U.S.A. 103, 9785–9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M.; Soll D. (2000) Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 69, 617–650. [DOI] [PubMed] [Google Scholar]
- Perona J. J.; Hou Y. M. (2007) Indirect readout of tRNA for aminoacylation. Biochemistry 46, 10419–10432. [DOI] [PubMed] [Google Scholar]
- Nozawa K.; O’Donoghue P.; Gundllapalli S.; Araiso Y.; Ishitani R.; Umehara T.; Soll D.; Nureki O. (2009) Pyrrolysyl-tRNA synthetase-tRNA(Pyl) structure reveals the molecular basis of orthogonality. Nature 457, 1163–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto J. K.; Adams K. L.; Xiang Z.; Wang L. (2009) Improving orthogonal tRNA-synthetase recognition for efficient unnatural amino acid incorporation and application in mammalian cells. Mol. Biosyst. 5, 931–934. [DOI] [PubMed] [Google Scholar]
- Hu Q.; Noll R. J.; Li H.; Makarov A.; Hardman M.; Graham Cooks R. (2005) The Orbitrap: a new mass spectrometer. J. Mass Spectrom. 40, 430–443. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800. [Google Scholar]
- Collaborative Computational Project, Number 4. (1994) The CCP4 suite: programs for protein crystallography Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Brunger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. [Google Scholar]
- Rodriguez-Hernandez A.; Bhaskaran H.; Hadd A.; Perona J. J. (2010) Synthesis of Glu-tRNAGln by engineered and natural aminoacyl-tRNA synthetases. Biochemistry 49, 6727–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.