

Abstract
Platelets recruit leukocytes and mediate interactions between leukocytes and endothelial cells. Platelets have been long described as markers of transplant rejection, but the contribution of platelets to transplant rejection has not been critically examined. We now demonstrate that following T-cell initiation of allograft rejection platelets contribute to T-cell recruitment, increased plasma inflammatory mediators, and accelerate T-cell meditated skin graft rejection. Prior work from our lab has shown that platelets secrete glutamate when activated, which then induces platelet thromboxane production by signaling through platelet expressed ionotropic glutamate receptors. Glutamate receptor antagonists therefore represent novel inhibitors of platelet accelerated inflammation. We have found that plasma glutamate is increased in mice that receive skin grafts and that mice treated with glutamate receptor antagonists have improved graft survival and decreased plasma thromboxane, platelet factor 4 (PF4/CXCL4) and IFNγ. Taken together, our work now demonstrates that subsequent to T-cell initiation of skin graft rejection, platelets contribute to further T-cell recruitment and that by blunting glutamate mediated platelet activation, graft survival is improved.
Introduction
According to the National Organ Procurement and Transplantation Network approximately 23,000 Americans received an organ transplant in 2008. Current immune suppression protocols have greatly improved transplant survival, but despite many therapeutic advances allograft rejection continues. Platelets have key roles in the recruitment of immune cells and can accelerate vascular inflammatory diseases (1–5). We have implicated platelets in amplifying alloantibody mediated transplant inflammation (6). However, the role of platelets in T-cell directed immune responses in general, and transplant rejection in particular, have not been explored fully.
Early pathological descriptions of transplants recognized the presence of platelets in kidney transplants (7) and a number of studies followed in which platelets were labeled with indium to track their accumulation in renal allografts (8, 9). These studies led to the general observation that platelets accumulate in renal allografts and may be markers of graft rejection. Recent studies have also identified the presence of prominent intravascular platelet aggregates in experimental and clinical transplants that undergo antibody-mediated rejection (10–13). Kirk and colleagues have gone beyond these more observational studies and made a critical finding that CD154 (CD40L) shed from platelets can serve as a co-stimulatory molecule remote from the transplant to induce rejection of murine cardiac allografts (14). Using a skin transplant model we demonstrated that platelets have a key role in increasing leukocyte trafficking and graft vascular inflammation (6). These studies set the stage for a deeper investigation into platelets not only as markers of transplant vascular injury, but as mediators contributing to the pathogenesis of graft rejection.
Platelets can recruit lymphocytes to the site of the inflammation through contact dependent and independent mechanisms. T-cells express PSGL-1 that interacts with P-selectin expressed by activated platelets. Platelets may also interact with T-cells through CD154 on platelets. In experiments of ischemia-reperfusion injury to the liver, platelets augmented the recruitment of CD4+ T-cells to hepatic sinusoids (15). Activated platelets also enhance the intrahepatic accumulation of cytotoxic T-lymphocytes in murine models of viral hepatitis (16). Platelet and T-cell interactions may do more than just localize T-cells, they may also augment T-cell immune responses. CD154 on platelets can augment the help delivered by low levels of CD4+ T cells for germinal center development and isotype switching to IgG antibody production (17). We have found that the platelet derived chemokine platelet factor 4 (PF4/CXCL4) can increase T-cell CXCR3 expression and T-cell trafficking in a model of neurovascular inflammation (18). These studies provide a rationale to consider the poorly explored role of platelets in T-cell mediated allograft rejection.
Cyclooxygenase (COX) products, such as prostaglandins and thromboxane, have important roles in stimulating and shaping acquired immune responses. Platelets are a major source of these pro-inflammatory molecules. Receptors for COX products are expressed by many immune cells, including T-cells. Recent work has demonstrated that some prostaglandins, such as prostaglandin E2 (PGE2), acting on T-cells facilitate Th1 cell differentiation and amplify IL-23 mediated Th17 cell expansion (19). The same investigators had found earlier that the binding activity for thromboxane receptors (TP) is high in T cells, but not in B cells, suggesting that thromboxane-TP signaling may modulate peripheral T-cell immune responses (20, 21). Studies specific to transplantation have demonstrated that TP−/− mice had reduced immune-mediated tissue injury following cardiac transplantation, showing that thromboxane augments cellular immune responses to transplants and inflammatory tissue injury (22).
We have recently demonstrated that platelets have an important role in alloantibody induced innate immune responses to skin grafts (6). Platelets support leukocyte and endothelial cell interactions and contribute pro-inflammatory molecules that sustain leukocyte recruitment (23). We have also demonstrated that platelets contain and release large concentrations of glutamate upon activation. Platelets express ionotropic glutamate receptors including AMPA and Kainate type receptors (24). Glutamate promotes platelet activation by signaling via platelet glutamate receptors leading to COX activation, the production of thromboxane, and amplification of platelet activation (25). Glutamate induced platelet thromboxane production may do more than just support platelet activation and thrombus growth, it may also promote immune activation, including T-cell driven transplant rejection.
We now demonstrate that platelets have a role in transplant rejection. Using a skin graft model, we found that following T-cell initiation of graft rejection platelets are activated leading to increased plasma glutamate and platelet thromboxane production. Glutamate receptor antagonists demonstrate therapeutic potential in reducing T-cell mediated transplant rejection.
Materials and Methods
Reagents
PF4, TNFα and IFNγ ELISA kits were purchased from R&D Systems. Thromboxane B2 ELISA kits were purchased from Cayman Chemicals. Glutamate concentration was determined using a Glutamate Oxidase kit from Invitrogen. CNQX (6-cyano-7-nitroquinoxaline-2, 3-dione) was purchased from Sigma.
Platelet depletion antibodies and its control IgG were purchased from Emfret Analytics. The depleting antibodies are a mixture of rat monoclonal antibodies directed against mouse GPIb (CD42b). This is a platelet specific receptor and results in the depletion of only platelets. At the concentrations we used (1 μg/g) platelet depletion is sustained for no more than 3 days (Supplementary Figure 1). Monoclonal antibodies to the T-cell receptor (TCR) beta chain (clone H57-597), CD4, CD8 and CD14 were purchased from BD Pharmingen (San Diego, CA).
Transplants
All mouse studies were performed under protocols approved by the University of Rochester Animal Care and Use Committee using procedures we have published (6). Briefly, B6 nude mice (Taconic) were anesthesized with ketamine and xylazine (80/13 mg/kg) and transplant beds prepared by removal of the epidermis and dermis. The thin skin from ears of H-2 incompatible B10.A mice (H-2Kk, Jackson Labs) were grafted into fitted beds on 10–12 week old C57BL/6 nude mice (H-2Kb). Mice were bandaged for 1 week to allow for graft healing and establishment of vascular connections.
Immunohistochemistry
Harvested transplants were fixed in methanol-water-acetic acid (60%-30%-10%). Tissue was then embedded, sectioned, and immuohistochemistry performed using protocols and procedures described previously with antibodies to CD3 (26, 27).
T-cell Isolations
T-cells were isolated from the spleen and lymph nodes of control C57BL/6 mice using a negative selection T-cell enrichment kit (StemCell Technologies). On the initial study day, 1×106 T-cells were injected intravenously via the retro-orbital plexus into graft recipients.
To quantify T-cell infiltrates in the skin grafts we harvested skin from the center of the transplant tissue and placed it in 3 mL of RPMI media with 5% FBS for 1 hr. The base of the graft was then gently scraped with a razor blade, the skin then sectioned into small pieces, passed in and out of an 18 gauge needle and vortexed vigorously to dissociate cells. The cells were transferred into a 15 mL tube, 1 mL of Percoll added, mixed and centrifuged at 1300 × g for 30 mins to isolate a mononuclear cell layer at the interface. Mononuclear cells were then washed before incubating with a monoclonal antibody and the number of cells as per cent total mononuclear cells determined by flow cytometry.
Statistical Methods
Data are expressed as means ± standard deviation. Statistical comparisons between two groups were performed using Student’s t-test. Graft survival was analyzed using a log-rank test.
Results
To determine the role of platelets in T-cell dependent transplant rejection, we used a skin transplant model in which the thin skin from an ear of a B10.A mouse (H-2Kk) is grafted onto the flank of a C57BL/6 nude mouse (H-2Kb). After allowing 7 days for graft healing and establishment of vascular connections, bandages were removed and mice were reconstituted with 1×106 T-cells isolated from naïve wild type C57Bl6 mice. Using this model in our prior published work we have demonstrated that the skin grafts are vascularized and we have imaged both platelet and white blood cell perfusion of the grafts (23). Other have also demonstrated that vessels of the host dermis and the skin graft connect by 7 days after transplantation and that endothelial cells within the graft are of donor origin until about 21 days (28). The time of T-cell reconstitution is referred to as study day 0.
To establish that platelets were activated following T-cell reconstitution and T-cell initiation of graft rejection, plasma was isolated from control ungrafted nude mice, mice that received skin grafts but no T-cells, and from mice that were given skin grafts and were T-cell reconstituted. PF4 is a platelet specific chemokine and was used as a plasma marker of platelet activation. Mice that received skin grafts but no T-cells did not have significantly increased plasma PF4 compared to control nude mice (Fig. 1A, white vs grey bars). However, T-cell reconstituted mice had a large increase in plasma PF4 that was sustained over 4 days after T-cell reconstitution (Fig. 1A, black vs white bars), indicating that T-cells are needed to initiate platelet activation. T-cell reconstitution in the absence of a skin allograft had no effect on platelet activation (Supplemental Fig. 2).
Figure 1.

Activated Platelets Accelerate Transplant Rejection. A. Platelets are activated early in graft rejection. Plasma PF4 was measured in mice without skin grafts, control skin grafted mice (no T-cells) and skin grafted mice that were T-cell reconstituted (n=5 ± S.D. *P<0.03). B. Platelet depleted mice have improved graft survival. Representative skin grafts on study day 7. C. Day 7 graft survival. Mice were T-cell reconstituted and treated with control IgG or T-cell reconstituted and platelet depleted by treatment with monoclonal antibodies to GP1b (*P<0.01). D. Graft Survival Curve. Mice were platelet depleted on day 0 and Day 4 after T-cell reconstitution and graft rejection was monitored daily (*P<0.03).
To determine whether platelet activation plays a role in graft rejection, mice that had received skin grafts were T-cell reconstituted and on study day 0 mice were either treated with control IgG or a mixture of monoclonal antibodies (1 μg/g) against mouse GPIb (CD42b) to deplete platelets. This treatment greatly reduces platelet numbers (Supplemental Fig 1), but has no effect on other cell populations (18, 23, 29). Skin graft survival was then evaluated on day 7 using visual parameters. Total rejection was manifested by scab formation and graft retraction (Fig. 1B). Approximately 50% of control IgG treated mice had visible evidence of graft rejection, whereas less than 20% of platelet depleted mice had rejected skin grafts (Fig. 1C). To look at graft survival over a longer time, we also followed the grafts for 10 days after T-cell reconstitution. In contrast to Figure 1C, to achieve sustained platelet depletion mice were treated with control IgG or platelet depleting antibody on days 0 and 4. By day 10 control graft survival was only 11% (Fig. 1D, white diamonds). In contrast, mice made platelet deficient had 50% graft survival on day 10 (Fig. 1D, black squares). These data demonstrate that following initiation of skin graft inflammation by T-cells, platelets have a significant role in accelerating rejection.
Platelets have well described roles in promoting monocyte trafficking across sites of vascular inflammation (1–3), but the role of platelets and platelet derived inflammatory mediators in supporting T-cell trafficking is less well understood. On study day 7, a large number of CD3 positive cells were noted infiltrating skin grafts (Fig. 2A, left side). However, the number of CD3+ cells was decreased in platelet depleted mice (Fig. 2A, right side). To determine whether platelets increase T-cell trafficking into transplanted tissue, skin grafts from control and platelet depleted mice were collected on study day 5, and mononuclear cell infiltrates were isolated from the graft followed by density centrifugation. T-cells were quantified by flow cytometry with antibodies to TCR (Supplemental Fig. 3). Approximately 35% of the mononuclear cells isolated from the control skin grafts were TCR positive, compared to only 25% in platelet depleted mice (Fig. 2B). These data demonstrate that platelets increase T-cell infiltration into skin grafts.
Figure 2.
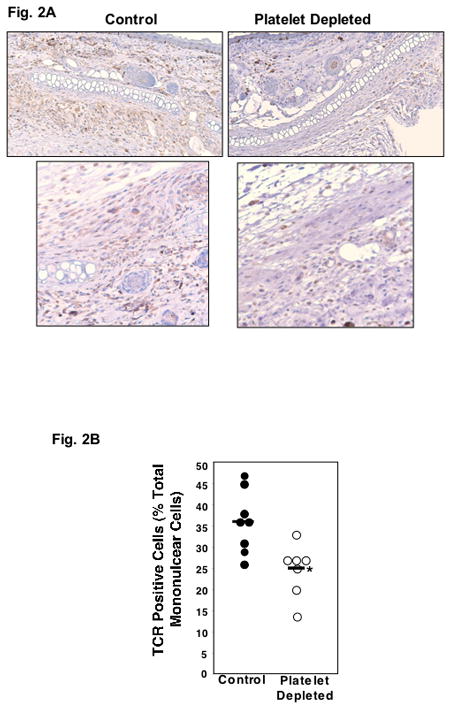
Platelets Promote Graft Inflammation. A. Immunohistochemistry. Skin grafts were immunostained with anti-CD3 antibody. Left side represent control mice, right side platelet depleted mice. B. T-cell Recruitment. On study day 5 mononuclear cells were isolated from control and platelet depleted skin grafts and the number of T-Cell Receptor (TCR) positive cells quantified by FACS analysis (*P<0.03).
Not only may platelets augment T-cell recruitment, they may also influence cytokine production. To examine this, plasma from control and platelet depleted mice was collected on study day 5, and TNFα and INFγ measured using an ELISA for each. Mice that are platelet depleted have significantly less TNFα (Fig. 3A) and INFγ (Fig. 3B) compared to control mice.
Figure 3.
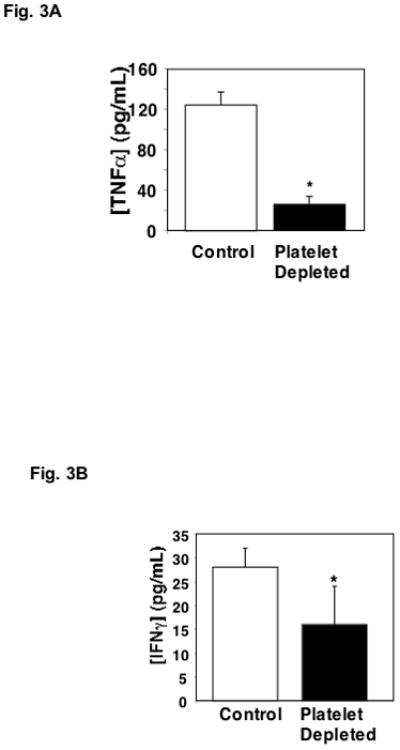
Platelets Promote Cytokine Production. A. Plasma TNFα. Plasma was isolated from control and platelet depleted mice on day 5 and TNFα was measured by ELISA (n=5 ± S.D. *P<0.01). B. Plasma IFNγ. Plasma was isolated from control and platelet depleted mice on day 5 and IFNγ was determined by ELISA (n=5 ± S.D. *P<0.05).
Much past and emerging research has demonstrated the important role of COX products, including thromboxane, in augmenting vascular inflammation and directing T-cell immune responses (20, 22). Platelets are a major source of thromboxane. To determine whether platelets contribute to plasma thromboxane production during graft rejection skin grafted mice were treated with platelet depleting antibody or control IgG at the time of T-cell reconstitution. Plasma was then isolated on day 5 and the stable thromboxane breakdown product TXB2 was measured by ELISA. Mice that were skin grafted and T-cell reconstituted had greatly increased plasma TXB2 compared to control ungrafted mice (Fig. 4A, black vs white). Nude mice with skin grafts but not T-cell reconstituted have the same plasma TXB2 as control mice (Supplemental Fig. 4). Platelet depletion abrogated the increased plasma TXB2 found in T cell reconstituted and skin grafted mice (Fig. 4A, grey vs white) indicating that platelets are an important source of thromboxane during skin graft rejection.
Figure 4.
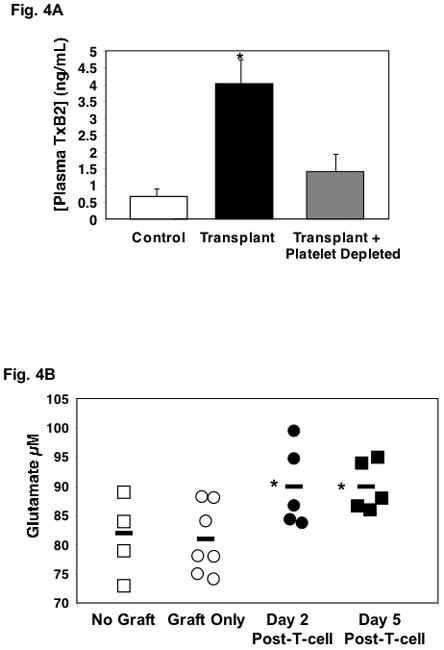
Glutamate Drives Thromboxane Production. A. Platelets are a major source of plasma thromboxane. On study day 5 plasma samples were collected from control mice (no graft) and skin grafted mice that were T-cell reconstituted with or without platelet depletion. TXB2 was quantified by ELISA (n=5–6 ± S.D. *P<0.01 vs control). B. Plasma glutamate is increased transplanted mice. Plasma samples were collected from control mice (no skin graft), mice with skin grafts only (no T-cells) and mice with skin grafts plus T-cells. Glutamate was quantified by an enzymatic assay (*P<0.05).
Platelets release glutamate upon activation (24, 30), and we have shown that glutamate increases platelet reactivity and thromboxane production through platelet ionotropic glutamate receptors (24, 25). Our recent work demonstrated that this is in part due to glutamate mediated stimulation of platelet thromboxane production (25). To determine whether plasma glutamate is increased in skin graft recipients, plasma was isolated from control ungrafted nude mice, mice that were skin grafted but not T-cell reconstituted, and mice that were skin grafted and T-cell reconstituted. Using an enzymatic colorimetric assay, glutamate concentration was determined. Similar to plasma PF4, a skin graft alone did not increase plasma glutamate, however with initiation of rejection, plasma glutamate was significantly increased 2 and 5 days after T cell reconstitution (Fig. 4B).
Because glutamate drives platelet thromboxane production, glutamate receptor antagonists, such as CNQX, may be beneficial in improving graft survival. Our past work has demonstrated that AMPAR antagonists decrease platelet activation, in part by blunting agonist induced thromboxane production (31, 32). Based on these data, glutamate driven platelet thromboxane production may be very important in graft rejection, and CNQX is a potentially valuable compound to ameliorate these effects. To test this, we treated skin graft recipients with either control PBS or 2 mg/kg of CNQX daily by intraperitoneal (ip) injection and graft survival was monitored for 7 days. Similar to our preceding experiments, control PBS treated mice had approximately 65% graft rejection (Fig. 5A, white bar). In contrast, mice treated with CNQX had much improved graft survival, with approximately 35% of skin grafts rejecting (Fig. 5A, black bar). Graft rejection was also observed over a longer time and mice treated with CNQX had significantly improved graft survival (Fig. 5B).
Figure 5.
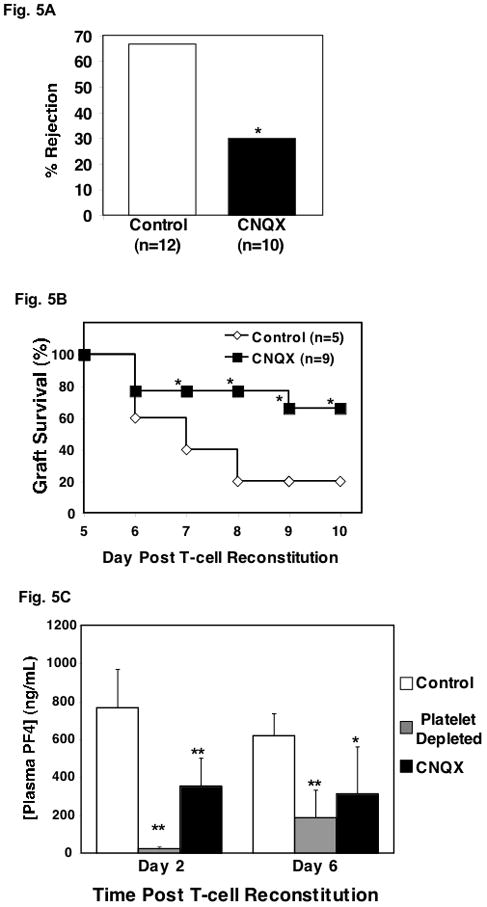
Glutamate Receptor Antagonist Improves Graft Survival. A. T cell reconstituted mice treated with glutamate receptor antagonist have improved skin graft survival on day 7 (*P<0.01). B. Graft survival curve (*P<0.05). C. CNQX reduced platelet activation as measured by PF4 in T cell reconstituted mice with skin grafts. Plasma was isolated from control mice, mice that were platelet depleted and mice treated with CNQX (n=5 ± S.D. *P<0.05, **P<0.01 vs control).
We have reported previously that glutamate receptor signaling helps augment platelet activation and that CNQX is an effective platelet antagonist (24). To determine whether the protective effect of CNQX is in part be due to platelet inhibition, plasma was isolated from control mice, platelet depleted mice, and CNQX treated mice. All of these mice were skin grafted and T-cell reconstituted. As expected at early (day 2) and late (day 6) time points after T-cell reconstitution control mice had elevated plasma PF4 concentrations (Fig. 5C). Platelet depleted mice had greatly reduced plasma PF4 at day 2 and as platelet numbers rebound, the PF4 concentration increased by day 6 (Fig. 5C). CNQX treated mice had significantly reduced plasma PF4 compared to control mice indicating that its protective effect may in part be due to platelet inhibition (Fig. 5C).
Similar to platelet depletion studies the delay in transplant rejection was also reflected in decreased T-cell infiltrates. Staining for CD3 positive T-cells demonstrated fewer T-cells in CNQX treated skin grafts compared to grafts on untreated control mouse grafts (Fig. 6A). Mononuclear cells were also isolated from control and CNQX treated mouse skin grafts, and TCR positive cells as percent of total mononuclear cells was determined by flow cytometry. Approximately 25% of isolated mononuclear cells from control mice were TCR positive (Fig. 6B). The number of infiltrating T-cells was greatly reduced in mice treated with CNQX (Fig. 6B). We also further defined the types of cellular infiltrates by quantifying CD4, CD8 T-cell subsets and macrophages (CD14 positive) by flow cytometry. Platelet depleted mice had reduced numbers of CD4, CD8 and CD14 positive cells, but only CD8+ cells were significantly reduced compared to control skin graft mice (Fig. 6C, grey vs white). All types of cells were significantly reduced in mice treated with CNQX (Fig. 6C, black vs white).
Figure 6.

Glutamate Receptor Antagonist Reduced T-cell Recruitment and Inflammation. A. CNQX treated mice have fewer T-cell infiltrates. Immunohistochemistry with anti-CD3 antibody. B. Quantification of T-cell Infiltrates (*P<0.05). C. CD4, CD8 and CD14 positive cell infiltrates by flow cytometry (n=5 ± S.D. *P<0.05 vs Control). D. Plasma TXB2 (n=5 ± S.D. *P<0.03). E. Plasma IFNγ (n=5 ± S.D. *P<0.03).
If AMPAR antagonists increase graft survival in part by blunting glutamate mediated platelet thromboxane production and immune stimulation, we predicted that CNQX treated mice would have reduced plasma thromboxane and T-cell derived cytokines. To determine this, we isolated plasma from control and CNQX treated mice and measured TXB2 and IFNγ. CNQX significantly reduced plasma TXB2 (Fig. 6D) and IFNγ (Fig. 6E) compared to control mice. These data indicate that inhibition of platelet glutamate receptors increased graft survival in part by directly or indirectly reducing plasma pro-inflammatory mediators.
Discussion
Platelets are dynamic cells with important hemostasis and immune regulatory functions. Our past work used a model in which allo-antibody initiated inflammation in skin grafts to demonstrate an important role for platelets in leukocyte recruitment (6). The present experiments demonstrate that platelets themselves do not initiate skin graft inflammation and rejection, but with T-cell reconstitution and T-cell initiated graft rejection, platelets are activated leading to an increase in plasma thromboxane, IFNγ, augmented T cell infiltrates and graft rejection. Whether platelets are activated by T-cell driven graft vascular inflammation or by direct T-cell interactions is not clear in this study, but our data support a role for platelets in sustaining T-cell recruitment and graft rejection. We also demonstrated that plasma glutamate is increased in mice that received skin grafts and that glutamate receptor antagonists improve graft survival and blunt T-cell responses. These data point to a novel platelet mediated mechanisms of transplant rejection, glutamate mediated platelet COX activation, and a potential clinical utility for glutamate receptor antagonists in prolonging graft survival.
Platelet interactions with innate immune cells are much better defined than platelet and T-cell interactions. However, platelet interactions with T-cells may be just as important. Platelets express numerous surface receptors for contact dependent interactions with T-cells and platelets also secrete a large number of chemokines, cytokines and other inflammatory molecules with the potential to increase T-cell responses in a contact independent manner. Our data indicate that platelets, perhaps via glutamate mediated thromboxane release, may contribute to allograft rejection. It is clear that platelet depleted mice and mice treated with glutamate receptor antagonists have reduced plasma IFNγ. This may indicate an important role for platelets in sustaining T-cell recruitment and activation, and a role for platelets in enhancing IFNγ production in the cycle of allograft inflammation (33, 34). IFNγ results in endothelial cell activation and with it, more platelet activation, T-cell recruitment, and continued IFNγ production. Therefore, by blunting platelet activation this cycle with IFNγ as a central player may be broken.
We have found that as early as 2 days after reconstitution with unsensitized T-cells there is vascular inflammation and platelet activation. Studies have indicated that within 48 hrs after transfer, T-cells may undergo as may as 3 rounds of division (35), and as many as 20% of T-cells may recognize alloantigen (36). Fairchild and co-workers have demonstrated that endogenous CD8 memory T cells in nonsensitized recipients infiltrate cardiac allografts within 24 h of reperfusion (37). In addition, the nude mouse can make IgM alloantibodies that may crosslink MHC and activate complement prior to T-cell reconstitution, helping to rapidly localize T cells to the graft.
In this work we present data to demonstrate a potential role for platelet glutamate receptor signaling stimulation of thromboxane production in transplant recipient mice. This in turn may assist in T-cell recruitment, activation, and transplant rejection. Glutamate has long been known to be an important signaling molecule in the central nervous system, but its role in the periphery is becoming better appreciated. Glutamate receptors are expressed on non-CNS tissue including platelets, and we have shown that platelet glutamate receptors have a role in platelet activation and thromboxane production (24, 25, 30, 38). We have also demonstrated that in a large cohort of platelet donors a block of polymorphisms in the Kainate type glutamate receptor is associated with a decrease in patient plasma thromboxane and platelet aggregation (25). These data demonstrate a potentially important clinical relevance for this class of receptors. COX enzymes are expressed by many cells, but platelets are a major source of COX derived inflammatory molecules that contribute to vascular inflammatory diseases such as atherosclerosis. Thomboxane receptor signaling has also been shown to be important in graft rejection as TP receptor knockout mice have improved heart allograft survival (22, 39).
Thromboxane represents a single platelet derived inflammatory molecule that may have a role in further T-cell recruitment. Other platelet derived inflammatory molecules may be just as important. For example, PF4 alone or in concert with other chemokines such as CCL5 (RANTES) might also increase T cell infiltrates into the skin grafts. Platelet α-granules also contain numerous cytokines such as IL-1β, TGF-β and TNF-α. These cytokines have multiple effects in transplants, and of particular relevance to platelets are the effects of these cytokines on vascular endothelial cells. For example, TNF-α stimulates the exocytosis of von Willebrand factor and P-selectin from Weible-Palade storage granules in endothelial cells (40, 41). The increased expression of these adhesion molecules promotes platelet activation and attachment that may then increase T-cell recruitment and trafficking.
Based on these data we propose that in our model T-cells initiate vascular inflammation resulting in platelet recruitment, platelet activation, elaboration of glutamate, platelet thromboxane production, and sustained platelet activation and T-cell recruitment. Despite some attention in the 1970’s and early 1980’s, the role of platelets in transplant rejection has not been closely examined. Platelets are much more numerous than other immune cells and when stimulated release or produce large quantities of pro-inflammatory molecules. Because of their vital role in hemostasis this important function has been greatly overlooked. There are few platelet antagonists currently in clinical use. These are used mainly for people at risk of heart attack or stroke, and platelet inhibitors are now a major part of reducing the risk of cardiovascular events. Our work demonstrates a potential role for platelet derived thromboxane in T-cell recruitment and graft rejection, further underscoring the possible utility of common drugs like aspirin and ibuprofen and perhaps glutamate receptor antagonists to block COX upstream signaling. Thromboxane and other COX products such as prostaglandins are known to direct T-cell responses (19, 20). One of the largest and most rapid sources for induction of thromboxane and prostaglandins is platelets. Our work implies that this may be an important area for future clinical study, and novel platelet inhibitors such as glutamate receptor antagonists may be of potential clinical utility in targeting diverse vascular inflammatory diseases, including transplantation.
Although small and anucleate, platelets are dynamic cells capable of diverse functions in vascular biology. Our studies indicate platelets may have a role in enhancing T-cell allograft immune responses. Further basic science and clinical studies are needed to fully elucidate the most relevant pathways, but this work indicates that inhibiting platelet glutamate receptor signaling may be one pathway of note.
Supplementary Material
Acknowledgments
Funding Sources: R01HL093179, R01HL093179-02S109 and R01HL094547 to CNM; WMB is supported by P01AI087586-01.
References
- 1.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 2.Huo Y, Ley KF. Role of platelets in the development of atherosclerosis. Trends Cardiovasc Med. 2004;14:18–22. doi: 10.1016/j.tcm.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003;101:2661–2666. doi: 10.1182/blood-2002-07-2209. [DOI] [PubMed] [Google Scholar]
- 4.Smyth SS, McEver RP, Weyrich AS, Morrell CN, Hoffman MR, Arepally GM, French PA, Dauerman HL, Becker RC. Platelet functions beyond haemostasis. J Thromb Haemost. 2009 doi: 10.1111/j.1538-7836.2009.03586.x. [DOI] [PubMed] [Google Scholar]
- 5.Kirk AD, Morrell CN, Baldwin WM., 3rd Platelets influence vascularized organ transplants from start to finish. Am J Transplant. 2009;9:14–22. doi: 10.1111/j.1600-6143.2008.02473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrell CN, Murata K, Swaim AM, Mason E, Martin TV, Thompson LE, Ballard M, Fox-Talbot K, Wasowska B, Baldwin WM., 3rd In Vivo Platelet-Endothelial Cell Interactions in Response to Major Histocompatibility Complex Alloantibody. Circ Res. 2008 doi: 10.1161/CIRCRESAHA.107.170332. [DOI] [PubMed] [Google Scholar]
- 7.Porter KA. Morphological Aspects Of Renal Homograft Rejection. Br Med Bull. 1965;21:171–175. doi: 10.1093/oxfordjournals.bmb.a070388. [DOI] [PubMed] [Google Scholar]
- 8.Fenech A, Nicholls A, Smith FW. Indium (111In)-labelled platelets in the diagnosis of renal transplant rejection: preliminary findings. Br J Radiol. 1981;54:325–327. doi: 10.1259/0007-1285-54-640-325. [DOI] [PubMed] [Google Scholar]
- 9.Oluwole S, Wang T, Fawwaz R, Satake K, Nowygrod R, Reemtsma K, Hardy MA. Use of indium-111-labeled cells in measurement of cellular dynamics of experimental cardiac allograft rejection. Transplantation. 1981;31:51–55. doi: 10.1097/00007890-198101000-00012. [DOI] [PubMed] [Google Scholar]
- 10.Wasowska BA, Qian Z, Cangello DL, Behrens E, Van Tran K, Layton J, Sanfilippo F, Baldwin WM., 3rd Passive transfer of alloantibodies restores acute cardiac rejection in IgKO mice. Transplantation. 2001;71:727–736. doi: 10.1097/00007890-200103270-00007. [DOI] [PubMed] [Google Scholar]
- 11.Ota H, Fox-Talbot K, Hu W, Qian Z, Sanfilippo F, Hruban RH, Baldwin WM., 3rd Terminal complement components mediate release of von Willebrand factor and adhesion of platelets in arteries of allografts. Transplantation. 2005;79:276–281. doi: 10.1097/01.tp.0000146195.76904.d3. [DOI] [PubMed] [Google Scholar]
- 12.Nakashima S, Qian Z, Rahimi S, Wasowska BA, Baldwin WM., 3rd Membrane attack complex contributes to destruction of vascular integrity in acute lung allograft rejection. J Immunol. 2002;169:4620–4627. doi: 10.4049/jimmunol.169.8.4620. [DOI] [PubMed] [Google Scholar]
- 13.Wehner J, Morrell CN, Reynolds T, Rodriguez ER, Baldwin WM., 3rd Antibody and complement in transplant vasculopathy. Circ Res. 2007;100:191–203. doi: 10.1161/01.RES.0000255032.33661.88. [DOI] [PubMed] [Google Scholar]
- 14.Xu H, Zhang X, Mannon RB, Kirk AD. Platelet-derived or soluble CD154 induces vascularized allograft rejection independent of cell-bound CD154. J Clin Invest. 2006;116:769–774. doi: 10.1172/JCI27155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khandoga A, Hanschen M, Kessler JS, Krombach F. CD4+ T cells contribute to postischemic liver injury in mice by interacting with sinusoidal endothelium and platelets. Hepatology. 2006;43:306–315. doi: 10.1002/hep.21017. [DOI] [PubMed] [Google Scholar]
- 16.Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, Chisari FV, Ruggeri ZM, Guidotti LG. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat Med. 2005;11:1167–1169. doi: 10.1038/nm1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elzey BD, Sprague DL, Ratliff TL. The emerging role of platelets in adaptive immunity. Cell Immunol. 2005;238:1–9. doi: 10.1016/j.cellimm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 18.Srivastava K, I, Cockburn A, Swaim A, Thompson LE, Tripathi A, Fletcher CA, Shirk EM, Sun H, Kowalska MA, Fox-Talbot K, Sullivan D, Zavala F, Morrell CN. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe. 2008;4:179–187. doi: 10.1016/j.chom.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, Sugimoto Y, Narumiya S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med. 2009;15:633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 20.Narumiya S. Prostanoids in immunity: roles revealed by mice deficient in their receptors. Life Sci. 2003;74:391–395. doi: 10.1016/j.lfs.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 21.Ushikubi F, Aiba Y, Nakamura K, Namba T, Hirata M, Mazda O, Katsura Y, Narumiya S. Thromboxane A2 receptor is highly expressed in mouse immature thymocytes and mediates DNA fragmentation and apoptosis. J Exp Med. 1993;178:1825–1830. doi: 10.1084/jem.178.5.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas DW, Rocha PN, Nataraj C, Robinson LA, Spurney RF, Koller BH, Coffman TM. Proinflammatory actions of thromboxane receptors to enhance cellular immune responses. J Immunol. 2003;171:6389–6395. doi: 10.4049/jimmunol.171.12.6389. [DOI] [PubMed] [Google Scholar]
- 23.Morrell CN, Murata K, Swaim AM, Mason E, Martin TV, Thompson LE, Ballard M, Fox-Talbot K, Wasowska B, Baldwin WM., 3rd In vivo platelet-endothelial cell interactions in response to major histocompatibility complex alloantibody. Circ Res. 2008;102:777–785. doi: 10.1161/CIRCRESAHA.107.170332. [DOI] [PubMed] [Google Scholar]
- 24.Morrell CN, Sun H, Ikeda M, Beique JC, Swaim AM, Mason E, Martin TV, Thompson LE, Gozen O, Ampagoomian D, Sprengel R, Rothstein J, Faraday N, Huganir R, Lowenstein CJ. Glutamate mediates platelet activation through the AMPA receptor. J Exp Med. 2008;205:575–584. doi: 10.1084/jem.20071474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun H, Swaim A, Herrera JE, Becker D, Becker L, Srivastava K, Thompson LE, Shero MR, Perez-Tamayo A, Suktitpat B, Mathias R, Contractor A, Faraday N, Morrell CN. Platelet Kainate Receptor Signaling Promotes Thrombosis by Stimulating Cyclooxygenase Activation. Circ Res. 2009 doi: 10.1161/CIRCRESAHA.109.198861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, Wasowska BA, Baldwin WM, 3rd, Pober JS, Lowenstein CJ. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc Natl Acad Sci U S A. 2007;104:1301–1306. doi: 10.1073/pnas.0602035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murata K, Fox-Talbot K, Qian Z, Takahashi K, Stahl GL, Baldwin WM, 3rd, Wasowska BA. Synergistic deposition of C4d by complement-activating and non-activating antibodies in cardiac transplants. Am J Transplant. 2007;7:2605–2614. doi: 10.1111/j.1600-6143.2007.01971.x. [DOI] [PubMed] [Google Scholar]
- 28.de Waal RM, Bogman MJ, Cornelissen IM, Vermeulen AN, Koene RA. Expression of donor class I major histocompatibility antigens on the vascular endothelium of mouse skin allografts. Transplantation. 1986;42:178–183. doi: 10.1097/00007890-198608000-00015. [DOI] [PubMed] [Google Scholar]
- 29.Kisucka J, Butterfield CE, Duda DG, Eichenberger SC, Saffaripour S, Ware J, Ruggeri ZM, Jain RK, Folkman J, Wagner DD. Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc Natl Acad Sci U S A. 2006;103:855–860. doi: 10.1073/pnas.0510412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aliprandi A, Longoni M, Stanzani L, Tremolizzo L, Vaccaro M, Begni B, Galimberti G, Garofolo R, Ferrarese C. Increased plasma glutamate in stroke patients might be linked to altered platelet release and uptake. J Cereb Blood Flow Metab. 2005;25:513–519. doi: 10.1038/sj.jcbfm.9600039. [DOI] [PubMed] [Google Scholar]
- 31.Sun H, Swaim A, Herrera JE, Becker D, Becker L, Srivastava K, Thompson LE, Shero MR, Perez-Tamayo A, Suktitipat B, Mathias R, Contractor A, Faraday N, Morrell CN. Platelet kainate receptor signaling promotes thrombosis by stimulating cyclooxygenase activation. Circ Res. 2009;105:595–603. doi: 10.1161/CIRCRESAHA.109.198861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrell CN, Sun H, Ikeda M, Beique JC, Swaim AM, Mason E, Martin TV, Thompson LE, Gozen O, Ampagoomian D, Sprengel R, Rothstein J, Faraday N, Huganir R, Lowenstein CJ. Glutamate mediates platelet activation through the AMPA receptor. J Exp Med. 2008 doi: 10.1084/jem.20071474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koh KP, Wang Y, Yi T, Shiao SL, Lorber MI, Sessa WC, Tellides G, Pober JS. T cell-mediated vascular dysfunction of human allografts results from IFN-gamma dysregulation of NO synthase. J Clin Invest. 2004;114:846–856. doi: 10.1172/JCI21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Burns WR, Tang PC, Yi T, Schechner JS, Zerwes HG, Sessa WC, Lorber MI, Pober JS, Tellides G. Interferon-gamma plays a nonredundant role in mediating T cell-dependent outward vascular remodeling of allogeneic human coronary arteries. Faseb J. 2004;18:606–608. doi: 10.1096/fj.03-0840fje. [DOI] [PubMed] [Google Scholar]
- 35.Min B, Paul WE. Endogenous proliferation: burst-like CD4 T cell proliferation in lymphopenic settings. Semin Immunol. 2005;17:201–207. doi: 10.1016/j.smim.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166:973–981. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- 37.Schenk AD, Nozaki T, Rabant M, Valujskikh A, Fairchild RL. Donor-reactive CD8 memory T cells infiltrate cardiac allografts within 24-h posttransplant in naive recipients. Am J Transplant. 2008;8:1652–1661. doi: 10.1111/j.1600-6143.2008.02302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrarese C, Tremolizzo L, Rigoldi M, Sala G, Begni B, Brighina L, Ricci G, Albizzati MG, Piolti R, Crosti F, Dalpra L, Frattola L. Decreased platelet glutamate uptake and genetic risk factors in patients with Parkinson’s disease. Neurol Sci. 2001;22:65–66. doi: 10.1007/s100720170049. [DOI] [PubMed] [Google Scholar]
- 39.Rocha PN, Plumb TJ, Crowley SD, Coffman TM. Effector mechanisms in transplant rejection. Immunol Rev. 2003;196:51–64. doi: 10.1046/j.1600-065x.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 40.Jurd KM, Stephens CJ, Black MM, Hunt BJ. Endothelial cell activation in cutaneous vasculitis. Clin Exp Dermatol. 1996;21:28–32. [PubMed] [Google Scholar]
- 41.Sun RJ, Muller S, Wang X, Zhuang FY, Stoltz JF. Regulation of von willebrand factor of human endothelial cells exposed to laminar flows: an in vitro study. Clin Hemorheol Microcirc. 2000;23:1–11. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.