

Abstract
Estrogen metabolism and growth factor signaling pathway genes play key roles in breast cancer development. We evaluated associations between breast cancer and tagging SNPs of 107 candidate genes of these pathways using single allele- and haplotype-based tests. We first sought concordance of associations between two study populations: the Nashville Breast Cohort (510 cases, 988 controls), and the Cancer Genetic Markers of Susceptibility breast cancer study (1,145 cases, 1,142 controls). Findings across the two study populations were concordant at tagging SNPs of six genes, and at previously published SNPs of FGFR2. We sought further replication of results for EGFR, NCOA7, and FGFR2 in the independent Collaborative Breast Cancer Study (1,552 cases, 1,185 controls). Associations at NCOA7 and FGFR2 replicated across all three studies. The association at NCOA7 on 6q22.32, detected by a haplotype spanning the initial protein-coding exon (5′ - rs9375411, rs11967627, rs549438, rs529858, rs490361, rs17708107 - 3′), has not been previously reported. The haplotype had a significant inverse association with breast cancer in each study (ORHet 0.69 (NBC), 0.76 (CGEMS), 0.79 (CBCS)), and a meta-analysis ORHet of 0.75 (95% CI 0.65-0.87, P = 1.4 × 10-4) in the combined study populations. The haplotype frequency was 0.07 among cases, and 0.09 among controls; homozygotes were infrequent and each ORHom was not significant. NCOA7 encodes a nuclear receptor co-activator that interacts with estrogen receptor α to modulate its activity. These observations provide consistent evidence that genetic variants at the NCOA7 locus may confer a reduced risk of breast cancer.
Keywords: Breast cancer, estrogen, genetic susceptibility, polymorphism, association, growth factor, benign breast disease
Introduction
Breast cancer is one of the most common malignancies among women worldwide. Epidemiological studies show that first-degree female relatives of women with breast cancer are at approximately two-fold increased risk of developing the disease, and twin studies suggest that 27% of risk for breast cancer is heritable (1). The comprehensive identification of both risk and protective alleles may yield important risk prediction models (2, 3). Mendelian, high- and moderate-penetrance breast cancer susceptibility genes together account for approximately one fifth of the observed heritable risk for breast cancer (4-13). Susceptibility alleles identified in genome-wide association studies (GWAS) have smaller effects on relative breast cancer risk (1.2 to 1.7 fold relative risks), but have a relatively high population prevalence with allele frequencies of 5% to 50% (12). At present, these account for approximately 8% of observed heritable risk (13-22). Several variants identified by GWAS of breast cancer are in genes of growth factor signaling and estrogen metabolism pathways, pathways previously known to play key roles in the development of breast cancer.
We describe an ongoing multi-stage association study of breast cancer risk investigating candidate genes of growth factor signaling and estrogen metabolism pathways. We employed a tagging SNP approach to efficiently detect genetic variation associated with breast cancer based upon known patterns of linkage disequilibrium among regional SNPs. In contrast to prior studies using a single SNP allele approach, we additionally employed a systematic haplotype-based analytic strategy to better capture ancestral genetic diversity for tests of association. We inferred the extant haplotypes of the study population at a given region of the genome, representing the ancestral versions of the region, any of which could harbor a disease-associated genetic variant. Haplotypes can more directly mark a disease variant than individual SNP alleles, and enable more powerful tests of association. Statistical multiple comparisons were addressed through sequential replication of observations across independent study populations. Our principal study population is a cohort of women with pathologically defined forms of benign breast disease, among them women with benign proliferative breast disease at increased risk of subsequently developing invasive breast cancer. This enabled correlation of identified genetic risk factors for breast cancer with presence of potential histological precursor lesions.
Materials and Methods
Overview
We conducted a nested case-control investigation of women within the Nashville Breast Cohort (NBC) (23-26), a longitudinal study of women diagnosed with benign breast disease, a subset of whom later developed invasive breast cancer. We evaluated associations between breast cancer and common SNPs in 107 candidate genes in the NBC study population, and compared the results to parallel analyses from the Cancer Genetic Markers of Susceptibility (CGEMS) breast cancer study data (16), seeking concordant observations. We further replicated the concordant association of variants at three genes with breast cancer risk in the independent Collaborative Breast Cancer Study (CBCS) (27). Finally, we investigated potential interaction of the genetic and benign proliferative breast disease risk factors among women of the NBC.
Study Subjects
The Nashville Breast Cohort (NBC) is an ongoing, retrospective cohort study of 17,030 predominantly Caucasian women who underwent a breast biopsy that revealed benign parenchyma or fibroadenoma at Vanderbilt, St. Thomas, and Baptist Hospitals in Nashville, Tennessee from 1954 to the present. Benign proliferative breast lesions carry an increased risk for subsequent invasive breast cancer, and are believed to be a non-obligate precursor lesion (28-30). Roughly a third of the subjects of the cohort had this form of benign breast disease. Extensive histopathological data and benign breast disease biopsies accompany each of the subjects of the NBC. The source of germline DNA for NBC subjects is the archival (FFPE) benign tissue biopsy. Additional details on the NBC have been published elsewhere (24, 26). Entry biopsy FFPE blocks and complete follow up were available for 8,897 women of the cohort, of whom 575 developed incident breast cancer and were Caucasian. The mean age of cases of the NBC at benign breast biopsy was 45 (range 16 to 75) and at breast cancer diagnosis was 61 (range 32 to 96); 80% were postmenopausal at breast cancer diagnosis. We performed a nested case-control study, as previously described (23). Briefly, for each case we selected two controls from the risk set of women who had not been diagnosed with breast cancer in a similar period of observation. These controls were selected without replacement. Controls were matched to cases by age, race, and year of entry biopsy. DNA extraction was done by standard paraffin removal, proteinase K digestion, phenol/chloroform extraction, and ethanol precipitation. Successful DNA extractions from benign archival entry biopsy specimens were performed for 549 of the 575 Caucasian cases (95.5%). A total of 510 of the 549 cases were matched to controls for whom DNA extractions were also successful. Successful DNA extraction was achieved for 988 of the 1020 selected controls (96.9%). The study included a total of 478 trios (1 case matched to 2 controls), and 32 pairs (1 case matched to 1 control). The original archival FFPE biopsy blocks (benign tissue) that were used for DNA extraction generally were quite old: 13.3% date to the 1950s, 28.3% to the 1960s, 39.3% to the 1970s, 15.5% to the 1980s, 3.5% to the 1990s, and 0.1% to the 2000s (2002 was the most recent).
Two independent Caucasian study populations were used for replication of significant associations identified in the NBC, both well described within the published literature. The first is the CGEMS GWAS with 1,145 postmenopausal breast cancer cases and 1,142 controls from the Nurses' Health Study (16). This was an existing data set available for our analysis. Age at diagnosis for CGEMS cases was provided in five-year intervals, with a median interval of 65-69. The second study population is the Collaborative Breast Cancer Study, for which buccal cell DNA samples were available for our genotyping for 1,552 cases and 1,185 controls (27). The mean age at diagnosis of cases of the CBCS was 54 (range 28-73); 59% were postmenopausal at breast cancer diagnosis.
Selection of Candidate Genes and Variants
We selected genes in estrogen metabolism pathways and growth factor signaling pathways using databases of gene function, of biochemical pathways, and of protein-protein interaction (BioCarta, KEGG, OMIM, Entrez Gene, BIND, and HPRD). These searches yielded a set of 870 candidate genes in the targeted pathways. Among these, 107 were selected for this first-round investigation (others remain under ongoing study). We selected tagging SNPs based upon HapMap data for Caucasian (CEU) subjects (31). We identified HapMap SNPs within a gene and 20 kb flanking intervals, omitting SNPs with a minor allele frequency < 0.05 or within non-unique sequences. SNPs were assigned to linkage disequilibrium (LD) bins using LDSelect (r2 ≥ 0.8) (32). A SNP categorized as a candidate functional SNP or represented on Illumina GWAS chips was preferentially selected as the tag for a given LD bin. Tagging SNPs for each gene are provided in SupplementaryTable 1.
Genotyping of NBC and CBCS Subjects
Genotypes of CGEMS subjects became available as an in silico dataset from the National Institutes of Health during the course of this study, while genotypes of NBC and CBCS subjects were directly assayed. Genotyping was accomplished using the Illumina GoldenGate assay (Illumina, San Diego, CA). GoldenGate chemistry combines allele-specific primer extension, adapter ligation, and universal amplification of a highly multiplexed assay on a bead-based microarray system (33, 34). Genotyping of 1,613 SNPs was attempted in NBC subjects, encompassing the set of 107 genes; 1,469 SNPs (91%) successfully converted for assay (given in Supplementary Table 1). Saliva DNA samples were genotyped for a subset of 56 living NBC study subjects for comparison to the genotype of the original FFPE benign breast disease tissue block (dating from 1963 to 1994) of the same subjects. FFPE DNA genotyping fidelity was estimated to be 99.5%: 81,692 of 82,096 saliva DNA / FFPE DNA genotype pairs were concordant. On average, each 96-well plate of DNA that was genotyped contained 3.3 duplicate DNA samples of these pairs. For CBCS study DNA samples, each 96 well plate contained an average of 5.4 duplicate DNA samples (231 duplicate pairs in total), and 20,534 of 20,585 genotype pairs (99.8%) were concordant. We successfully obtained 2,628,086 of 2,636,946 (99.7%) attempted genotypes of the NBC and CBCS study subjects.
Statistical Analysis
We implemented BEAGLE version 3.1.0 (35, 36) within custom software to impute missing genotypes and determine haplotypes for each sample, across each gene. Systematic tests of association were then performed for sliding windows of SNPs across each gene. These windows varied in width from 1 to 10 SNPs; “sliding” involved scanning a given window consisting of a fixed number of SNPs along a gene one SNP at a time. At each step a test of association with breast cancer was performed. Thus both single allele- (one SNP) and haplotype-based analyses (two to ten SNPs) were conducted. Haplotype-based analytic strategies can be more powerful when the disease variant is not directly tagged (37-40). Overlapping windows are typically observed to redundantly detect a given association with this range of window sizes. Where a given genotype assay failed for a NBC or CBCS study subject, we imputed the missing data if it could be assigned with probability of 1.0. After imputation, only 0.16% of genotypes were missing in these subjects. Subjects with missing genotypes for a given haplotype were excluded from analysis. Genotypes of SNPs assayed in the NBC and CBCS studies, but not directly assayed in the CGEMS study, were imputed for CGEMS subjects by BEAGLE with a mean probability of 0.94 (35, 36). To accomplish this, BEAGLE used the known genotypes for NBC, CGEMS and reference HapMap CEU trios for a given gene, as well as HapMap genotypes of 500 SNPs to each flank as additional input.
Conditional logistic regression analyses of the NBC data were used to estimate breast cancer odds ratios (ORs), adjusted for age at entry biopsy and year of entry biopsy. Unconditional logistic regression was used to calculate these ORs in the CGEMS and CBCS studies, adjusted for age. The ORs, 95% confidence intervals (CIs), and P-values were derived under a model that included two β parameters to concurrently assess the risk in those heterozygous or homozygous for the allele or haplotype under consideration. Associations between genotype and breast cancer were considered to be nominally significant if the associated two-sided P value was less than 0.05. The associations of particular interest in this paper were those that were significant in all three study populations. Note that haplotypes consist of alleles of multiple SNPs on the same chromosome. Subjects who are homozygous for a haplotype are homozygous at each SNP of the haplotype. Subjects who are heterozygous have one copy of the haplotype and one of a distinct haplotype; the subject must be heterozygous at a least one of the haplotype's SNPs. The combined analyses of all three data sets were performed in two ways. First, we combined all three studies together for each haplotype using a fixed-effects meta-analysis (41). An accompanying test of inter-study heterogeneity for all odds ratios of FGFR2 and of NCOA7 was not significant, but was significant for six of eighteen odds ratios of EGFR (Table 2). These tests of heterogeneity used a χ2 statistic derived by the metan program (42). Second, we analyzed data of all three studies combined using unconditional logistic regression, adjusting for age and study site. For NBC women in this combined analyses, individual matching of cases to controls was ignored. This second analysis was highly consistent with the first analysis, with somewhat more significant P values.
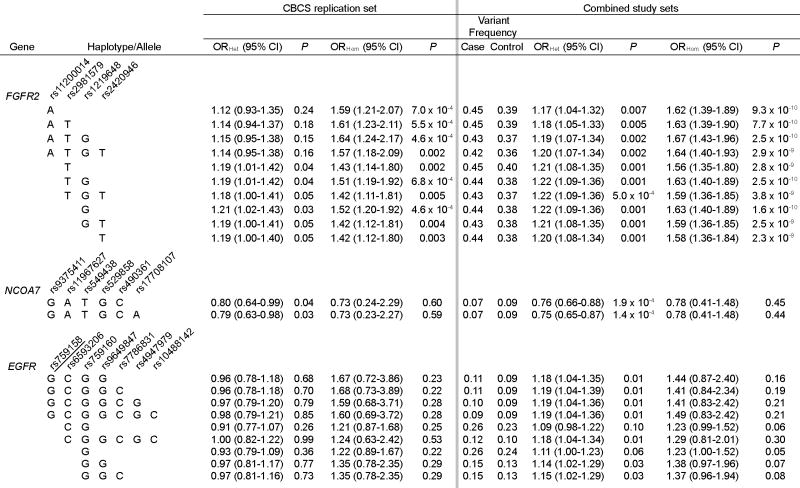
Table 2. Associations between variants of FGFR2, NCOA7, and EGFR and breast cancer risk: CBCS, and combined studies (NBC, CGEMS, CBCS).
|
Each table row for a given gene presents a haplotype/allele for which tests of association were concordantly significant in the NBC and CGEMS study populations. The underlined SNP of EGFR was not genotyped in the CGEMS study, but was imputed with probability 0.79. Results of the combined studies are derived from meta-analysis.
A variant that concordantly replicated as a breast cancer risk factor within the NBC, CGEMS, and CBCS studies was then further analyzed for evidence of statistical interaction with a history of benign proliferative breast disease. Benign breast disease histology was uniquely available for subjects of the NBC, all of whom had forms of biopsy-proven benign breast disease. We derived the combined effects on breast cancer risk of haplotype and proliferative disease (PD) in the patient's entry biopsy. These models contained two parameters for the heterozygous or homozygous haplotype under consideration, a parameter for PD, two interaction parameters for the joint effects of heterozygous and homozygous haplotypes with PD, age at entry biopsy, and year of entry biopsy.
Results
We employed sliding window single allele- and haplotype-based tests to systematically seek concordant associations with breast cancer risk in both the NBC and CGEMS breast cancer studies. We evaluated tagging SNPs of 107 candidate genes, as well as previously published GWAS SNPs in FGFR2 and ESR1. Figure 1 presents the log-transformed significance of all sliding window tests conducted within the NBC and CGEMS studies. These included all single allele- and haplotype-based tests at each gene. Genetic variants of six genes (EGFR, ESRRG, NCOA2, NCOA7, PRMT8, and VDR), as well as the GWAS SNPs in FGFR2, were associated with breast cancer risk in both studies (Figure 1, red highlight for CGEMS, green highlight for NBC). ESR1 GWAS SNPs (rs10872676, rs2046210, and rs3020314) were not significant in either study population. All concordant results of NBC and CGEMS study subjects among these 107 candidate genes are detailed in Table 1 and in Supplementary Table 2.
Figure 1.

Association of breast cancer with evaluated estrogen and growth factor signaling pathway genes. Data of the Nashville Breast Cohort (NBC) and of the CGEMS breast cancer study are jointly illustrated. A Manhattan plot shows −log10(P-values) from sliding-window analysis of tagging SNPs of 107 genes, and of published GWAS SNPs in ESR1 and FGFR2. Windows ranged in width from 1 SNP (single allele analysis) to 10 SNPs (haplotype analysis). All statistical tests conducted in NBC and CGEMS study data are plotted. A given data point depicted at a SNP may represent either a single allele test, or a test of a haplotype beginning at the SNP and that extends to the right (corresponding to the window width). Data points include both those for the heterozygous and for the homozygous state for each variant. Data points plotted above the horizontal midline correspond to a test with an OR >1, while those plotted below it correspond to a test with an OR <1. Highlighted data points in 7 genes (named in solid boxes) each designate a genetic variant that was significant within NBC data (green) and also within the corresponding analysis of CGEMS data (red). Black/blue shading distinguishes adjacent genes of the figure.
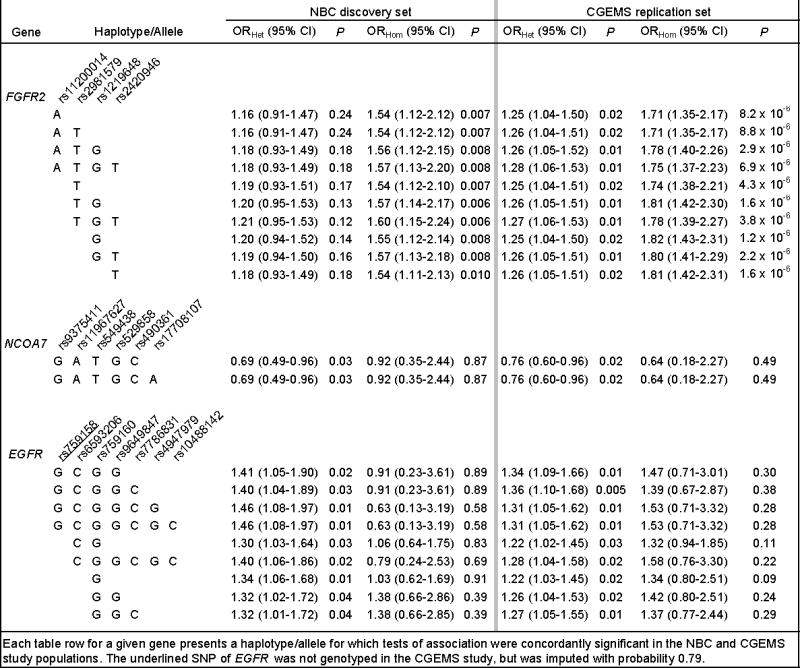
Table 1. Associations between variants of FGFR2, NCOA7, and EGFR and breast cancer risk: NBC, and CGEMS.
|
Each table row for a given gene presents a haplotype/allele for which tests of association were concordantly significant in the NBC and CGEMS study populations. The underlined SNP of EGFR was not genotyped in the CGEMS study, but was imputed with probability 0.79.
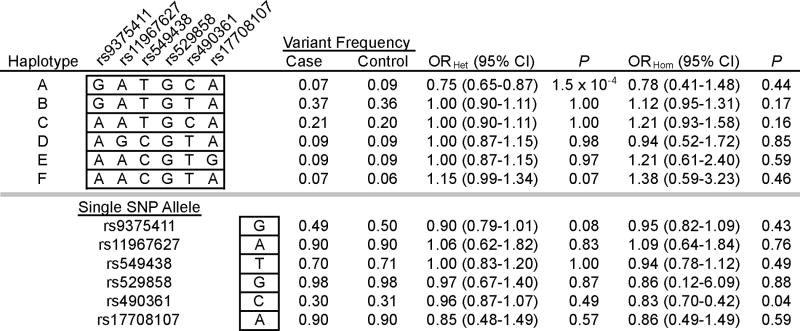
We completed analysis of tagging SNPs of two of the six genes, EGFR and NCOA7, as well as the GWAS SNPs of FGFR2, in the independent CBCS population. The remaining four genes shown in Supplementary Table 2 are pending further investigation. Table 1 presents nominally significant tests of association for EGFR, NCOA7, and FGFR2 in the NBC and CGEMS studies; Table 2 presents results for the CBCS study, and for a meta-analysis of all three studies combined. Only FGFR2 and NCOA7 were associated with breast cancer in the independent CBCS study population. FGFR2 carries known susceptibility variants confirmed in prior GWAS (rs11200014, rs2981579, rs1219648, and rs2420946), whereas the association at NCOA7 on 6q22.32 has not been previously reported. Six common NCOA7 haplotypes are detected by rs9375411, rs11967627, rs549438, rs529858, rs490361, and rs17708107; one haplotype was consistently more common among controls than among cases (Tables 1, 2, and 3). This haplotype spans the initial protein-coding exon and its flanking introns, in a region of intermediate LD that extends to the neighboring gene (HEY2 and HINT3, see Supplementary Figure 1). This NCOA7 haplotype had a significant inverse association with breast cancer in each study (ORHet of 0.69 (NBC), 0.76 (CGEMS), 0.79 (CBCS)); meta-analysis combining all three studies yielded an ORHet of 0.75 (P = 1.4 × 10-4). The test of heterogeneity of these study odds ratios studies was not significant (P = 0.77). An alternative logistic regression analysis of the combined data of these study populations that adjusted for age and for study site provided similar results (for example, the NCOA7 haplotype GATGCA yielded an ORHet of 0.75 (95% CI 0.65-0.87, P = 1.4 × 10-4). A test for trend based on a simple additive logistic regression model of the combined data of the studies, adjusted for age, yielded an OR of 0.77 (95% CI 0.68-0.88, P = 1.4 × 10-4). With a control frequency of 0.09, homozygotes for the NCOA7 haplotype were infrequent and our power was limited to accurately estimate the effect of the homozygous state.
Table 3.
NCOA7 haplotype and single SNP analyses.
|
Results are those of a meta-analysis of the combined NBC, CGEMS and CBCS study populations.
We investigated the hypothesis that either of two replicating genetic variants (NCOA7 GATGCA haplotype heterozygote or the FGFR2 rs1219648 G allele homozygote) might alter risk of breast cancer associated with a history of PD in NBC subjects. Women with a history of PD had a 66% excess risk of breast cancer (OR = 1.66; 95% CI, 1.32 – 2.10; P = 1.7×10-5). Among women without PD, those who were heterozygous for the NCOA7 haplotype had an OR of 0.53 (95% CI 0.32 – 0.89, P = 0.017) compared to women who did not inherit the haplotype. Women with both PD and the heterozygous NCOA7 haplotype had an OR of 0.83 (95% CI, 0.45-1.53; P = 0.54) compared to women without PD and without the haplotype. This suggests that the haplotype attenuated the excess risk associated with PD. Among women without PD, those who were homozygous for the risk allele of FGFR2 had an OR of 1.42 (95% CI 0.90–2.25, P = 0.13) compared to women homozygous for the alternative allele. Women with PD who were homozygous for the risk allele of FGFR2 had an OR of 2.40 (95% CI, 1.20 – 4.81; P = 0.013) compared to women without PD and who were homozygous for the alternative allele. The tests of interaction (i.e. departure from the multiplicative model) between PD and NCOA7, and between PD and FGFR2 were not significant.
Discussion
This multi-stage study investigated the potential role of candidate genes of estrogen metabolism pathways and growth factor signaling pathways in breast cancer risk within three well-characterized cohorts. Our current results identify a novel haplotype at NCOA7 that is associated with a reduction in breast cancer risk. The NCOA7 haplotype had a consistent significant inverse association with breast cancer in each of three independent study populations. Each of the individual SNPs that comprise the haplotype failed to detect the association, and illustrate the potential for improved detection of genetic associations through haplotype-based analysis (see Table 3). Our results also replicated the known risk association of SNPs of the second intron of FGFR2. At FGFR2, the four genotyped SNPs (each previously published) distinguish only two common haplotypes of equally significant opposite effect. However, CGEMS data of this region of FGFR2 includes additional SNPs (seven, from rs11200014 to rs2420946) that discern four common haplotypes. Among them, a haplotype that is inversely associated with breast cancer is the most significant (data not shown). Thus, at both NCOA7 and FGFR2, prominent haplotypes were observed to be inversely associated with breast cancer.
Among the set of genes explored, a number of additional genes were also associated with breast cancer in both the NBC and CGEMS (detailed in Supplementary Table 2). Each of these genes is plausibly linked to breast cancer risk, including: ESRRG (encoding an estrogen-related receptor), NCOA2 (encoding a nuclear receptor co-activator), PRMT8 (a member of a gene family, some known to act as steroid receptor co-activators), and VDR (a gene with a long investigational history in epidemiological studies of cancer risk). Further study in the CBCS and additional populations is needed to discern true from false positive observations among them. NCOA7 encodes a nuclear receptor co-activator that directly interacts with estrogen receptor α to modulate its activity. NCOA7 is recruited to estrogen receptor α target gene promoters following estradiol treatment in a dynamic fashion, similar to other co-activators. But NCOA7 is not similar to other co-activators in sequence or structure, and may represent a novel co-activator class. The ability of NCOA7 to bind ERβ, TRβ, PPARγ, and RARα has also been demonstrated (43). Variants affecting NCOA7 co-activator functions could impact hormone-induced gene transcription. Alternatively, NCOA7 may function in the prevention of oxidative DNA damage (44) by estrogen quinones as a mechanism for carcinogenesis (reviewed in (45)).
The associated NCOA7 haplotype encompassed the initial protein-encoding exon and was not efficiently tagged by any of the individual SNPs evaluated within the gene and its flanking sequence (20 kb upstream or downstream). A maximum r2 of 0.20 was observed between the haplotype and tagging SNP rs490361 among NBC subjects. In HapMap data of Caucasians, the 140 kb gene resides within a larger region of moderate linkage disequilibrium, spanning approximately 250 kb (Supplementary Figure 1). The NCOA7 haplotype is not in notable linkage disequilibrium with other individual HapMap SNPs of the 250 kb interval (maximum r2 =0.16 with rs674859). It is possible that the association signal that we detected at NCOA7 may be attributable to variation anywhere within the larger region of linkage disequilibrium. The identification of candidate functional variants will require additional investigation of this interval, including the genes HEY2, NCOA7, and HINT3.
The first stage of this study was conducted using samples from the Nashville Breast Cohort, whose participants all have a history of benign breast disease. Studies have shown that those with benign proliferative breast disease have an approximately two-fold increase in breast cancer risk (24-26, 28, 29, 46). Epithelial hyperplastic breast lesions are thought to represent an initial step in a non-obligate path that can result in invasive breast cancer (28, 30). Given this, one might anticipate that this histologic risk factor could interact with additional genetic risk factors for the development of breast cancer. We observed that the joint effects of the genetic (FGFR2 or NCOA7) and histologic risk factors on breast cancer risk appear to be multiplicative. It is possible that proliferative breast disease is on a causal pathway between genotype and breast cancer, or that genotype directly affects the risk of both proliferative breast disease and breast cancer. Our nested case-control study was not designed to distinguish between these two hypotheses.
We initially conducted an exploratory analysis of the NBC of 1,469 tagging SNPs from 107 genes associated with estrogen metabolism and growth factor signaling pathways. The source of DNA from the NBC was from archival FFPE benign breast tissue. DNA from this source was not amenable to current chip-based GWAS techniques, but was amenable to alternative moderate-throughput methods. In our sliding window analytic approach we evaluated 199,937 overlapping single allele and haplotype windows in the NBC. The extent of this overlap meant that many of these tests were highly correlated and were effectively evaluating the same variation in allele frequencies between cases and controls. Nevertheless, the risk of spurious findings in our original analysis due to multiple comparisons artifacts was very high. To address this, we replicated our results in the CGEMS study population, and then again in the CBCS study population. The fact that an NCOA7 haplotype was significantly associated with breast cancer risk in all three of these studies markedly increases the likelihood that this association is real. However, further investigation in independent study populations is warranted.
An association at NCOA7 has not previously been described by GWAS of breast cancer. Five GWAS investigations of breast cancer have identified twenty-three susceptibility loci to date. Each of these studies have conducted single-allele analyses as the principal analytic strategy, and have sought sequential replication of observations to discern true from false positive results. One of the discovered loci resides at 6q22.33, initially detected by the SNP rs2180341 (20, 47, 48). This locus is 1.4 Mb telomeric to NCOA7; the two loci are not in linkage disequilibrium. Single-allele analysis of tagging SNPs at NCOA7 failed to detect an association in the NBC, CGEMS, or CBCS breast cancer study populations. Only haplotype-based analysis detected the association within these study populations. Given this observation, single allele analysis within each of the GWAS studies might not be likely to detect the association at NCOA7. CGEMS GWAS data was accessible to us for haplotype-based investigation, and was concordant.
The goal of this project has been to identify genetic factors that permit the identification of women who are at particular risk for breast cancer and for whom informed clinical decisions regarding prevention are valuable. The risks conferred by individual common variants identified to date are small. Risk profiles generated by these alleles may be useful in stratification of population risk, but they do not yet have sufficient positive predictive value for individual assessment (2, 3). The portfolio of genetic variants inherited by an individual includes both risk and protective alleles. More comprehensive knowledge of risk-modifying genetic variants is needed for development of clinically useful risk-stratification tests. Such tests may in the future improve clinical care by identifying women who would most benefit from more intensive screening, those who may be at particularly increased risk if they were to take hormone replacement therapy, or those who may benefit from prophylactic treatment.
Supplementary Material
Supplementary Figure 1. Linkage disequilibrium (LD) plot across a 434 kb interval surrounding the NCOA7 locus for CEU subjects of the HapMap. The region of the breast-cancer associated haplotype is designated by the green box. The plot presents the pairwise LD measure D′, with each diamond indicating the magnitude of LD (Red, D′ = 1 (LOD ≥ 2); blue, D′ = 1 (LOD < 2); pink, D′ < 1 (LOD ≥ 2); white, D′ < 1 (LOD < 2)).
Acknowledgments
We wish to acknowledge the contributions of the participating study subjects, as well as that of investigators of the Cancer Genetic Markers of Susceptibility Study, and additional investigators of the Collaborative Breast Cancer Study, Drs. Montserrat Garcia-Closas and Linda T. Titus-Ernstoff. This study was supported by NIH grants CA098131, CA050468, CA068485, 1UL 1RR024975, by a MERIT grant from the US Department of Veterans Affairs, and by an award from the V Foundation. The CBCS was supported by NIH grants CA105197, CA47147, CA47305, and CA69664.
Footnotes
There are no financial disclosures.
References
- 1.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 2.Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Polygenes, risk prediction, and targeted prevention of breast cancer. N Engl J Med. 2008;358:2796–803. doi: 10.1056/NEJMsa0708739. [DOI] [PubMed] [Google Scholar]
- 3.Wacholder S, Hartge P, Prentice R, Garcia-Closas M, Feigelson HS, Diver WR, et al. Performance of common genetic variants in breast-cancer risk models. N Engl J Med. 2010;362:986–93. doi: 10.1056/NEJMoa0907727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garber JE, Goldstein AM, Kantor AF, Dreyfus MG, Fraumeni JF, Jr, Li FP. Follow-up study of twenty-four families with Li-Fraumeni syndrome. Cancer Res. 1991;51:6094–7. [PubMed] [Google Scholar]
- 5.Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12:3209–15. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 6.Masciari S, Larsson N, Senz J, Boyd N, Kaurah P, Kandel MJ, et al. Germline E-cadherin mutations in familial lobular breast cancer. J Med Genet. 2007;44:726–31. doi: 10.1136/jmg.2007.051268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nevanlinna H, Bartek J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene. 2006;25:5912–9. doi: 10.1038/sj.onc.1209877. [DOI] [PubMed] [Google Scholar]
- 8.Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–7. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–5. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 10.Saal LH, Gruvberger-Saal SK, Persson C, Lovgren K, Jumppanen M, Staaf J, et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet. 2008;40:102–7. doi: 10.1038/ng.2007.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–41. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 12.Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nat Genet. 2008;40:17–22. doi: 10.1038/ng.2007.53. [DOI] [PubMed] [Google Scholar]
- 13.Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010;42:504–7. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox A, Dunning AM, Garcia-Closas M, Balasubramanian S, Reed MW, Pooley KA, et al. A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet. 2007;39:352–8. doi: 10.1038/ng1981. [DOI] [PubMed] [Google Scholar]
- 15.Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–93. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–4. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stacey SN, Manolescu A, Sulem P, Rafnar T, Gudmundsson J, Gudjonsson SA, et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007;39:865–9. doi: 10.1038/ng2064. [DOI] [PubMed] [Google Scholar]
- 18.Zheng W, Long J, Gao YT, Li C, Zheng Y, Xiang YB, et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet. 2009;41:324–8. doi: 10.1038/ng.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmed S, Thomas G, Ghoussaini M, Healey CS, Humphreys MK, Platte R, et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat Genet. 2009;41:585–90. doi: 10.1038/ng.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gold B, Kirchhoff T, Stefanov S, Lautenberger J, Viale A, Garber J, et al. Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci U S A. 2008;105:4340–5. doi: 10.1073/pnas.0800441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stacey SN, Manolescu A, Sulem P, Thorlacius S, Gudjonsson SA, Jonsson GF, et al. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2008;40:703–6. doi: 10.1038/ng.131. [DOI] [PubMed] [Google Scholar]
- 22.Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, Cox DG, et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1) Nat Genet. 2009;41:579–84. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dupont WD, Breyer JP, Bradley KM, Schuyler PA, Plummer WD, Sanders ME, et al. Protein phosphatase 2A subunit gene haplotypes and proliferative breast disease modify breast cancer risk. Cancer. 2010;116:8–19. doi: 10.1002/cncr.24702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dupont WD, Page DL. Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med. 1985;312:146–51. doi: 10.1056/NEJM198501173120303. [DOI] [PubMed] [Google Scholar]
- 25.Dupont WD, Parl FF, Hartmann WH, Brinton LA, Winfield AC, Worrell JA, et al. Breast cancer risk associated with proliferative breast disease and atypical hyperplasia. Cancer. 1993;71:1258–65. doi: 10.1002/1097-0142(19930215)71:4<1258::aid-cncr2820710415>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 26.Page DL, Dupont WD, Rogers LW, Rados MS. Atypical hyperplastic lesions of the female breast. A long-term follow-up study. Cancer. 1985;55:2698–708. doi: 10.1002/1097-0142(19850601)55:11<2698::aid-cncr2820551127>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Newcomb PA, Egan KM, Titus-Ernstoff L, Chanock S, Welch R, et al. Genetic polymorphisms in base-excision repair pathway genes and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:353–8. doi: 10.1158/1055-9965.EPI-05-0653. [DOI] [PubMed] [Google Scholar]
- 28.Hartmann LC, Sellers TA, Frost MH, Lingle WL, Degnim AC, Ghosh K, et al. Benign breast disease and the risk of breast cancer. N Engl J Med. 2005;353:229–37. doi: 10.1056/NEJMoa044383. [DOI] [PubMed] [Google Scholar]
- 29.London SJ, Connolly JL, Schnitt SJ, Colditz GA. A prospective study of benign breast disease and the risk of breast cancer. JAMA. 1992;267:941–4. [PubMed] [Google Scholar]
- 30.Lakhani SR. The transition from hyperplasia to invasive carcinoma of the breast. J Pathol. 1999;187:272–8. doi: 10.1002/(SICI)1096-9896(199902)187:3<272::AID-PATH265>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 31.Altshuler D, Brooks LD, Chakravarti A, Collins FS, Daly MJ, Donnelly P. A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–20. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen R, Fan JB, Campbell D, Chang W, Chen J, Doucet D, et al. High-throughput SNP genotyping on universal bead arrays. Mutation Research. 2005;573:70–82. doi: 10.1016/j.mrfmmm.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 34.Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, et al. Highly parallel SNP genotyping. Cold Spring Harbor Symposia on Quantitative Biology. 2003;68:69–78. doi: 10.1101/sqb.2003.68.69. [DOI] [PubMed] [Google Scholar]
- 35.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–23. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Bakker PI, Yelensky R, Pe'er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–23. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 38.Breyer JP, Sanders ME, Airey DC, Cai Q, Yaspan BL, Schuyler PA, et al. Heritable variation of ERBB2 and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2009;18:1252–8. doi: 10.1158/1055-9965.EPI-08-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fallin D, Schork NJ. Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am J Hum Genet. 2000;67:947–59. doi: 10.1086/303069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yaspan BL, Breyer JP, Cai Q, Dai Q, Elmore JB, Amundson I, et al. Haplotype analysis of CYP11A1 identifies promoter variants associated with breast cancer risk. Cancer Res. 2007;67:5673–82. doi: 10.1158/0008-5472.CAN-07-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sterne JA. Meta-analysis in Stata: an updated collection from the Stata Journal. College Station TX: Stata Press; 2009. [Google Scholar]
- 42.Harris R, Bradburn M, Deeks J, Harbord R, Altman D, Sterne J. Metan: fixed- and random-effects meta-analysis. Stata J. 2008;8:3–28. [Google Scholar]
- 43.Shao W, Halachmi S, Brown M. ERAP140, a conserved tissue-specific nuclear receptor coactivator. Mol Cell Biol. 2002;22:3358–72. doi: 10.1128/MCB.22.10.3358-3372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durand M, Kolpak A, Farrell T, Elliott NA, Shao W, Brown M, et al. The OXR domain defines a conserved family of eukaryotic oxidation resistance proteins. BMC Cell Biol. 2007;8:13. doi: 10.1186/1471-2121-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolton JL, Thatcher GR. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol. 2008;21:93–101. doi: 10.1021/tx700191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palli D, Rosselli del Turco M, Simoncini R, Bianchi S. Benign breast disease and breast cancer: a case-control study in a cohort in Italy. Int J Cancer. 1991;47:703–6. doi: 10.1002/ijc.2910470513. [DOI] [PubMed] [Google Scholar]
- 47.Hemminki K, Muller-Myhsok B, Lichtner P, Engel C, Chen B, Burwinkel B, et al. Low-risk variants FGFR2, TNRC9 and LSP1 in German familial breast cancer patients. Int J Cancer. 2010;126:2858–62. doi: 10.1002/ijc.24986. [DOI] [PubMed] [Google Scholar]
- 48.Kirchhoff T, Chen ZQ, Gold B, Pal P, Gaudet MM, Kosarin K, et al. The 6q22.33 locus and breast cancer susceptibility. Cancer Epidemiol Biomarkers Prev. 2009;18:2468–75. doi: 10.1158/1055-9965.EPI-09-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Linkage disequilibrium (LD) plot across a 434 kb interval surrounding the NCOA7 locus for CEU subjects of the HapMap. The region of the breast-cancer associated haplotype is designated by the green box. The plot presents the pairwise LD measure D′, with each diamond indicating the magnitude of LD (Red, D′ = 1 (LOD ≥ 2); blue, D′ = 1 (LOD < 2); pink, D′ < 1 (LOD ≥ 2); white, D′ < 1 (LOD < 2)).