

Abstract
The effect of hypercapnia on cerebral metabolic rate of oxygen consumption (CMRO2) has been a subject of intensive investigation and debate. Most applications of hypercapnia are based on the assumption that a mild increase in partial pressure of carbon dioxide has negligible effect on cerebral metabolism. In this study, we sought to further investigate the vascular and metabolic effects of hypercapnia by simultaneously measuring global venous oxygen saturation (SvO2) and total cerebral blood flow (tCBF), with a temporal resolution of 30 seconds using magnetic resonance susceptometry and phase-contrast techniques in 10 healthy awake adults. While significant increases in SvO2 and tCBF were observed during hypercapnia (P<0.005), no change in CMRO2 was noted (P>0.05). Additionally, fractional changes in tCBF and end-tidal carbon dioxide (R2=0.72, P<0.005), as well as baseline SvO2 and tCBF (R2=0.72, P<0.005), were found to be correlated. The data also suggested a correlation between cerebral vascular reactivity (CVR) and baseline tCBF (R2=0.44, P=0.052). A CVR value of 6.1%±1.6%/mm Hg was determined using a linear-fit model. Additionally, an average undershoot of 6.7%±4% and 17.1%±7% was observed in SvO2 and tCBF upon recovery from hypercapnia in six subjects.
Keywords: carotid artery, cerebral blood flow measurement, cerebral hemodynamics, energy metabolism, MRI
Introduction
Carbon dioxide (CO2) is a byproduct of oxidative metabolism and is an important cellular signaling molecule in all organisms. The arterial partial pressure of carbon dioxide (pCO2) is one of the major regulators of cerebral circulation. Increased pCO2 (hypercapnia) causes vasodilatation that, in turn, results in a drop in vascular resistance and a concomitant increase in cerebral blood flow (CBF) when autoregulation pathways are intact (Brian, 1998). Carbon dioxide likely mediates its effects by lowering perivascular pH which, in turn, is known to drive vascular tone. However, the exact mechanism by which this modulation occurs is not completely understood (Sharabi et al, 2009).
Since hypercapnia is a common pathophysiologic condition accompanying various cardiorespiratory diseases (e.g., chronic obstructive pulmonary disease, hypoventilation syndromes), understanding its effects on CBF and metabolism are of pivotal clinical importance. The effect of hypercapnia-induced hyperperfusion is used as a diagnostic tool to measure cerebrovascular reactivity (CVR), providing information on the integrity of cerebral autoregulation and is used to assess intracerebral hemodynamics (Muller and Schimrigk, 1996). Cerebrovascular reactivity also serves as a predictor of cerebrovascular disease progression (Liem et al, 2009), and its impairment is regarded as a risk factor for future vascular events such as stroke.
While the vascular effects of hypercapnia on CBF have long been recognized (Kety and Schmidt, 1948), its effects upon central nervous system activity and cerebral metabolism are not well established. Diagnostic and experimental applications of hypercapnia to the study of CVR are based on the assumption that mild alterations of pCO2 have negligible effect on cerebral metabolism.
The first quantitative report of the effect of hypercapnia on cerebral metabolic rate of oxygen consumption (CMRO2), which appeared almost 60 years ago, suggested conservation of cerebral metabolism upon inhalation of 5% to 7% CO2 (Kety and Schmidt, 1948). Since then, numerous groups have investigated neuronal and metabolic implications of hypercapnia and reported a wide spectrum of responses ranging from increase (Jones et al, 2005), decrease (Kliefoth et al, 1979; Xu et al, 2011), and stasis of CMRO2 (Chen and Pike, 2010; Kety and Schmidt, 1948; Massik et al, 1989). Electrophysiological studies investigating the effect of hypercapnia have not been conclusive either, with results ranging from extreme cortical depression to extreme arousal (with some even reporting seizures) (Woodbury and Karler, 1960). Differences in methodologies such as strength and duration of CO2 stimulus, use of anesthetic agents, and species studied, etc., have confounded direct comparison and interpretation of these results.
Even though there is no complete consensus regarding the metabolic effects of hypercapnia, constant CMRO2 is often assumed during increased pCO2 for normalizing blood oxygen-level dependent response in functional magnetic resonance imaging (Davis et al, 1998; Kim et al, 1999). The functional blood oxygen-level dependent effect (increase in signal amplitude during functional activation due to decreased transverse relaxation rate, R2*) is a complex function of CBF, cerebral blood volume (CBV), and CMRO2. With the assumption of constant CMRO2, the decrease in R2* during hypercapnia results solely from an increase in CBF and commensurate reduction in oxygen extraction fraction. On the other hand, functional activation paradigms, such as visual stimulation, have been shown to cause a smaller relative decrease in R2*, the difference being ascribed to greater oxygen extraction due to increased CMRO2 (Kim et al, 1999). Hence, hypercapnia has been used to ‘calibrate' blood oxygen-level dependent signal against purely hemodynamic effects without concomitant metabolic changes.
The primary motivation for the present research was to further elucidate the effect of hypercapnia on CMRO2 using a more advanced methodology. The current work builds on some of the authors' recently conceived method for simultaneously measuring venous oxygen saturation (SvO2) and total cerebral inflow (tCBF) to ensure efficient, accurate, and reliable CMRO2 quantification, with a temporal resolution of about two measurements per minute (Jain et al, 2010). The method measures venous blood oxygen saturation in the superior sagittal sinus (SSS) via magnetic resonance (MR) susceptometry, yielding the intravascular bulk magnetic susceptibility relative to surrounding tissue, and simultaneously measures arterial flow in the major inflow vessels (internal carotid and vertebral arteries) by phase-contrast MR imaging (MRI).
Materials and methods
Ten nonsmoking, healthy volunteers (males, age: 28±5 years) were recruited for the study after giving informed consent. The subjects were judged to be healthy on the basis of their medical history and physical examination. The particular population demographic was chosen to ensure maximal subject compliance to the hypercapnia challenge protocol. The protocol was approved by the Institutional Review Board of University of Pennsylvania.
Stimulus Protocol
Moderate hypercapnia was induced by administering 5% CO2 gas mixed in room air (20% O2; 75% N2) through a 100-L Douglas bag to allow resistance-free supply of air via a mouthpiece. A closed loop was formed between the subject's lungs and the 100-L Douglas bag, which was accomplished by employing a nose clip and having the subjects inhale and exhale from and into a large-diameter short-length tube attached to the Douglas bag. The concentration of gases being exhaled into the Douglas bag (5% to 6%) thus approximately matched that in the bag. Additionally, the small volume of exhaled air rapidly equilibrated with the substantially large volume of the Douglas bag, thereby causing negligible change in the FiCO2 (inspired CO2) delivered throughout our experimental paradigm. The capnometer (Precess, Invivo, Orlando, FL, USA) for end-tidal CO2 (EtCO2) sampling was attached to the above-mentioned mouthpiece tubing. Additionally, heart rate and SaO2 (arterial oxygen saturation) were monitored throughout the experiment using a digital pulse oximeter (Precess, Invivo). The paradigm consisted of three phases: normocapnia (baseline), hypercapnia, and normocapnia (recovery) for durations of 3, 3, and 5 minutes, respectively. For the hypercapnic stimulus, a duration of 3 minutes was chosen to be comparable to the duration employed in previous MRI studies (Bulte et al, 2009; Chen and Pike, 2010). Additionally, a 3-mL venous blood sample was drawn from the antecubital vein of the subjects to determine hematocrit (Hct), hemoglobin, and mean corpuscular hemoglobin concentration (constants used in the computation of HbO2) on the day of the MRI exam.
Measurement Principle
The SvO2 quantification relies on the relative magnetic susceptibility difference, Δχ, of intravascular blood and surrounding tissue. The vessel of interest is modeled as a long paramagnetic cylinder (Fernández-Seara et al, 2006; Haacke et al, 1997) and oxygen saturation (%HbO2) is determined as
 |
where Δφ is the average phase difference between intravascular blood and surrounding tissue and is measured as
ΔB is the incremental field inside the vessel relative to that of the adjacent tissue and ΔTE is the interecho spacing between two gradient-recalled echoes. Δχdo=4π(0.27) (Spees et al, 2001) is the susceptibility difference in SI units between fully deoxygenated and oxygenated erythrocytes, Hct is the volume fraction of the packed erythrocytes in whole blood, and θ is the tilt angle of the vessel with respect to the main field Bo.
Total CBF was quantified by using phase-contrast MRI. The method utilises motion sensitizing gradient waveforms to encode information about velocity into the phase of the MR signal. Specifically, the pulse sequence involves two interleaves; both having null zeroth gradient moment along the direction of blood flow ( 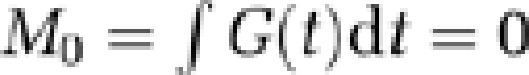 ) but nonzero first gradient moments (
) but nonzero first gradient moments ( 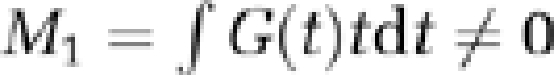 ). The latter determines the sensitivity of the accrued phase difference between the two interleaves, Δφ, to the velocity of the flowing spins and is expressed as,
). The latter determines the sensitivity of the accrued phase difference between the two interleaves, Δφ, to the velocity of the flowing spins and is expressed as,
where ΔM1 is the difference in first moment between the two interleaves and is dictated by a user-defined parameter VENC, defined as
VENC represents the velocity that causes a net phase accumulation of π radians and is typically chosen to be 30% higher than the maximum velocity expected.
Finally, CMRO2 was calculated via Fick's equation,
where CMRO2 is in μmol/min per 100 g, CBF is the total cerebral blood inflow to the brain in mL/100g per minute, SaO2 and SvO2 represent the arterial and venous oxygen saturation levels, respectively, Ca is the oxygen concentration in moles of O2 per 100 mL of blood and was determined in each subject based on their hemoglobin level assuming an O2 carrying capacity of 1.39 mL of O2 per gram of hemoglobin (West, 2007).
Magnetic Resonance
An interleaved gradient-echo pulse sequence was used for simultaneous measurement of SvO2 and tCBF at two different anatomic locations. Specifically, SvO2 was quantified at the level of SSS using MR susceptometry based oximetry and tCBF was determined in the internal carotid and vertebral arteries via phase-contrast MR described above. Additional details on the pulse sequence is given in some of the authors' previous work (Jain et al, 2010).
Images were acquired on a 3T Siemens Tim Trio system (Siemens Medical Solutions, Erlangen, Germany) using a 12-channel head and neck coil. A vendor provided sagittal FLASH (fast low angle shot) sequence was used to obtain an angiogram of head and neck vessels. The sequence is based on a fast rephased–dephased gradient-echo sequence and generates two echoes per excitation to provide images where static tissue is eliminated while signal from flowing blood is preserved. Sequence parameters: 280 × 280, resolution: 1.5 × 1.5 × 40 mm3, slices=3, TR=38 milliseconds, TE=7.2 milliseconds, flip angle=15°, total scan time=22 seconds. This, in conjunction with gradient-echo axial localizer scans was used to choose the straightest section of vessels of interest (SSS, internal carotid and vertebral arteries) and estimate the tilt angle θ of the SSS with respect to Bo from the coordinates of the centroid of the vessel (Δz∼4 cm) (Langham et al, 2009).
Scan parameters
Voxel size=1 × 1 × 5 mm3, flip angle=25°, TR=35 milliseconds, echo spacing=3.5 milliseconds for SvO2 quantification and voxel size=0.85 × 0.85 × 5 mm3, VENC=60 cm/s (normocapnia) and 100 cm/s (hypercapnia) for tCBF quantification, total scan time=28 seconds. Additionally, a T1-weighted 3D magnetization-prepared rapid gradient-echo image data set (voxel size=1 × 1 × 1 mm3) was used to estimate intracranial volume (Mugler and Brookeman, 1990). Total brain volume was obtained using a semiautomated region-growing algorithm (ITKSnap; Yushkevich et al, 2006).
It is noted that the typical tilt angles of SSS in our study were <15°. Since the induced field outside a cylinder falls off as the inverse square of the distance and the square of the sine of the tilt angle (Haacke et al, 1999), the magnetic field outside the SSS was negligibly perturbed (note in Figures 2C–2E the sharp drop in the net accumulated phase outside the SSS and the near constancy of the field in the reference tissue). A region of interest was chosen within the SSS and in the surrounding homogenous brain tissue at a distance on the order of the diameter of the vessel and Δφ was calculated by subtracting the net accumulated phase in the two regions.
A linear model for cerebral vascular response to hypercapnia was assumed and CVR was estimated as the slope of the %ΔtCBF versus ΔEtCO2 curve. Analysis of variance was used to test for differences in group means of SvO2, tCBF, and CMRO2 between the three test conditions (baseline, hypercapnia, and recovery).
Results
Inhalation of 5% CO2 gas mixture produced similar changes in EtCO2 across all subjects. Figure 1 shows the time course of various physiological parameters in one of the subjects (heart rate, SaO2, and EtCO2) recorded during the experiment. Group means for all subjects are listed in Table 1. Figure 2 displays the magnitude and phase-difference images at the level of the SSS (Figures 2B–2E) and velocity maps of the neck vessels (Figures 2F–2I). Notice the expected decrease in phase contrast for the SSS, indicating increased SvO2 levels and the enhanced blood velocity in neck vessels during hypercapnia.
Figure 1.

Time course plots of various physiological parameters: heart rate (HR), arterial oxygen saturation (SaO2), and end-tidal CO2 (EtCO2) during baseline, hypercapnia (5% CO2 shaded), and recovery in a representative subject.
Table 1. Summary data of various parameters.
| SaO2 (%) | SvO2 (%) | tCBF (mL/100 g per minute) | CMRO2 (μmol/100 g per minute) | HR (b.p.m.) | EtCO2 (mm Hg) | |
|---|---|---|---|---|---|---|
| Baseline | 97±1 | 65±6 | 49±6 | 134±10 | 65±13 | 39±1 |
| Hypercapnia | 97±1 | 78±5* | 80±13* | 130±13 | 73±13 | 47±2* |
| Recovery | 97±1 | 64±5 | 47±6 | 132±8 | 66±13 | 38±2 |
CMRO2, cerebral metabolic rate of oxygen consumption; EtCO2, end-tidal CO2; HR, heart rate; SaO2, arterial oxygen saturation; SvO2, venous oxygen saturation; tCBF, total cerebral blood flow.
Group means and s.d. of various measured and physiological parameters.
*Indicates statistically significant changes (P<0.005).
Figure 2.

Images from which venous oxygen saturation (SvO2) and total cerebral blood flow (tCBF) during baseline, hypercapnia, and recovery were derived. (A) Sagittal localizer angiogram indicating the locations for SvO2 (top) and tCBF (bottom) measurements; (B) axial magnitude; (C–E) phase-difference images for SvO2 quantification highlighting superior sagittal sinus (SSS); (F) axial magnitude and (G–I) velocity images of the neck showing the major cerebral inflow vessels (internal carotid and vertebral arteries). Note the change in contrast in vessels during hypercapnia.
The initial tCBF and SvO2 response to hypercapnia is estimated to be <30 seconds, consistent with previously reported results. The response stabilized after ∼1 to 2 minutes for all subjects (Figure 3). Additionally, an average undershoot of 6.7%±4% and 17.1%±7% was observed in SvO2 and tCBF values upon recovery from hypercapnia in six subjects (Figure 3).
Figure 3.

Plot of time-resolved measurements of total cerebral blood flow (tCBF) and venous oxygen saturation (SvO2) during normocapnia and hypercapnia (shaded) in a representative subject. Note the undershoot during recovery from hypercapnia.
The group averages for the various measured parameters during the three experimental conditions are given in Table 1. A significant linear correlation was observed between percent ΔtCBF and ΔEtCO2 (R2=0.72, P<0.005), and CVR, computed as the slope of the above curve, was 6.1%±1.6%/mm Hg (Figure 4A). Further, CVR was negatively correlated with baseline tCBF, even though this relationship was only marginally significant (R2=0.44, P=0.052; Figure 4B). Baseline SvO2 and tCBF were correlated as expected (R2=0.72, P<0.005; Figure 4C). Finally, significant changes in both SvO2 and tCBF were observed during hypercapnia (both P<0.005; Figures 5A and 5B), whereas global CMRO2 remained unchanged (P=0.78; Figure 5C).
Figure 4.

(A) Percent change in total cerebral blood flow (CBF) versus change in end-tidal CO2 (ΔEtCO2) in response to hypercapnia. The slope of the curve represents cerebrovascular reactivity (CVR); relationship between (B) intersubject CVR and baseline tCBF and (C) baseline venous oxygen saturation (SvO2) versus tCBF.
Figure 5.

Scatter plot of functional parameters for all subjects during baseline, hypercapnia (shaded), and recovery: (A) total cerebral blood flow (tCBF); (B) venous oxygen saturation (SvO2); (C) cerebral metabolic rate of oxygen consumption (CMRO2). The vertical span of each diamond represents the 95% confidence interval (CI) of the group means, symbols refer to individual subjects.
Discussion
The effect of increased blood CO2 on neuronal activity and cerebral oxidative metabolism has been a subject of intensive investigation during the past six decades. In this study, we report simultaneous measurement of metabolic (SvO2) and hemodynamic (tCBF) parameters in response to mild hypercapnia (5% inspired CO2), in healthy, awake, adult subjects by means of quantitative MRI. Our results support the notion of negligible change in CMRO2 with a mild hypercapnic stimulus. Previous human studies with different methodologies, but also invoking Fick's principle for CMRO2 quantification (Chen and Pike, 2010; Kety and Schmidt, 1948; Massik et al, 1989), as well as in vivo small animal studies measuring neuronal metabolites of oxidative phosphorylation (adenosine triphosphate, adenosine diphosphate, and adenosine monophosphate), have reported similar results upon exposure to mild hypercapnia (Buckweitz et al, 1980; Folbergrova et al, 1974).
In distinction to prior work, our experimental approach allows for rapid, simultaneous measurement of SvO2, tCBF, and EtCO2. The ability to simultaneously quantify two key parameters in Fick's equation (equation 5) gives us particular confidence in the veracity of our CMRO2 estimates as opposed to all prior work requiring sequential measurements (Chen and Pike, 2010; Xu et al, 2011). Additionally, our method enables point-by-point temporal analysis of vascular and metabolic responses to hypercapnia with a temporal resolution of 30 seconds. This improves the experimental sensitivity to visualize short time scale responses to pCO2 changes (e.g., detection of temporal lags in achieving a steady-state response due to abrupt induction and cessation of hypercapnia). Even the most recently conducted hypercapnia studies have precluded such an evaluation (Chen and Pike, 2010; Xu et al, 2011).
The observed experimental CVR value of 6.1%±1.6%/mm Hg, obtained using a linear-fit model, is in the range of previously reported values (4% to 7%/mm Hg) (Chen and Pike, 2010; Noth et al, 2006; Pollock et al, 2009). The between-subject variability of ∼21% in the individual CVR measurements is in good agreement with coefficients of variation reported previously (Kassner et al, 2010; Totaro et al, 1998). While the underlying mechanism for this variation in CVR is not fully understood, we conjecture that it might be explainable, at least in part, by intersubject variations in baseline tCBF. Support for such an interpretation is the observation of a moderate negative correlation, approaching statistical significance, between vascular response to hypercapnia (CVR) and baseline tCBF (R2=0.44, P=0.052). This association suggests that the capacity for vasodilatation by a hypercapnic stimulus might be a function of baseline cerebral flow. A similar negative correlation (though not statistically significant) between regional cerebral vascular response to hypercapnia and baseline CBF values in the occipital cortex had been noted in a positron emission tomography-based study by Ito et al (2008).
Of interest, our experimental data demonstrate concordance with many known principles of human physiology. For example, a strong positive correlation was observed between baseline SvO2 and tCBF, analogous to prior reports by Xu et al (2009), and more recently by some of the present authors (Jain et al, 2010). Under stable resting cerebral physiologic conditions, changes in flow are unlikely to change the rate of metabolism. This concept is based on the assumption that in a resting awake healthy human brain, the metabolic demand should be constant (Lajtha et al, 2007). As expected during hypercapnia, the observed increase in SvO2 was accompanied by a commensurate increase in tCBF, suggesting cerebral activity to remain invariant.
Our data indicate that the steady-state tCBF hypercapnic response is attained with some delay. Prior work has shown that the bulk of this delay is caused by the time required for the build-up of CO2 in arterial blood; which in turn is a function of the level of ventilation, CO2 diffusion time and pulmonary blood flow (Shapiro et al, 1965, 1966). However, studies have also noted that tCBF equilibration lags behind arterial pCO2 stabilization. Therefore, we posit that the delay we observed might also be due to CO2 mediating its physiologic effects via alterations in perivascular pH. Many animal studies have noted that decreases in pH result in vasodilatation and, conversely, increases cause vasoconstriction. These effects have been thought to be mediated by secondary messengers regulating smooth muscle vascular tone (Kontos et al, 1977).
In our experimental paradigm it is plausible to assume that as arterial pCO2 increases during presentation of the hypercapnic stimulus, CO2 diffuses into the brain parenchyma, eventually attaining a steady-state value. The initial changes in pH are likely offset, at least in part, by local tissue buffering at the intracellular and extracellular level (Celotto et al, 2008). However, as the hypercapnic stimulus persists and the steady-state parenchymal pCO2 value is reached, these local homoeostatic measures are overwhelmed. The ensuing decrease in pH likely then triggers the production of secondary intracellular mediators at end arterioles, thereby lowering cerebral vascular resistance and producing the observed increase in blood flow (Brian, 1998). As these events depend on a series of processes occurring, it is reasonable to assume that the observed steady-state response would develop over a period of minutes rather than instantaneously. In our study, the initial changes in CBF were observed in <30 seconds; however, the time to achieve steady-state CBF was on the order of 1 to 2 minutes. These time scales are consistent with previous reports (Poulin et al, 1996; Shapiro et al, 1965, 1966).
We also noted an average of 17% decrease in CBF below the baseline after cessation of the hypercapnic stimulus in 6 of the 10 subjects (Figure 3). Other investigators have also reported similar undershoots in tCBF during recovery from hypercapnia. Previous work by Claassen et al (2007) and Poulin et al (1996), using transcranial doppler ultrasound, as well as more recent work by Chen and Pike (2010), using arterial spin labeling (ASL), indicate similar trends in tCBF data.
Although the exact physiological mechanism of this undershoot in CBF is unclear, we hypothesize that this response might have been mediated by a combination of rapid respiratory egress of pCO2 via hyperventilation, in conjunction with a slower reequilibration of cellular buffering mediators (such as tissue bicarbonate and inorganic bases). So, even after cessation of the hypercapnic stimulus, arterial pCO2 likely remains above the normal physiologic range. This interpretation is supported by the presence of transiently elevated EtCO2 (above baseline) even upon abrupt removal of the hypercapnic stimulus. Consequently, hyperventilation would persist as central pH-sensitive chemoreceptors in the respiratory centers of the pons and medulla, as well as peripheral receptors for pH and CO2 in the carotid and aortic bodies continue to fire (Speck, 1992). However, as the respiratory clearance of CO2 quickly ensues (Shapiro et al, 1965, 1966), the cellular chemical buffering mediators (initiated at the onset of hypercapnia) are likely to take longer to equilibrate, resulting in a brief period of tissue alkalosis. The proposed brief period of tissue alkalosis during recovery from hypercapnia has also been reported in a 31P-Nuclear Magnetic Spectroscopy-based rat study (Nishimura et al, 1989). This transient increase in pH may produce vasoconstriction at local end arterioles resulting in the observed undershoot in tCBF. It is noted that hyperventilation-induced hypocapnia is unlikely to be the reason for the observed undershoot as a similar trend in EtCO2 data was not observed.
As pointed out earlier, numerous studies have investigated the vascular, neuronal, and metabolic implications of hypercapnia. While a review of the hypercapnia literature in its entirety is beyond the scope of the current article, two recent studies utilising MRI-based methods for CMRO2 quantification and a study protocol somewhat similar to ours are of particular relevance, even though they are based on a different methodology for quantifying SvO2 and flow. Our results are in good agreement with those reported by Chen and Pike (2010). Specifically, they reported a conservation of CMRO2 during mild hypercapnia and a CVR value very similar to ours (5.9 versus 6.1). Additionally, an undershoot in tCBF values was also observed in the presented tCBF time series plots. However, our results are at variance with those reported by Xu et al (2011), who observed a decrease in CMRO2 during hypercapnia with the effect being dose dependent.
A few methodological differences could potentially have contributed to the discrepant results. Typically, including in our study, CMRO2 is calculated as the product of whole brain blood flow and difference in oxygen saturation between arterial and SSS blood. However, Xu et al measured CBF in the SSS as representative of tCBF. Under physiological alterations such as hypercapnia, flow through the SSS would be a valid alternative for tCBF as long as the proportion of cerebral venous drainage remained constant. However, it is conceivable that in order to accommodate the dramatic increases in flow prompted by hypercapnia, other venous conduits might be recruited beyond the SSS. It is noted that the cerebral venous system is valveless; and several routes for drainage exist. The extent of venous drainage might vary under physiological challenges as the cerebral veins and sinuses are low compliance vessels with a fixed capacity for drainage. Hence, there might be great variability in the distribution of venous return when faced with increases in CBF such as the present experimental paradigm. The above could lead to underestimation of flow changes in response to hypercapnia when approximating tCBF from a measurement in the SSS.
We note that quantification of SvO2 in the SSS as representative of global SvO2 to be of lesser concern. Positron emission tomography data demonstrated little interregional variation in SvO2 during resting conditions in human subjects. Hence the SSS, which receives a large portion of the cerebral venous drainage, is likely to be a good indicator of global SvO2 (Lebrun-Grandie et al, 1983). Direct spectrophotometric measurements in anesthetized cats have given similar results (Buckweitz et al, 1980).
While our experiment lends support for negligible change in CMRO2 in response to hypercapnia, our measurements do not preclude the possibility of regional variations in CMRO2. In fact, evidence for such regional heterogeneities in neuronal response to hypercapnia have been reported previously (Biswal et al, 1997; Chen and Pike, 2010; St John, 1981). Such observations withstanding, we sought to evaluate only global metabolic response to hypercapnia in our experiment. Additionally, we investigated only the effects of mild and acute exposure to increased CO2 levels. It is probable that a stronger hypercapnic stimulus, or a longer exposure might alter the CMRO2. There is some evidence for such CMRO2 changes based on previous animal studies (Zappe et al, 2008). Finally, it is possible that the small population size studied might have limited our ability to evaluate changes in CMRO2 in response to hypercapnia. However, while designing the study, our analysis indicated that a subject size (N) of 10 would allow detection of 7% to 8% relative difference in any of the measured parameters, with 80% power and 5% significance. Reproducibility of CMRO2 at 3T was previously found to be on the order of 8% (Jain et al, 2010).
In summary, this study sought to investigate the effect of mild hypercapnia (∼9 mm Hg) on global CMRO2 by simultaneously measuring tCBF and global SvO2. The simultaneous measurement scheme is likely to improve accuracy and reliability of measuring CMRO2 as opposed to performing sequential measurements. Additionally, the achieved temporal resolution (∼0.5 minutes) allows probing the temporal dynamics of vascular and metabolic effects of hypercapnia.
Acknowledgments
The project described was supported by Grant Number UL1RR024134 from the National Center For Research Resources. Also, we would like to thank Dr Andrew Cucchiara for help with statistics.
The authors declare no conflict of interest.
Footnotes
This work was supported by grants NIH R01-MH080892 and NIH R21-HL088182.
References
- Biswal B, Hudetz AG, Yetkin FZ, Haughton VM, Hyde JS. Hypercapnia reversibly suppresses low-frequency fluctuations in the human motor cortex during rest using echo-planar MRI. J Cereb Blood Flow Metab. 1997;17:301–308. doi: 10.1097/00004647-199703000-00007. [DOI] [PubMed] [Google Scholar]
- Brian JE., Jr Carbon dioxide and the cerebral circulation. Anesthesiology. 1998;88:1365–1386. doi: 10.1097/00000542-199805000-00029. [DOI] [PubMed] [Google Scholar]
- Buckweitz E, Sinha AK, Weiss HR. Cerebral regional oxygen consumption and supply in anesthetized cat. Science. 1980;209:499–501. doi: 10.1126/science.7394515. [DOI] [PubMed] [Google Scholar]
- Bulte DP, Drescher K, Jezzard P. Comparison of hypercapnia-based calibration techniques for measurement of cerebral oxygen metabolism with MRI. Magn Reson Med. 2009;61:391–398. doi: 10.1002/mrm.21862. [DOI] [PubMed] [Google Scholar]
- Celotto AC, Capellini VK, Baldo CF, Dalio MB, Rodrigues AJ, Evora PR. Effects of acid-base imbalance on vascular reactivity. Braz J Med Biol Res. 2008;41:439–445. doi: 10.1590/s0100-879x2008005000026. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Pike GB. Global cerebral oxidative metabolism during hypercapnia and hypocapnia in humans: implications for BOLD fMRI. J Cereb Blood Flow Metab. 2010;30:1094–1099. doi: 10.1038/jcbfm.2010.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claassen JA, Zhang R, Fu Q, Witkowski S, Levine BD. Transcranial Doppler estimation of cerebral blood flow and cerebrovascular conductance during modified rebreathing. J Appl Physiol. 2007;102:870–877. doi: 10.1152/japplphysiol.00906.2006. [DOI] [PubMed] [Google Scholar]
- Davis TL, Kwong KK, Weisskoff RM, Rosen BR. Calibrated functional MRI: mapping the dynamics of oxidative metabolism. Proc Natl Acad Sci USA. 1998;95:1834–1839. doi: 10.1073/pnas.95.4.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Seara M, Detre JA, Techawiboonwong A, Wehrli FW. MR susceptometry for measuring global brain oxygen extraction. Magn Reson Med. 2006;55:967–973. doi: 10.1002/mrm.20892. [DOI] [PubMed] [Google Scholar]
- Folbergrova J, Ponten U, Siesjo BK. Patterns of changes in brain carbohydrate metabolites, amino acids and organic phosphates at increased carbon dioxide tensions. J Neurochem. 1974;22:1115–1125. doi: 10.1111/j.1471-4159.1974.tb04344.x. [DOI] [PubMed] [Google Scholar]
- Haacke EM, Brown RW, Thompson MR, Venkatesan R. Magnetic Resonance Imaging: Physical Principles and Sequence Design. New York: John Wiley & Sons; 1999. [Google Scholar]
- Haacke EM, Lai S, Reichenbach JR, Kuppusamy K, Hoogenraad FGC, Takeichi H, Lin W. In vivo measurement of blood oxygen saturation using magnetic resonance imaging: a direct validation of the blood oxygen level-dependent concept in functional brain imaging. Human Brain Mapping. 1997;5:341–346. doi: 10.1002/(SICI)1097-0193(1997)5:5<341::AID-HBM2>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Ito H, Kanno I, Ibaraki M, Suhara T, Miura S. Relationship between baseline cerebral blood flow and vascular responses to changes in PaCO2 measured by positron emission tomography in humans: implication of inter-individual variations of cerebral vascular tone. Acta Physiol (Oxf) 2008;193:325–330. doi: 10.1111/j.1748-1716.2008.01847.x. [DOI] [PubMed] [Google Scholar]
- Jain V, Langham MC, Wehrli FW. MRI estimation of global brain oxygen consumption rate. J Cereb Blood Flow Metab. 2010;30:1598–1607. doi: 10.1038/jcbfm.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M, Berwick J, Hewson-Stoate N, Gias C, Mayhew J. The effect of hypercapnia on the neural and hemodynamic responses to somatosensory stimulation. Neuroimage. 2005;27:609–623. doi: 10.1016/j.neuroimage.2005.04.036. [DOI] [PubMed] [Google Scholar]
- Kassner A, Winter JD, Poublanc J, Mikulis DJ, Crawley AP. Blood-oxygen level dependent MRI measures of cerebrovascular reactivity using a controlled respiratory challenge: reproducibility and gender differences. J Magn Reson Imaging. 2010;31:298–304. doi: 10.1002/jmri.22044. [DOI] [PubMed] [Google Scholar]
- Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest. 1948;27:484–492. doi: 10.1172/JCI101995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG, Rostrup E, Larsson HB, Ogawa S, Paulson OB. Determination of relative CMRO2 from CBF and BOLD changes: significant increase of oxygen consumption rate during visual stimulation. Magn Reson Med. 1999;41:1152–1161. doi: 10.1002/(sici)1522-2594(199906)41:6<1152::aid-mrm11>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Kliefoth AB, Grubb RL, Jr, Raichle ME. Depression of cerebral oxygen utilization by hypercapnia in the rhesus monkey. J Neurochem. 1979;32:661–663. doi: 10.1111/j.1471-4159.1979.tb00404.x. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Raper AJ, Patterson JL. Analysis of vasoactivity of local pH, PCO2 and bicarbonate on pial vessels. Stroke. 1977;8:358–360. doi: 10.1161/01.str.8.3.358. [DOI] [PubMed] [Google Scholar]
- Lajtha A, Gibson GE, Dienel GA.2007Handbook of Neurochemistry and Molecular Neurobiology: Brain Energentics. Integration of Molecular and Cellular Processes3rd ed.New York: Springer [Google Scholar]
- Langham MC, Magland JF, Epstein CL, Floyd TF, Wehrli FW. Accuracy and precision of MR blood oximetry based on the long paramagnetic cylinder approximation of large vessels. Magn Reson Med. 2009;62:333–340. doi: 10.1002/mrm.21981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun-Grandie P, Baron JC, Soussaline F, Loch'h C, Sastre J, Bousser MG. Coupling between regional blood flow and oxygen utilization in the normal human brain. A study with positron tomography and oxygen 15. Arch Neurol. 1983;40:230–236. doi: 10.1001/archneur.1983.04050040060010. [DOI] [PubMed] [Google Scholar]
- Liem MK, Lesnik Oberstein SA, Haan J, Boom R, Ferrari MD, Buchem MA, Grond J. Cerebrovascular reactivity is a main determinant of white matter hyperintensity progression in CADASIL. AJNR Am J Neuroradiol. 2009;30:1244–1247. doi: 10.3174/ajnr.A1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massik J, Jones MD, Jr, Miyabe M, Tang YL, Hudak ML, Koehler RC, Traystman RJ. Hypercapnia and response of cerebral blood flow to hypoxia in newborn lambs. J Appl Physiol. 1989;66:1065–1070. doi: 10.1152/jappl.1989.66.3.1065. [DOI] [PubMed] [Google Scholar]
- Mugler JP, III, Brookeman JR. Three-dimensional magnetization-prepared rapid gradient-echo imaging (3D MP RAGE) Magn Reson Med. 1990;15:152–157. doi: 10.1002/mrm.1910150117. [DOI] [PubMed] [Google Scholar]
- Muller M, Schimrigk K. Vasomotor reactivity and pattern of collateral blood flow in severe occlusive carotid artery disease. Stroke. 1996;27:296–299. doi: 10.1161/01.str.27.2.296. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Johnson DC, Hitzig BM, Okunieff P, Kazemi H. Effects of hypercapnia on brain pHi and phosphate metabolite regulation by 31P-NMR. J Appl Physiol. 1989;66:2181–2188. doi: 10.1152/jappl.1989.66.5.2181. [DOI] [PubMed] [Google Scholar]
- Noth U, Meadows GE, Kotajima F, Deichmann R, Corfield DR, Turner R. Cerebral vascular response to hypercapnia: determination with perfusion MRI at 1.5 and 3.0 Tesla using a pulsed arterial spin labeling technique. J Magn Reson Imaging. 2006;24:1229–1235. doi: 10.1002/jmri.20761. [DOI] [PubMed] [Google Scholar]
- Pollock JM, Deibler AR, Whitlow CT, Tan H, Kraft RA, Burdette JH, Maldjian JA. Hypercapnia-induced cerebral hyperperfusion: an underrecognized clinical entity. AJNR Am J Neuroradiol. 2009;30:378–385. doi: 10.3174/ajnr.A1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin MJ, Liang PJ, Robbins PA. Dynamics of the cerebral blood flow response to step changes in end-tidal PCO2 and PO2 in humans. J Appl Physiol. 1996;81:1084–1095. doi: 10.1152/jappl.1996.81.3.1084. [DOI] [PubMed] [Google Scholar]
- Shapiro E, Wasserman AJ, Patterson JL., Jr Human cerebrovascular response time to elevation of arterial carbon dioxide tension. Arch Neurol. 1965;13:130–138. doi: 10.1001/archneur.1965.00470020020003. [DOI] [PubMed] [Google Scholar]
- Shapiro W, Wasserman AJ, Patterson JL., Jr Mechanism and pattern of human cerebrovascular regulation after rapid changes in blood CO2 tension. J Clin Invest. 1966;45:913–922. doi: 10.1172/JCI105406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharabi K, Lecuona E, Helenius IT, Beitel GJ, Sznajder JI, Gruenbaum Y. Sensing, physiological effects and molecular response to elevated CO2 levels in eukaryotes. J Cell Mol Med. 2009;13:4304–4318. doi: 10.1111/j.1582-4934.2009.00952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck DF. Respiratory Control. Lexington: University Press of Kentucky; 1992. [Google Scholar]
- Spees WM, Yablonskiy DA, Oswood MC, Ackerman JJ. Water proton MR properties of human blood at 1.5 Tesla: magnetic susceptibility, T(1), T(2), T*(2), and non-Lorentzian signal behavior. Magn Reson Med. 2001;45:533–542. doi: 10.1002/mrm.1072. [DOI] [PubMed] [Google Scholar]
- St John WM. Respiratory neuron responses to hypercapnia and carotid chemoreceptor stimulation. J Appl Physiol. 1981;51:816–822. doi: 10.1152/jappl.1981.51.4.816. [DOI] [PubMed] [Google Scholar]
- Totaro R, Barattelli G, Quaresima V, Carolei A, Ferrari M. Evaluation of potential factors affecting the measurement of cerebrovascular reactivity by near-infrared spectroscopy. Clin Sci (Lond) 1998;95:497–504. [PubMed] [Google Scholar]
- West JB.2007Pulmonary Physiology and Pathophysiology: An Integrated, Case-Based Approach2nd ed.Philadelphia, PA: Lippincott Williams & Wilkins [Google Scholar]
- Woodbury DM, Karler R. The role of carbon dioxide in the nervous system. Anesthesiology. 1960;21:686–703. doi: 10.1097/00000542-196011000-00012. [DOI] [PubMed] [Google Scholar]
- Xu F, Ge Y, Lu H. Noninvasive quantification of whole-brain cerebral metabolic rate of oxygen (CMRO2) by MRI. Magn Reson Med. 2009;62:141–148. doi: 10.1002/mrm.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Uh J, Brier MR, Hart J, Jr, Yezhuvath US, Gu H, Yang Y, Lu H. The influence of carbon dioxide on brain activity and metabolism in conscious humans. J Cereb Blood Flow Metab. 2011;31:58–67. doi: 10.1038/jcbfm.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 2006;31:1116–1128. doi: 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Zappe AC, Uludag K, Oeltermann A, Ugurbil K, Logothetis NK. The influence of moderate hypercapnia on neural activity in the anesthetized nonhuman primate. Cereb Cortex. 2008;18:2666–2673. doi: 10.1093/cercor/bhn023. [DOI] [PMC free article] [PubMed] [Google Scholar]