

Abstract
Dominant mutations in superoxide dismutase cause amyotrophic lateral sclerosis (ALS), an adult-onset neurodegenerative disease characterized by loss of motor neurons. Using mice carrying a deletable mutant gene, diminished mutant expression in astrocytes did not affect onset, but delayed microglial activation and sharply slowed later disease progression. These findings demonstrate that mutant astrocytes are viable targets for therapies to slow progression of non-cell-autonomous killing of motor neurons in ALS.
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease, characterized by a progressive, fatal loss of motor neurons. Dominant mutations in the gene for superoxide dismutase (SOD1) are the most frequent cause of inherited ALS. Ubiquitous expression of mutant SOD1 in rodents leads to progressive, selective motor neuron degeneration due to an acquired toxic property(ies). The exact mechanism responsible for motor neuron degeneration in ALS, however, is not known1, 2. Mutant damage within the vulnerable motor neurons is a key determinant of disease onset3, while accumulating evidence supports an active role of non-neuronal cells in motor neuron degeneration3–7. Evidence with selective gene excision3 or bone marrow grafting5 has demonstrated that mutant SOD1-derived damage within microglia accelerates later disease progression. Despite the importance of astrocyte function, the role of mutant action within astrocytes in disease has not been tested in vivo.
To examine whether mutant SOD1 damage within astrocytes contributes to disease, LoxSOD1G37R mice3, carrying a mutant SOD1 gene that can be deleted by the action of the Cre recombinase, were mated with GFAP-Cre mice (Supplementary Fig. 1) which express both Cre recombinase and β-galactosidase (LacZ) under the control of the human GFAP promoter8. Although this GFAP-Cre transgene is expressed within a subset of neurons in the cerebellum and hippocampus during embryogenesis9, measurement of β-galactosidase activity (by deposition of a blue reaction product after addition of the X-gal substrate) demonstrated that Cre-mediated recombination was restricted within the spinal cord to GFAP-reactive astrocytes (Fig. 1a,b). Efficiency of mutant gene excision within cultured astrocytes from newborn LoxSOD1G37R/ GFAP-Cre+ mice was ~76% (Fig. 1d,e), determined by quantitative PCR for human SOD1 transgene number (Fig. 1d) and immunoblotting for mutant SOD1 levels (Fig. 1e). Neither detectable Cre-activity nor mutant gene excision was observed within microglia (Fig.1c; Supplementary Fig. 2).
Figure 1. Selective Cre-mediated gene excision demonstrates mutant SOD1 action within astrocytes to be a primary determinant of later disease progression.

(a,b) β-galactosidase (β–gal) activity within astrocytes in (a) whole or (b) anterior horn region of the lumbar spinal cord section of GFAP-Cre/Rosa26 reporter mice visualized with X-gal and immunostaining with GFAP antibody. (Inset) Magnified image of the boxed area in (b). Arrows indicate β–Gal/GFAP-Cre expressing astrocytes. (c, d) LoxSOD1G37R transgene levels (n=3 for each group) in (c) primary microglia or (d) astrocytes from LoxSOD1G37R/GFAP-Cre+ and LoxSOD1G37R mice using real-time PCR. (e) SOD1G37R and mouse SOD1 content determined by immunoblotting in extracts from isolated primary astrocytes of (lanes 1, 2) LoxSOD1G37R/GFAP-Cre+ and (lanes 3–5) a dilution series of a comparable extract from LoxSOD1G37R astrocytes representing 25%, 50% and 100% of the protein amounts loaded in lanes 1 and 2. Ages at which (f) early disease phase (to 10% weight loss) (p=0.76) or (g) end-stage (p<0.0001) was reached for (red) LoxSOD1G37R/GFAP-Cre+ mice and (blue) LoxSOD1G37R littermates. Mean ages ± standard deviation were provided. (h–j) Mean onset (p=0.47) (h), mean duration of early disease (p=0.35) (i) (from onset to 10% weight loss) and a later disease (p<0.0001) (j) (from 10% weight loss to end-stage) for (red) LoxSOD1G37R/GFAP-Cre+ and (blue) LoxSOD1G37R littermates. At each time point, p value was determined by unpaired t test. Error bars denote standard deviation.
A simple, objective measure of disease onset and early disease was applied by initiation of weight loss, itself reflecting denervation-induced muscle atrophy. Reduction of SOD1G37R in astrocytes did not slow disease onset nor early disease (GFAP-Cre+: 341.6 ± 48.9 days; GFAP-Cre−: 337.0 ± 35.8 days; Fig. 1f,h). However, late disease progression (from early disease to end-stage) was sharply delayed, providing a mean extension of survival by 48 days (Cre+: 87.4 days; Cre−: 39.5 days; Fig. 1j). Progression from onset to early disease was more modestly slowed by 14 days (Cre+: 99.3 days; Cre−: 85.2 days; Fig. 1i). Overall survival was extended by 60 days (Cre+: 436.5 ± 38.8 days; Cre−: 376.5 ± 26.9 days; Fig. 1g). This contrasts with delayed disease onset from diminished mutant synthesis solely within motor neurons (with a VAChT-Cre transgene carrying the motor neuron specific vesicular acetylcholine transporter promoter) without affecting disease progression (Supplementary text and Supplementary Fig. 3), just as reported previously with an Islet1-Cre transgene that is expressed within motor neurons and some peripheral tissues3.
Astrocytic and microglial cell activation is a well accepted feature of SOD1 mutant mediated ALS1,2. Appearance of an elevated proportion of GFAP-positive astrocytes initiated prior to disease onset (Fig. 2a) in LoxSOD1G37R mice. This astrogliosis was progressive and was readily apparent by onset (Fig. 2b) and more prominent during disease progression (Fig. 2c). Despite substantial mutant reduction, astrogliosis was not, however, different in comparing disease-matched LoxSOD1G37R/GFAP-Cre+ mice (Fig. 2d,e) and LoxSOD1G37R/Cre− mice (Fig. 2b,c).
Figure 2. Selective downregulation of mutant SOD1 in astrocytes significantly inhibits microglial activation.
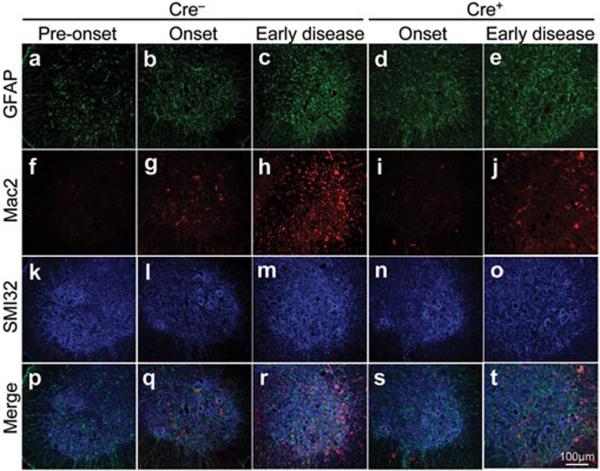
(a–e) GFAP detected within astrocytes, (f–j) Mac2-positive activated microglia, and (k–o) motor neurons identified with the neurofilament antibody SMI-32 staining in the lumbar spinal cord of a LoxSOD1G37R mouse (a, f, k, p) prior to disease onset, (b, g, l, p) at disease onset, or (c, h, m, r) at early disease, together with LoxSOD1G37R/GFAP-Cre+ mice (d, i, n, s) at onset or (e, j, o, t) early disease. (p–t) Merged images.
Microglial activation initiated at earliest disease onset in Cre− mice (Fig. 2g) and was progressively more prominent during disease progression (Fig. 2h). Microglial activation was, however, significantly delayed from onset through early disease in the GFAP-Cre+ mice when mutant SOD1 levels were reduced only in astrocytes (Fig. 2i,j). By exploiting the presence of β-galactosidase to mark astrocytes with diminished SOD1 mutant synthesis, examination of sections throughout lumbar spinal cords of symptomatic LoxSOD1G37R /GFAP-Cre+ mice revealed an inverse relationship (Fig. 3a–g) between the number of astrocytes with reduced mutant SOD1 (Cre+) and activated microglia (correlation coefficient: r = −0.868, p<0.001), despite comparable astrocytic activation. Thus, microglial activation was most prominent in areas with the highest mutant SOD1-expressing astrocyte concentration.
Figure 3. Mutant-expressing astrocytes enhance microglial activation and induction of iNOS.

(a–f) Images of (a, d) β-galactosidase, (b, e) Mac2, and (c, f) GFAP staining from a (a–c) left and (d–f) right lumbar spinal cord section from a 12-month-old LoxSOD1G37R/GFAP-Cre+ mouse. (a, d) GFAP-Cre+ astrocytes are marked by β-galactosidase. (g) Inverted correlation between the number of Cre-positive astrocytes and Mac2-positive microglia in LoxSOD1G37R/GFAP-Cre+ mice lumbar spinal cord sections. Correlation coefficient r= −0.868 (p<0.001). (h–m) Lumbar spinal cord sections from (h–j) LoxSOD1G37R and (k–m) LoxSOD1G37R/GFAP-Cre+ mice at early disease stage immunostained with antibodies to (h, k) iNOS, (i, l) Mac2, and (j, m) GFAP. (n) Quantification of iNOS-positive cells within anterior horn from lumbar spinal cord of symptomatic LoxSOD1G37R mice. Averaged percent of iNOS+/Mac2+ (red), iNOS+/GFAP+ (blue), and iNOS+/other cell type (black) per total iNOS+ cells was plotted. (o–r) Magnified images of anterior horn from lumbar spinal cord of symptomatic LoxSOD1G37R mice stained with (o) iNOS, (p) Mac2, and (q) GFAP. (r) Merged image illustrates that iNOS positive cells are Mac2-positive microglia.
Elevated production of nitric oxide by upregulated inducible nitric oxide synthase (iNOS) synthesis has been reported in mutant SOD1 mice10, albeit deletion of the iNOS gene has modest11 or no12 effect on SOD1-mediated disease. It is not known in which glial cells this nitric oxide is produced in in vivo models of ALS, although both microglia and astrocytes have an ability to produce it when stimulated in vitro13. Triple staining of lumbar spinal cord sections with iNOS, Mac2, and GFAP antibodies revealed that almost all iNOS-positive cells were Mac2-positive microglia (Fig. 3n–r; Supplementary Fig. 4), indicating that activated microglia are the primary cell type producing nitric oxide in this SOD1 mouse model. Diminishing mutant synthesis within astrocytes inhibited iNOS induction in disease matched, symptomatic SOD1 mice (Fig. 3h,k), in accord with significant inhibition of microglial activation (Fig. 3i,l).
A role of astrocytes in inherited ALS has been previously raised in several contexts. Mutant expressing astrocytes produce and release one or more as yet uncharacterized components that can accelerate motor neuron death in vitro6, 7. Focal loss of the astrocytic EAAT2 glutamate transporter within affected regions14 (Supplementary Fig. 5) and the failure to increase glutamate uptake in SOD1G93A astrocytes in vitro15 support glutamate dependent excitotoxicity as a component of disease. Nevertheless, diminished mutant SOD1 synthesis within most astrocytes did not affect disease dependent loss of EAAT2 from those astrocytes (Supplementary Fig. 5), demonstrating that reduction in glutamate transport reflects non-cell autonomous damage to astrocytes, in part, from mutant SOD1 synthesized by other cells. Our use of selective gene excision has now demonstrated that mutant SOD1 damage within both microglia3 and astrocytes (Fig. 1g–j), accelerates later disease progression, without affecting initiation of motor neuron degeneration and phenotypic disease onset. Discovery that damage within astrocytes determines the timing of microglial activation and infiltration provides a further insight that beyond any direct effect of mutant astrocytes on motor neurons such astrocytes amplify an inflammatory response from microglia (through production of nitric oxide and toxic cytokines), leading to further damage to the motor neurons and accelerated disease progression through a non-cell autonomous mechanism (Supplementary Fig. 6). These findings validate therapies, including astrocytic stem cell replacement approaches, to slow disease progression in ALS by supplementing healthy astrocytes or modulating toxicity within astrocytes to control an inflammatory response of microglia.
Supplementary Material
Acknowledgement
This work was supported by NIH Grant (NS 27036) and a grant from the Packard ALS Center at Johns Hopkins (to D.W.C.), as well as MDA Developmental Grant, Uehara Memorial Foundation, Nakabayashi Trust for ALS Research, and Grant-in-aid for Scientific Research (19591021) and on Priority Area (19044048) from MEXT, Japan (to K.Y.). Salary support for D.W.C is provided by the Ludwig Institute for Cancer Research. S.B. is a recipient of a Fondation pour la Recherche Medical fellowship, an INSERM fellowship, and a MDA Developmental Grant.
References
- 1.Pasinelli P, Brown RH. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 2.Boillee S, Vande Velde C, Cleveland DW. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 3.Boillee S, et al. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 4.Clement AM, et al. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- 5.Beers DR, et al. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagai M, et al. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bajenaru ML, et al. Molecular and cellular biology. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fraser MM, et al. Cancer research. 2004;64:7773–7779. doi: 10.1158/0008-5472.CAN-04-2487. [DOI] [PubMed] [Google Scholar]
- 10.Almer G, Vukosavic S, Romero N, Przedborski S. J Neurochem. 1999;72:2415–2425. doi: 10.1046/j.1471-4159.1999.0722415.x. [DOI] [PubMed] [Google Scholar]
- 11.Martin LJ, et al. The Journal of comparative neurology. 2007;500:20–46. doi: 10.1002/cne.21160. [DOI] [PubMed] [Google Scholar]
- 12.Son M, Fathallah-Shaykh HM, Elliott JL. Ann Neurol. 2001;50:273. doi: 10.1002/ana.1104. [DOI] [PubMed] [Google Scholar]
- 13.Barbeito LH, et al. Brain research. 2004;47:263–274. doi: 10.1016/j.brainresrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Howland DS, et al. Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vermeiren C, et al. J Neurochem. 2006;96:719–731. doi: 10.1111/j.1471-4159.2005.03577.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.