

Abstract
Myostatin (MSTN) is a member of the transforming growth factor-β superfamily of cytokines and is a negative regulator of skeletal muscle mass. Compared with MSTN+/+ mice, the extensor digitorum longus muscles of MSTN−/− mice exhibit hypertrophy, hyperplasia, and greater maximum isometric force production (Fo), but decreased specific maximum isometric force (sFo; Fo normalized by muscle cross-sectional area). The reason for the reduction in sFo was not known. Studies in myotubes indicate that inhibiting myostatin may increase muscle mass by decreasing the expression of the E3 ubiquitin ligase atrogin-1, which could impact the force-generating capacity and size of muscle fibers. To gain a greater understanding of the influence of myostatin on muscle contractility, we determined the impact of myostatin deficiency on the contractility of permeabilized muscle fibers and on the levels of atrogin-1 and ubiquitinated myosin heavy chain in whole muscle. We hypothesized that single fibers from MSTN−/− mice have a greater Fo, but no difference in sFo, and a decrease in atrogin-1 and ubiquitin-tagged myosin heavy chain levels. The results indicated that fibers from MSTN−/− mice have a greater cross-sectional area, but do not have a greater Fo and have a sFo that is significantly lower than fibers from MSTN+/+ mice. The extensor digitorum longus muscles from MSTN−/− mice also have reduced levels of atrogin-1 and ubiquitinated myosin heavy chain. These findings suggest that myostatin inhibition in otherwise healthy muscle increases the size of muscle fibers and decreases atrogin-1 levels, but does not increase the force production of individual muscle fibers.
Keywords: growth differentiating factor-8, permeabilized muscle fiber contractility, atrogin-1, muscle atrophy F-box
myostatin (growth differentiating factor-8) is a member of the transforming growth factor-β (TGF-β) superfamily of cytokines and is a negative regulator of skeletal muscle mass. Both targeted inhibition of myostatin and naturally occurring loss-of-function mutations in the myostatin gene result in an up to twofold increase in skeletal muscle mass (2, 9, 18, 20, 35). Conversely, treatment with recombinant myostatin results in cachexia and muscle atrophy (38). Due to the profound increase in skeletal muscle mass that occurs as a result of the inhibition of myostatin, much interest has focused on myostatin inhibition for the treatment of muscle injuries and muscle wasting diseases.
While the inhibition of myostatin results in a clear muscle mass phenotype, arguably with as great an impact as any other single growth factor, the full range of consequences of myostatin deficiency on muscle contractility is not known. Our laboratory previously showed that, compared with wild-type (MSTN+/+) mice, extensor digitorum longus (EDL) muscles of MSTN−/− mice displayed a 66% greater mass, a 61% increase in muscle fibers, 63% larger total muscle cross-sectional area (CSA), and a 23% increase in muscle fiber CSA (20). Consistent with the greater muscle mass, fiber number, and CSA, maximum isometric force (Fo) was 34% greater for EDL muscles of MSTN−/− mice compared with MSTN+/+ mice, but the specific Fo (sFo; Fo normalized by CSA) of MSTN−/− mice was 18% lower (20). The reason for this reduction in sFo was not known. Furthermore, when EDL muscles were subjected to lengthening contractions, those from MSTN+/+ mice had a 30% decrease in Fo, whereas those from MSTN−/− mice had a 45% decrease in Fo compared with preinjury values (20). The basis for the greater susceptibility to contraction-induced injury was not known, but we proposed this likely came about due to a dramatic increase in the stiffness of the tendons from MSTN−/− mice (19).
Although the role of myostatin in regulating muscle mass is well established, the molecular mechanisms mediating the response are not fully understood. Myostatin binds to the type IIB and type IB activin receptors and activates the intracellular p38 MAPK and Smad2/3 signal transduction pathways (1, 19, 25, 27). In cultured myotubes treated with myostatin, activated Smad2 inhibited Akt and p70S6K activation (32), and treatment of mice with an antibody against myostatin increased the activation of p70S6K and the rate of myofibrillar protein synthesis (36). While these effects on the Akt-p70S6K pathway suggest that myostatin functions, at least in part, to decrease protein synthesis, myostatin may also control proteolysis in skeletal muscle. The ubiquitin-proteasome system is important in regulating skeletal muscle mass and protein turnover (28). Atrogin-1 (muscle atrophy F-box) is an E3 ubiquitin ligase expressed in skeletal muscle that directs the polyubiquitination of proteins to target them for breakdown by the 26S proteasome (3, 11). Treatment of C2C12 myotubes with myostatin decreased the diameter of the myotubes, increased atrogin-1 gene expression and protein levels, and increased protein ubiquitination (17). Similarly, myostatin treatment of human myotubes caused atrophy, but atrogin-1 levels did not increase (32). In whole animal studies, young MSTN−/− mice had increased atrogin-1 expression in quadriceps muscles (22), but in a Cre-loxP model of postnatal inactivation of myostatin, atrogin-1 transcript levels in gastrocnemius muscles were not changed by myostatin knockdown (37).
To gain a greater understanding of the mechanisms of action of myostatin and its influence on muscle functions, we determined the impact of myostatin deficiency on the contractile properties of permeabilized single-muscle fibers. This approach allowed us to precisely measure the contractile properties of muscle fibers without the confounding influence of pennation angle and series elastic elements (the aponeurosis and tendon). Additionally, to gain a greater understanding of the mechanisms behind the increase in muscle mass that occurs following myostatin inhibition, we measured the expression of atrogin-1 and the content of ubiquitinated myosin heavy chain in whole muscle tissue. We hypothesized that, compared with MSTN+/+ mice, single fibers from MSTN−/− mice have a greater Fo, but no difference in sFo or power production, and that whole muscles from MSTN−/− mice have decreased atrogin-1 gene expression and protein content and a reduction in ubiquitinated myosin heavy chain protein.
MATERIALS AND METHODS
Animals.
All experiments were conducted in accordance with the guidelines of the University of Michigan Committee on the Use and Care of Animals, which approved this study. Mice were housed in specific-pathogen-free conditions and were provided food and water ad libitum. The MSTN−/− mice are of a C57Bl/6 background and were a kind gift of Dr. Se-Jin Lee. The null MSTN allele was generated by replacing a portion of the third exon of the MSTN gene that encodes the COOH-terminal region of the mature myostatin protein with a neo cassette (18). The genotype of mice was determined by PCR-based analysis of DNA samples obtained via tail biopsy, as previously described (20). Male mice aged 5–6 mo were used in this study. EDL muscles were carefully removed from mice that were deeply anesthetized with Avertin.
Muscle fiber isolation and storage.
EDL muscles used for muscle fiber contractility were immediately placed into cold skinning solution. Fiber bundles ∼4–6 mm in length and 0.5 mm in diameter were dissected from the muscle samples. Following dissection, bundles were immersed for 30 min in skinning solution with 0.5% Brij 58 and then placed in storage solution and maintained for 24 h at 4°C, followed by storage at −20°C for 1–3 mo before use. On the day of an experiment, fiber bundles were removed from storage solution and placed in relaxing solution on ice. Single fibers were pulled from the bundle with fine mirror-finished forceps and transferred to an experimental chamber containing relaxing solution maintained at 15°C.
Single-fiber contractility.
Single-fiber contractility experiments were performed as modified from Panchangam et al. (23) and Rader et al. (26). Force responses and motor position were acquired at a sampling rate of 5 kHz through a 16-bit A-D board (National Instruments, NI-6052) and displayed and stored on a personal computer using a custom-designed LabVIEW program (National Instruments). The position of the motor was updated at a rate of 10 kHz by the LabVIEW program via a D-A channel on the acquisition board.
One end of the fiber was secured to a force transducer (Aurora Scientific, model 403A) using two ties of 10–0 monofilament nylon suture. The other end of the fiber was attached in a similar manner to the lever arm of a servomotor (Aurora Scientific, model 322C). The solution-changing system (Aurora Scientific, model 802A) consisted of six separate glass-bottom chambers machined into a moveable, temperature-controlled stainless-steel plate. Movement of the plate with respect to the fiber was achieved by remote-control of two stepper motors: one to lower and raise the chamber array, and the other to translate the plate to a new chamber position. The length of the fiber was adjusted to obtain a sarcomere length of 2.5 μm, determined by projecting a laser diffraction pattern produced by the fiber onto a calibrated target screen. Fiber length (Lf) was then measured by first aligning the innermost tie at one end of the fiber with the crosshairs of a microscope eyepiece graticule, then translating the entire apparatus with respect to the microscope using a micrometer drive with digital readout, until the innermost tie at the other end of the fiber was aligned with the crosshairs.
Fiber CSA was estimated with fibers held at Lf using fiber width and depth measurements from high-magnification digital images of both top and side views of the fiber. Side views were obtained using a prism embedded in the side of the chamber. Five width-depth measurement pairs were obtained at 100-μm intervals along the midsection of the fiber. Fiber CSA was calculated for each width-depth pair, assuming an elliptical cross section, and overall CSA was estimated by averaging the five individual areas.
Relaxed single fibers were activated by first immersing them in a chamber containing a low-Ca2+ concentration ([Ca2+]) preactivating solution for 3 min and then immersing them in a chamber containing high-[Ca2+] activating solution (pCa ∼4.5) to elicit Fo. The preactivating solution was weakly buffered for Ca2+, resulting in very rapid activation and force development upon introduction of the activating solution (21). sFo was calculated by dividing Fo by CSA.
Susceptibility to contraction-induced injury was assessed by applying a single stretch to a fully activated fiber (Fig. 1). The stretch was equivalent in amplitude to 30% of Lf and was applied at a constant velocity of 0.5 Lf × s−1. Fibers were immediately returned to their original length at 0.5 Lf × s−1 and allowed to regenerate force until a new steady-state level was reached. The difference between the prestretch and poststretch steady-state forces was used to calculate force deficit as a percentage of prestretch force (Fig. 1).
Fig. 1.
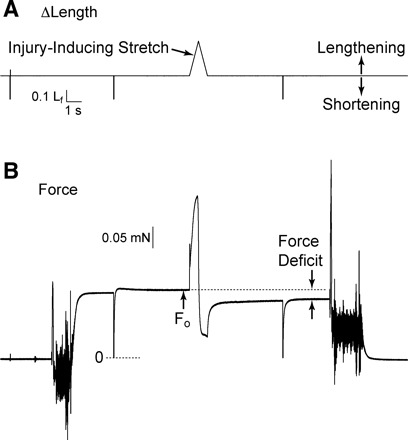
Determination of fiber susceptibility to injury caused by a lengthening contraction. A: fiber length (Lf) perturbations applied through control of servomotor position. B: force responses corresponding to the length perturbations. A lengthening movement of 30% of Lf was applied at a constant velocity of 0.5 Lf × s−1. Following the lengthening contraction, the fiber was immediately returned to original length at the same velocity. The large, rapid shortening movements evident in the records were sufficient in size to slacken the fiber and serve to indicate the zero force level in the force record. Fibers were allowed to remain slack for 30 ms before being rapidly returned to original length. Note the sustained reduction in force following stretch of the fully activated fiber. Δ, Change; Fo, maximum isometric force.
Force-velocity characteristics were evaluated by applying a series of step-ramp shortening movements to the fully activated fiber. The shortening movements were initiated from a length of Lf + 10% of Lf and had a total amplitude of 20% of Lf. The step amplitude was 4–5% of Lf, and its purpose was to discharge the strained series elasticity before the ramp, thereby reducing force to zero and decreasing the time required for the tension maintained during the ramp to reach a quasi-steady state. Reduction of active force to zero in response to the 4–5% step shortening also indicated that fiber end compliance was comparable to that reported in fibers with ends fixed using glutaraldehyde (8). Immediately following each ramp, an additional shortening step equivalent to 10% of Lf was applied to the fiber, followed 10 ms later by a step return to the original length. The final shortening step slackened the fiber briefly, and its purposes were to 1) indicate the location of the force baseline in the experimental recording; 2) ensure that the subsequent rapid reextension of the fully activated fiber to original length did not damage the fiber; and 3) maintain the homogeneity of the striation spacing by being functionally equivalent, when coupled with the reextension, to the rapid shortening, reextension cycle described by Brenner (5). After force regeneration was complete following the return to original length, the next step ramp was applied. Ramps were administered in sequence from fast to slow. The force maintained during shortening was measured at the time that the fiber had shortened by 0.1 Lf, and, consequently, its length was passing through Lf. The steady-state force that immediately preceded the step-ramp maneuver was taken as an index of the fiber's current Fo capability and the “F/Fo” terms in Eqs. 1 and 2 refer to the ratio of the force measured during shortening to that indicator of current isometric capability.
| (1) |
| (2) |
All forces were measured relative to the baseline revealed by the postramp step. This procedure resulted in 10 data points to which a rectangular hyperbola (Eq. 1) was fitted, where V is velocity of shortening, F′o is the intercept with the force axis, and a and b are the force and velocity asymptotes, respectively (12). The intersection of the fitted curve with the velocity axis was defined as Vmax (Lf × s−1), where Vmax is maximum velocity of shortening. The maximum power-generating capacity (Pmax, the peak of the force-power curve) was calculated from the parameters of the fitted curve according to Eq. 2 and then divided by fiber volume (Lf × CSA) to obtain normalized Pmax (W × l−1).
Maximum calcium-activated force measurements were made on N = 37 fibers from MSTN+/+ mice and 36 fibers from MSTN−/− mice. Of those fibers, the force-velocity analysis was performed on 11 from each experimental group, and the lengthening contractions were performed on 10 from each group. No individual fiber was subjected to both the force-velocity analysis and lengthening contractions.
Single-fiber solutions.
The skinning solution was composed of the following (in mM): 125 potassium propionate, 20 imidazole, 5 ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 2 MgCl2, and 2 ATP, pH 7.0. The composition of the storage solution was identical to that of skinning solution, with the exception that glycerol was substituted for 50% of the water volume. The relaxing solution (pCa ∼9.0) was composed of the following (in mM): 90 N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 10.3 Mg (total), 1.0 Mg2+, 50 EGTA, 8.0 ATP, 10.0 CrP, 1.0 NaN3, 36 Na (total), and 125 K (total), pH 7.1. Preactivating solution (pCa ∼8.0) was as follows (in mM): 90 HEPES, 8.50 Mg (total), 1.0 Mg2+, 0.10 EGTA, 50 1,6-diaminohexane-N,N,N′,N′-tetraacetic acid, 8.0 ATP, 10.0 CrP, 1.0 NaN3, 36 Na (total), and 125 K (total), pH 7.1. The activating solution (pCa ∼4.5) was as follows (in mM): 90 HEPES, 8.12 Mg (total), 1.0 Mg2+, 50 EGTA, 50 Ca2+ (total), 8.0 ATP, 10.0 CrP, 1.0 NaN3, 36 Na (total), and 125 K (total), pH 7.1. Potassium propionate was obtained from TCI America, and all other compounds were obtained from Sigma.
SDS-PAGE and immunoblot.
EDL muscles were homogenized in Laemmli's sample buffer with 1:20 β-mercaptoethanol, 1:20 protease inhibitor cocktail (Sigma), and 1:40 phosphatase inhibitor cocktail (Sigma) and then placed in boiling water for 5 min. Protein concentration of the samples was determined using an RC DC Protein Assay (Bio-Rad). Equal amounts of protein (10 μg) were loaded into 4% stacking, 7.5% resolving polyacrylamide gels and subjected to electrophoresis. Voltage was held at 80 V until samples entered the stacking gel, and voltage was subsequently increased to 150 V for the remainder of the run. To detect total myosin heavy chain, gels were stained with Coomassie brilliant blue (Bio-Rad). For immunoblots, proteins were transferred from gels to membranes at 100 V for 1 h and stained with Ponceau S to verify equal protein transfer. For ubiquitin immunoblots, 0.45-μm nitrocellulose membranes were blocked using casein (Vector Laboratories) and incubated with an horseradish peroxidase-conjugated anti-ubiquitin antibody (Santa Cruz Biotechnology). For atrogin-1 immunoblots, polyvinylidene difluoride membranes were blocked with 1% powdered milk and incubated with a rabbit anti-atrogin-1 antibody (ECM Biosciences) and horseradish peroxidase-conjugated goat anti-rabbit antibody (Santa Cruz Biotechnology). Membranes were developed with SuperSignal West Dura enhanced chemiluminescent reagents (Pierce Biotechnology) and visualized using a FluorChem chemiluminescent documentation system (Alpha Innotech).
RT-quantitative PCR.
RNA was isolated from samples using an RNeasy Fibrous Tissue kit (Qiagen) and treated with DNase I. Poly(A) mRNA was reverse transcribed using an Omniscript RT system (Qiagen) and oligo(dT)15 primers. cDNA was amplified using primers for atrogin-1 (forward: 5′-ATTCTACACTGGCAGCAGCA-3′; reverse: 5′-TGTAAGCACACAGGCAGGTC-3′) and β2-microglobulin (forward: 5′-ATGGGAAGCCGAACATACTG-3′; reverse: 5′-CAGTCTCAGTGGGGGTGAAT-3′) using a SYBR Green I PCR system (Qiagen) with Uracil DNA glycosylase (Invitrogen) in an Opticon 2 real-time thermal cycler (Bio-Rad). Quantitative PCR (qPCR) reactions were conducted in quadruplicate for each sample. Cycle threshold [C(t)] values for atrogin-1 were normalized to β2-microglobulin using the 2−ΔΔC(t) method (15). β2-Microglobulin was selected as a normalizing gene based on its stable expression in skeletal muscle tissue (16) and because of its stable expression relative to input RNA in our samples. The presence of single amplicons from qPCR reactions was verified by melting curve analysis, as well as electrophoresis using a 2% agarose gel. Genomic DNA contamination was not detected in qPCR reactions.
Statistical analysis.
Results are presented as means ± SE. KaleidaGraph 4.02 software was used to conduct statistical tests. Differences between MSTN+/+ and MSTN−/− mice were tested with Student's t-test with α = 0.05.
RESULTS
Single-fiber contractility.
The size and contractile properties data for single permeabilized fibers from MSTN+/+ and MSTN−/− mice are shown in Fig. 2. As measured from high-magnification digital images of both top and side views of the fiber, the CSA of permeabilized fibers from MSTN−/− mice was 21% larger than those from MSTN+/+ mice. Despite having a greater CSA, there was no difference in Fo between fibers from MSTN+/+ and MSTN−/− mice. Consequently, fibers from muscles of MSTN−/− mice had sFo values that were 21% lower than those from MSTN+/+. Following a lengthening contraction designed to cause contraction-induced injury to myofibrils, there was no difference in the postinjury Fo values between fibers from MSTN+/+ and MSTN−/− mice.
Fig. 2.
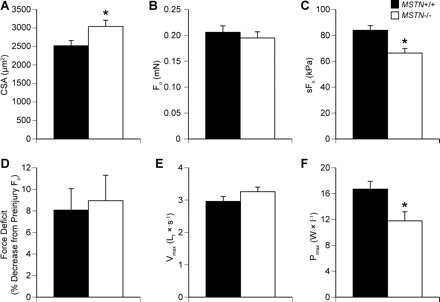
Contractile and morphological properties of permeabilized extensor digitorum longus (EDL) muscle fibers from myostatin MSTN+/+ and MSTN−/− mice. A: cross-sectional area (CSA) (MSTN+/+ N = 37; MSTN−/− N = 36). B: Fo (MSTN+/+ N = 37; MSTN−/− N = 36). C: specific Fo (sFo) (MSTN+/+ N = 37; MSTN−/− N = 36). D: decrease in Fo production following a 30% lengthening contraction (MSTN+/+ N = 10; MSTN−/− N = 10). E: maximum shortening velocity (Vmax) (MSTN+/+ N = 11; MSTN−/− N = 11). F: maximum power production (Pmax) (MSTN+/+ N = 11; MSTN−/− N = 11). Values are means ± SE. *Significantly different from MSTN+/+ (P < 0.05).
The force-velocity relationship and power-generating capacity of permeabilized fibers was also measured. There was no difference in the Vmax between MSTN+/+ and MSTN−/− mice (Fig. 2E). The force-velocity relationship (Fig. 3A) of fibers from MSTN−/− mice was virtually identical to that of MSTN+/+ mice. In contrast to the force-velocity relationship, the normalized power generated by fibers from the MSTN−/− mice was significantly lower than that of fibers from MSTN+/+ mice across a wide range of shortening velocities (Fig. 3B), a direct consequence of the reduced sFo observed in the MSTN−/− fibers.
Fig. 3.
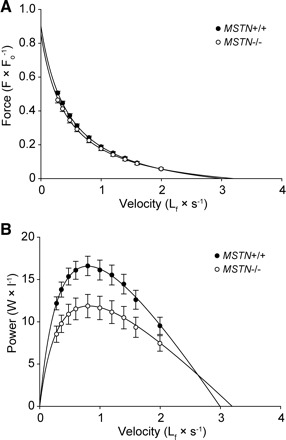
Force-velocity (A) and power-velocity (B) curves for EDL muscle fibers from MSTN+/+ and MSTN−/− mice. Note that the force-velocity relationship of fibers from MSTN−/− mice is not different from that of fibers from MSTN+/+ mice when the force axis is normalized by Fo, suggesting that cross-bridge kinetics are unchanged in the MSTN−/− phenotype. Power (the product of force and velocity), normalized by fiber volume and plotted as a function of shortening velocity, reveals that a reduction in normalized power is another consequence of reduced sFo in the MSTN−/− fibers. Each point represents mean ± SE; N = 11 fibers for each genotype.
Atrogin-1 and ubiquitinated myosin heavy chain content.
As myostatin was previously shown to induce the expression of atrogin-1 in C2C12 myotubes (17), and an increase in CSA without an associated increase in the ability to generate additional force could arise due to an accumulation of misfolded or damaged proteins that would otherwise be degraded by the ubiquitin-proteasome system, we measured the levels of atrogin-1 and ubiquitinated myosin heavy chain in EDL muscles from MSTN+/+ and MSTN−/− mice. Compared with MSTN+/+ mice, atrogin-1 protein (Fig. 4A) and gene expression (Fig. 4B) levels were lower in MSTN−/− mice. EDL muscles of MSTN−/− mice also displayed decreased levels of ubiquitinated myosin heavy chain compared with the levels observed for MSTN+/+ mice (Fig. 4A).
Fig. 4.
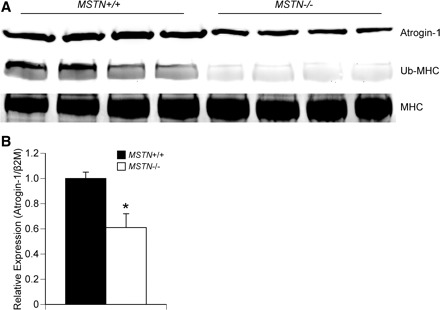
A: immunoblot showing that, compared with MSTN+/+ mice, EDL muscles from MSTN−/− mice have a decrease in atrogin-1 and ubiquitinated myosin heavy chain (Ub-MHC). Total myosin heavy chain (MHC) is shown as a loading control. N = 4 mice from each genotype. B: gene expression data showing that, compared with MSTN+/+ mice, EDL muscles from MSTN−/− mice have a decrease in atrogin-1 expression. N = 4 mice from each genotype. Values are means ± SE. *Significantly different from MSTN+/+ (P < 0.05).
DISCUSSION
The results of this study provide new insight into the role of myostatin in the determination of skeletal muscle contractility and morphology. In agreement with previous histology data from whole muscles (20), the CSA of permeabilized muscle fibers from MSTN−/− mice was greater than that from MSTN+/+ mice. Muscle fibers swell as a consequence of the permeabilization process (10), but the relative increase in CSA for permeabilized EDL fibers from MSTN−/− mice compared with MSTN+/+ mice was similar to that observed in muscle histology data (20), suggesting that the size difference is not caused by differential swelling of MSTN−/− and MSTN+/+ fibers during the permeabilization process. An increase in CSA is typically accompanied by an increase in Fo, reflecting an increased number of myofibrils acting in parallel. Despite having a greater CSA, the muscle fibers from MSTN−/− mice did not have higher Fo values than fibers from MSTN+/+ mice, suggesting that either the fibers from the MSTN−/− mice were accumulating noncontractile material, or individual myofibrils were generating lower forces.
In our laboratory's previous work on the contractility of whole muscles from MSTN+/+ and MSTN−/− mice, we showed that EDL muscles from MSTN−/− mice had a greater Fo, but lower sFo, than MSTN+/+ mice (20). Amthor and colleagues (2) also reported a reduction in sFo in EDL muscles of MSTN−/− mice, although no difference in Fo was observed. We hypothesized that the increase in whole muscle Fo for EDL muscles from MSTN−/− mice came about due to both the increase in muscle fiber CSA and the dramatic increase in the number of fibers present in the muscle (20). Based on the present observation of no difference in Fo between fibers from MSTN+/+ and MSTN−/− mice, the increase in whole muscle Fo in MSTN−/− mice appears to be due exclusively to hyperplasia, with no contributions from an increase in force production of individual muscle fibers. We also hypothesized that the decrease in whole muscle sFo in MSTN−/− mice compared with MSTN+/+ mice was due to an increase in the angle of pennation of fibers within the muscles of MSTN−/− mice (20). While large angles of pennation clearly result in lower sFo values for whole muscles, the small reduction in sFo attributable to altered pennation angle in EDL muscles from MSTN−/− mice (20) is minor compared with the reduction attributable to the lower sFo of individual muscle fibers.
Our laboratory has previously shown that, when subjected to lengthening contractions, the EDL muscles of MSTN−/− mice exhibit much greater force deficits than those from MSTN+/+ mice (20). We hypothesized at the time that that the greater force deficit for muscles from MSTN−/− mice was due to an increase in the stiffness of the aponeurosis and tendon (20), as a stiffer series elastic component results in individual muscle fibers undergoing greater strains relative to the entire muscle-tendon unit during a lengthening contraction. This hypothesis was supported by our laboratory's subsequent findings that the tendons of MSTN−/− mice are much stiffer than those of MSTN+/+ mice (19). In the present study, compared with MSTN+/+ mice, there was no difference in the force deficits of isolated single-muscle fibers from MSTN−/− mice following lengthening contractions. Since the aponeurosis, tendon, and components of the lateral force transmission pathways are eliminated in this system, the present findings provide further support for the view that the greater susceptibility to contraction-induced injury in whole muscles of MSTN−/− mice does not arise due to an inherent change within the myofibrils.
Activation of the canonical TGF-β pathway by other ligands might also play a role in the determination of muscle contractility, especially the Smad2 and Smad3 portion of the pathway. Treating MSTN−/− mice with a soluble form of type IIB activin receptor resulted in a further increase in muscle mass than what was brought about by the deficiency of myostatin alone (14). In mice that overexpress c-ski, a protooncogene that inhibits the ability of Smad2 and Smad3 to promote gene expression by sequestering these transcription factors to TGF-β inhibitory elements (31), fibers from the EDL muscle had an increased CSA, with no change in Fo and a subsequent decrease in sFo (7). Bruusgaard and colleagues (6) hypothesized that the c-ski-mediated increase in CSA and decrease in sFo was due to accumulation of nonfunctional proteins that had not yet been degraded. For iliofibularis muscles cultured from frogs, incubation of muscles with SB-431542, a compound that inhibits the receptor-mediated activation of Smad2 and Smad3, resulted in muscle hypertrophy, along with a decrease in sFo (34). These studies of components of the TGF-β signaling pathway are consistent with the findings of our present study.
The published data on the molecular mechanisms behind myostatin-mediated skeletal muscle atrophy are somewhat conflicting. While myostatin appears to inhibit protein synthesis, the ability of myostatin to regulate atrogin-1-mediated muscle atrophy is controversial. Our present work demonstrating that the absence of myostatin in muscles of MSTN−/− mice resulted in lower atrogin-1 gene and protein levels and lower levels of ubiquitinated myosin heavy chain is in agreement with McFarlane and colleagues (17), who showed that treatment of C2C12 myotubes with myostatin increased atrogin-1 transcript and protein levels and increased protein ubiquitination. In addition to studies in myotubes, our present work is in agreement with Sartori et al. (29), who demonstrated that transfection of a constitutively active form of the type I TGF-β receptor into mouse tibialis anterior muscle resulted in muscle fiber atrophy and a Smad3-dependent activation of the atrogin-1 promoter. Contrary to our findings and to those of McFarlane et al. (17) and Sartori et al. (29), Welle et al. (37) demonstrated that postnatal knockdown of myostatin did not change atrogin-1 expression, but Morissette and colleagues (22) showed an increase in atrogin-1 gene expression in MSTN−/− mice. Differences in our atrogin-1 gene expression data and those of Welle et al. (37) and Morrisette et al. (22) may be due to the choice of housekeeping gene. Our work supports the conclusions of McFarlane et al. (17) and Sartori et al. (29), that the increase in muscle mass resulting from the inhibition myostatin occurs, at least in part, by decreasing protein degradation. Amthor et al. (2) and Gentry et al. (9) reported finding tubular aggregates in some of fibers from the hindlimb muscles of MSTN−/− mice, and the accumulation of these tubular aggregates may be associated with inhibition of the normal proteolytic systems in muscle fibers (30). An accumulation of damaged or misfolded proteins that are not contributing to the force-generating capacity of muscle fibers due to a deficiency in the ubiquitin proteasome system is consistent with both our contractile data and molecular biology data from MSTN−/− mice. Further study is needed to understand the molecular mechanisms behind myostatin-mediated skeletal muscle atrophy.
As myostatin inhibition can result in a substantial increase in total muscle mass, there has been much interest in the development of therapeutic inhibitors of myostatin for the treatment of a wide variety of muscle-wasting diseases. While the results of the present study indicate that myostatin inhibition in healthy mice causes an increase muscle fiber size without increasing force production, this does not necessarily mean that therapeutic inhibition of myostatin cannot be beneficial in treating muscle-wasting diseases. Treatment of mdx mice, a murine model of Duchenne muscular dystrophy, with the propeptide of myostatin, increased both Fo and sFo of EDL muscles (4), but, in otherwise healthy muscle tissue, myostatin knockdown did not change whole muscle Fo or sFo (24). In a human clinical trial of a myostatin inhibitor, myostatin inhibition did not result in an improvement in whole muscle strength (33), but limited improvements in contractile properties of single fibers were observed (13). Obtaining sufficient quantities of single fibers from human muscle biopsy in patients with myopathies is challenging, and larger scale studies are necessary to evaluate the efficacy of myostatin inhibition in the treatment of muscle-wasting diseases. For injuries or diseases that involve an upregulation of atrogin-1 or other muscle atrophy genes, myostatin inhibition may help to reduce muscle atrophy and lessen strength loss; however, the inhibition of myostatin for purely ergogenic purposes in healthy individuals is not supported.
GRANTS
This study was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases Grants AR058920 and AR055624.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
REFERENCES
- 1. Allen DL, Unterman TG. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol 292: C188–C199, 2007 [DOI] [PubMed] [Google Scholar]
- 2. Amthor H, Macharia R, Navarrete R, Schuelke M, Brown SC, Otto A, Voit T, Muntoni F, Vrbóva G, Partridge T, Zammit P, Bunger L, Patel K. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc Natl Acad Sci USA 104: 1835–1840, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708, 2001 [DOI] [PubMed] [Google Scholar]
- 4. Bogdanovich S, Perkins KJ, Krag TOB, Whittemore LA, Khurana TS. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J 19: 543–549, 2005 [DOI] [PubMed] [Google Scholar]
- 5. Brenner B. Technique for stabilizing the striation pattern in maximally calcium-activated skinned rabbit psoas fibers. Biophys J 41: 99–102, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bruusgaard JC, Brack AS, Hughes SM, Gundersen K. Muscle hypertrophy induced by the Ski protein: cyto-architecture and ultrastructure. Acta Physiol Scand 185: 141–149, 2005 [DOI] [PubMed] [Google Scholar]
- 7. Chargé SBP, Brack AS, Hughes SM. Aging-related satellite cell differentiation defect occurs prematurely after Ski-induced muscle hypertrophy. Am J Physiol Cell Physiol 283: C1228–C1241, 2002 [DOI] [PubMed] [Google Scholar]
- 8. Chase PB, Denkinger TM, Kushmerick MJ. Effect of viscosity on mechanics of single, skinned fibers from rabbit psoas muscle. Biophys J 74: 1428–1438, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gentry BA, Ferreira JA, Phillips CL, Brown M. Hindlimb skeletal muscle function in myostatin-deficient mice. Muscle Nerve 43: 49–57, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Godt RE, Maughan DW. Swelling of skinned muscle fibers of the frog. Experimental observations. Biophys J 19: 103–116, 1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A 98: 14440–14445, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hill A. The heat of shortening and the dynamic constants of muscle. Proc R Soc Lond B Biol Sci 126: 136–195, 1938 [DOI] [PubMed] [Google Scholar]
- 13. Krivickas LS, Walsh R, Amato AA. Single muscle fiber contractile properties in adults with muscular dystrophy treated with MYO-029. Muscle Nerve 39: 3–9, 2009 [DOI] [PubMed] [Google Scholar]
- 14. Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad MEP, Tomkinson KN, Wright JF, Barker C, Ehrmantraut G, Holmstrom J, Trowell B, Gertz B, Jiang MS, Sebald SM, Matzuk M, Li E, Liang LF, Quattlebaum E, Stotish RL, Wolfman NM. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA 102: 18117–18122, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-delta delta C(T)] method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 16. Mahoney DJ, Carey K, Fu MH, Snow R, Cameron-Smith D, Parise G, Tarnopolsky MA. Real-time RT-PCR analysis of housekeeping genes in human skeletal muscle following acute exercise. Physiol Genomics 18: 226–231, 2004 [DOI] [PubMed] [Google Scholar]
- 17. McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, Smith H, Sharma M, Kambadur R. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J Cell Physiol 209: 501–514, 2006 [DOI] [PubMed] [Google Scholar]
- 18. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387: 83–90, 1997 [DOI] [PubMed] [Google Scholar]
- 19. Mendias CL, Bakhurin KI, Faulkner JA. Tendons of myostatin-deficient mice are small, brittle, and hypocellular. Proc Natl Acad Sci USA 105: 388–393, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mendias CL, Marcin JE, Calerdon DR, Faulkner JA. Contractile properties of EDL and soleus muscles of myostatin-deficient mice. J Appl Physiol 101: 898–905, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moisescu DG, Thieleczek R. Calcium and strontium concentration changes within skinned muscle preparations following a change in the external bathing solution. J Physiol 275: 241–262, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morissette MR, Cook SA, Buranasombati C, Rosenberg MA, Rosenzweig A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol 297: C1124–C1132, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Panchangam A, Claflin DR, Palmer ML, Faulkner JA. Magnitude of sarcomere extension correlates with initial sarcomere length during lengthening of activated single fibers from soleus muscle of rats. Biophys J 95: 1890–1901, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Personius KE, Jayaram A, Krull D, Brown R, Xu T, Han B, Burgess K, Storey C, Shah B, Tawil R, Welle S. Grip force, EDL contractile properties, and voluntary wheel running after postdevelopmental myostatin depletion in mice. J Appl Physiol 109: 886–894, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Philip B, Lu Z, Gao Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell Signal 17: 365–375, 2005 [DOI] [PubMed] [Google Scholar]
- 26. Rader EP, Cederna PS, Weinzweig J, Panter KE, Yu D, Buchman SR, Larkin LM, Faulkner JA. Contraction-induced injury to single permeabilized muscle fibers from normal and congenitally-clefted goat palates. Cleft Palate Craniofac J 44: 216–222, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol 23: 7230–7242, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 23: 160–170, 2008 [DOI] [PubMed] [Google Scholar]
- 29. Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, Sandri M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol 296: C1248–C1257, 2009 [DOI] [PubMed] [Google Scholar]
- 30. Sharma MC, Goebel HH. Protein aggregate myopathies. Neurol India 53: 273–279, 2005 [DOI] [PubMed] [Google Scholar]
- 31. Suzuki H, Yagi K, Kondo M, Kato M, Miyazono K, Miyazawa K. c-Ski inhibits the TGF-beta signaling pathway through stabilization of inactive Smad complexes on Smad-binding elements. Oncogene 23: 5068–5076, 2004 [DOI] [PubMed] [Google Scholar]
- 32. Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol 296: C1258–C1270, 2009 [DOI] [PubMed] [Google Scholar]
- 33. Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, Flanigan KM, Pestronk A, Tawil R, Wolfe GI, Eagle M, Florence JM, King WM, Pandya S, Straub V, Juneau P, Meyers K, Csimma C, Araujo T, Allen R, Parsons SA, Wozney JM, Lavallie ER, Mendell JR. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 63: 561–571, 2008 [DOI] [PubMed] [Google Scholar]
- 34. Watt KI, Jaspers RT, Atherton P, Smith K, Rennie MJ, Ratkevicius A, Wackerhage H. SB431542 treatment promotes the hypertrophy of skeletal muscle fibers but decreases specific force. Muscle Nerve 41: 624–629, 2010 [DOI] [PubMed] [Google Scholar]
- 35. Welle S, Bhatt K, Pinkert CA, Tawil R, Thornton CA. Muscle growth after postdevelopmental myostatin gene knockout. Am J Physiol Endocrinol Metab 292: E985–E991, 2007 [DOI] [PubMed] [Google Scholar]
- 36. Welle S, Burgess K, Mehta S. Stimulation of skeletal muscle myofibrillar protein synthesis, p70 S6 kinase phosphorylation, and ribosomal protein S6 phosphorylation by inhibition of myostatin in mature mice. Am J Physiol Endocrinol Metab 296: E567–E572, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Welle S, Burgess K, Thornton CA, Tawil R. Relation between extent of myostatin depletion and muscle growth in mature mice. Am J Physiol Endocrinol Metab 297: E935–E940, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ. Induction of cachexia in mice by systemically administered myostatin. Science 296: 1486–1488, 2002 [DOI] [PubMed] [Google Scholar]