

Summary
Deletions in mitochondrial DNA (mtDNA) have long been suspected to be involved in mammalian aging, but their role remains controversial. Recent research has demonstrated that relatively higher levels of mtDNA deletions correlate with premature aging in mtDNA mutator mice, which led to the conclusion that premature aging in these mice is driven by mtDNA deletions. However, it is reported here that the absolute level of deletions in mutator mice is quite low, especially when compared with the level of point mutations in these mice. It is thus argued that the available data are insufficient to conclude that mtDNA mutations drive premature aging in mtDNA mutator mice. It remains possible that clonal expansion of mtDNA deletions may result in sufficiently high levels to play a role in age-related dysfunction in some cells, but assessing this possibility will require studies of the distribution of these deletions among different cell types and in individual cells.
Introduction
Deletions in mitochondrial DNA (mtDNA) have long been suspected to be involved in mammalian aging. However, despite recent attempts to address this issue, the question remains open (Khrapko & Vijg, 2008). “mtDNA mutator” mice express a mitochondrial polymerase gamma lacking a proofreading 3′-exonuclease activity, causing greatly increased mtDNA mutation rates in association with various premature aging-like phenotypes (Zhang et al., 2000; Trifunovic et al., 2004; Kujoth et al., 2005; Bensch et al., 2007), and are thought to support the idea that mtDNA mutations may lead to mammalian aging (see, however, de Grey, 2004; Khrapko et al., 2006; Vermulst et al., 2007). In addition to significant loads of point mutations (up to 20–30 per mtDNA molecule) mutator mice also carry increased amounts of mtDNA deletions (Zhang et al., 2000; Trifunovic et al., 2004). In their recent article, Vermulst et al. (2008) for the first time reported the measurements of the relative amounts of mtDNA deletions in prematurely aging homozygous Polgamut/mut mutator mice in which both alleles of mtDNA polymerase gamma have been replaced by a proofreading-deficient variant. In these mutator mice, mtDNA mutations rapidly accumulated with age and by the end of their lifespan (about 15 months) the level of deletions was about 10 to 100 times higher (depending on the site assessed) than in age-matched wild-type controls. In the heterozygous Polga+/mut mice, which have lower levels of point mutations (about 4 per mtDNA) compared with the Polgamut/mut mice (though still increased relative to wild-type mice) there is no increase of mtDNA deletions. Interestingly, Polga+/mut mice do not age prematurely. Thus, the presence of mtDNA deletions correlates with premature aging. Based on these data, Vermulst et al. “identify mtDNA deletions as a driving force behind the premature aging phenotype of mitochondrial mutator mice.” They further suggest that since an increase in mtDNA deletions is associated exclusively with prematurely aging mice, then mtDNA deletions “seem to be the most important force behind the shortened lifespan of Polgamut/mut mice.” However, an increase of deletions in Polga mut/mut does not necessarily demonstrate a causal relationship, and may represent merely a correlation. More specifically, a concern is that the approach used to quantify deletions, the Random Mutation Capture, or RMC (Vermulst et al., 2008), does not estimate the absolute fractions of deletions in the mutator mouse tissues, but instead only measures the relative fractions of deletions in the Polgamut/mut tissue compared with control or Polga+/mut tissue. This tells us little about the potential impact of deletions on cellular physiology, because levels of deletions in control mice may be so low that even a many fold-increase may result in absolute levels that are low and therefore harmless. To address this limitation, we used the single molecule long range PCR approach (Kraytsberg & Khrapko, 2005) that estimates the absolute fraction of essentially all possible large mtDNA deletions.
Materials and methods
Tissue samples and DNA isolation
Polgamut/mut mutator mice tissues (15 months old) were provided by Drs. Marc Vermulst and Tomas Prolla. The mice were generated in Dr Tomas Prolla’s laboratory at the University of Wisconsin as described elsewhere (Kujoth et al., 2005). Tissues were snap frozen shortly upon dissection and stored at −80 °C. DNA was isolated following (Khrapko et al., 1999) by proteinase K/SDS lysis of the frozen tissue. About 10 mg of powdered frozen tissue was suspended in 0.2 mL of 10 mM EDTA, 0.5%SDS and 0.1 mg/mL proteinase K at 45 °C for 1 h. The lysate was diluted as needed (i.e. several orders of magnitude) with 10 mM Tris–HCl pH 8, 0.5 mM EDTA for smPCR and stored at −80 °C.
Estimation of the fraction of deletions by single molecule PCR (smPCR)
To estimate the fraction of mtDNA deletions in tissues of Polgamut/mut mice we used the long distance single molecule PCR (smPCR) approach (Kraytsberg & Khrapko, 2005). Briefly, DNA is diluted to the point (determined empirically) where there is about one amplifiable template DNA molecule per two PCR reactions, on average. PCR primers are arranged in such a way (one within the d-loop and another in the minor arch) that most types of mtDNA molecules with naturally occurring deletions, as well as the nondeleted wild-type mtDNA molecules would be amplified. Amplification of non-deleted molecules produces PCR fragments of about 14.5 kb in length, while deleted molecules produce shorter products (for example, a mtDNA with a 5 kb deletion would produce a 9.5 kb PCR product). Multiple PCR reactions were performed in parallel in multiwell PCR plates and PCR products were resolved by gel electrophoresis. Every reaction well initially contains one wild-type template, a deleted mtDNA, or neither (or, rarely, as a matter of chance, more than one molecule). Deleted mtDNA are readily distinguished by their smaller size (Fig. 1). Counting of PCR products of each type (wild-type vs. deletions) provides an estimate of the fraction of deleted mtDNA in the original DNA samples. To improve our ability to recognize a deleted smPCR product, we additionally digested PCR products with a set of restriction enzymes followed by a high-resolution gel separation to precisely determine the size of the deleted part. In this way, we were able to detect deleted mtDNA molecules with as few as 500 bp removed.
Fig. 1.
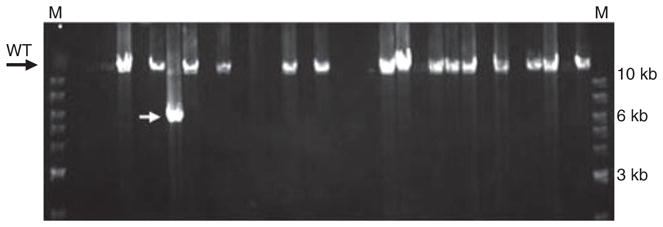
An output of a long range single molecule PCR of a Polgamut/mut sample visualized in 0.8% agarose gel. Each lane represents a single well of a smPCR reaction plate. Before PCR, each well contains either a normal length (wild-type) template molecule (resulting in a 14.5 kb product, as in lanes with DNA bands marked “wt”), or a molecule with deletion (lane with ~6 bk product marked with white arrow; deletions can be of various sizes), or no template at all (empty lanes). In this example, 16 reactions out of 32 yielded a PCR product, implying that there is about 0.7 templates per well on average (not 0.5 because some positive reactions may have contained two or more initial template molecules, which was accounted for quantitatively assuming Poisson distribution of templates). All positive reactions in this example with one exception contained nondeleted templates. One reaction contained a deletion (marked with an arrowhead). The entire study involved about 60 gels similar to this. M: marker lanes; a 1 kb DNA ladder.
Single molecule PCR
Nested smPCR was performed in two stages, in the volumes of 0.6 and 6 μL under mineral oil for the first and the second stage, respectively (reagents for the second stage was added to the same reaction well after completion of the first stage). Primers used were as follows: 2417F38/322R35 (first stage) and 2496F33/322R35 (second stage). In the name of a primer, the first number represents the’ position of the 5′-end of the primer, the letter F or R represents forward or reverse orientation with respect to conventional mtDNA sequence and the last number is the length. LA Taq (TaKaRa) thermostable DNA polymerase was used according to the manufacturer’s directions. There were 35 cycles in the first stage and 30 cycles in the second stage. Each PCR cycle was 20 s at 94 °C, 14.5 min at 68 °C.
Correction of mutant fractions
Uncorrected counting of deleted smPCR products, however, may result in significant overestimates of mutant fraction. This is because the probability that a single DNA template can be successfully replicated throughout its length by a thermostable DNA polymerase decreases with the length: longer molecules are more likely to carry impassable DNA lesions. This effect was first described and studied by Yakes and van Houten (Yakes & Van Houten, 1997). To account for this bias and therefore avoid overrepresentation of short PCR products, one needs to estimate the relative amplification probability of templates of different lengths. This was done by measuring the apparent number of copies in the same DNA sample using smPCR with fragments of different length. We determined that the probability of amplification of a 8-kb template is approximately five times higher, and the probability of amplification for 3-kb template is about 20-fold higher, than that for a full-length 14.5 kB template in the Polgamut/mut samples under investigation. We then binned all smPCR products shorter than 3 kb in length, and divided the count by 20. Similarly, we binned products between 3 and 8 kb and divided the count by 3. The count of deleted products between 8 kb and full-length was not corrected. As a result, our estimate of the relative number of deletions was an overestimate.
Results and discussion
We performed multiple smPCR reactions on DNA isolated from Polgamut/mut brain, duodenum and heart samples provided by Drs. Vermulst and Prolla. These samples included four from anterior frontal cortex, two from whole brain, two from duodenum and one from heart. The whole brain DNA samples were identical to those featured in a recent publication from Vermulst et al. (Vermulst et al., 2008). The results of smPCR are presented in Table 1. Note that the apparent sample-to-sample variability is due in part to the low numbers of deleted molecules. In summary, among over 1100 single molecules from brain samples we identified 27 molecules with large deletions, including 10 deleted PCR products over 8 kb in length (i.e. <6.5 kb deleted), 10 products from 8 to 3 kb, and 7 products shorter than 3 kb in length, which with correction (see Materials and Methods) for the length differences implies the presence of a deletion in about 1% of mtDNA molecules. This is likely an overestimation, because we used a conservative upper limit correction. Note that even without correction the overall fraction of deletions does not exceed a very modest fraction of 2.5%. There were no deletions among more than 320 amplified wild-type molecules from two duodenum DNA samples and among 144 molecules from a heart DNA sample from a Polgamut/mut mouse (all three DNA samples provided by Dr Vermulst). Apparently, levels of deletions in these tissues are even lower than those in the brain. These results are in qualitative agreement with the report by Kukat and Trifunovic, who also argued that the levels of mtDNA deletions in mtDNA mutator mice are low, though absolute levels of deletions were not determined (Kukat & Trifunovic, 2009).
Table 1.
Levels of mtDNA deletions in tissues of Polgamut/mut mice
| Tissue samples | Normal length molecules | Deletion molecules
|
Corrected mut. fraction | ||
|---|---|---|---|---|---|
| < 3 kb | 3–8 kb | > 8 kb | |||
| Whole brain | 296 | 1 | 1 | 3 | 0.011 |
| Whole brain | 187 | 1 | 0 | 0 | 0.0003 |
| Anterior frontal cortex | 169 | 2 | 2 | 2 | 0.016 |
| Anterior frontal cortex | 127 | 1 | 1 | 2 | 0.019 |
| Anterior frontal cortex | 135 | 2 | 6 | 1 | 0.023 |
| Anterior frontal cortex | 195 | 0 | 0 | 2 | 0.010 |
| All brain samples | 1109 | 7 | 10 | 10 | 0.013 |
| Duodenum | 201 | 0 | 0 | 0 | 0 |
| Duodenum | 120 | 0 | 0 | 0 | 0 |
| Heart | 144 | 0 | 0 | 0 | 0 |
Our estimates imply that in the homozygous Polgamut/mut mutator mouse, there are about 1000 point mutations for every one deleted mtDNA molecule. This is calculated based on our estimate of < 2% of deletions (i.e. < 0.02 deletions per genome) and 20–30 point mutations per mitochondrial genome. This represents about 800 additional point mutations in Polgamut/mut compared with Polga+/mut for every one additional deletion, since Polgamut/mut has been reported to contain about five times more point mutations compared with Polga+/mut (Vermulst et al., 2007).
To evaluate the role of mtDNA deletions in premature aging of the mutator mouse, one must consider the expected relative impact of deletions compared with point mutations, as well as absolute impact of deletions. Although not all point mutations are detrimental, a significant proportion of them, (D.M. Turnbull, unpublished data) are predicted to be just as detrimental as are mtDNA deletions. The huge overall excess of point mutations thus implies that if deletions were to play any role, they could do so only if they were concentrated to high mutant fractions in specific cells/cell types that represent a small proportion (no more than about 1%) of the tissue. Indeed, if deletions were distributed evenly across tissue, in each cell there would be 1 deletion per 1000 point mutations. It is highly unlikely that the presence of these occasional deletions would make any difference in the physiology of the cell. High concentration of mtDNA deletions in some but not other cells is usually achieved by clonal expansion of a single (or a few) initial deleted molecule in a cell. Thus to explore whether deletions present at low fraction may potentially be playing some role in the presence of an overwhelming excess of point mutations, it is necessary first to identify cell types that carry high fractions of clonally expanded mtDNA deletions. So far such cells have not been identified in mutator mice.
Another reason to expect that mtDNA deletions may cause premature aging only via clonal expansion to very high fractions in a small subpopulation of cells comes from the mito-mice, i.e. genetically engineered mice that bear various levels of a single type of mtDNA deletion throughout their bodies (Inoue et al., 2000). While mito-mice with a very high percentage of deletions (over ~70%) die early of renal failure, mito-mice with a lower percentage (~30% at the end of life) appear healthy and long-lived (Sato et al., 2005). In mito-mice, deletions appear to be relatively uniformly distributed among different tissues and among cells within tissue (Inoue et al., 2000). Thus, in a healthy mito-mouse with 30% deletions, essentially in every cell approximately 3 out of every 10 mtDNA molecules contains a deletion without causing any overt phenotype. This implies once again that only cells with high proportion of mtDNA deletions may potentially convey detrimental effects of increased deletions. Thus, in the mutator mouse, the search for such cells and evaluation of their functional significance for premature aging should precede conclusions about causative role of these deletions.
In addition to the mito-mouse, two other mouse models bear increased levels of mtDNA deletions. Unlike the mito-mouse, increased deletion levels in these other mouse models are caused not by the introduction of a single type of deletion into the zygote, but by increased generation of random deletions, i.e. in a manner more similar to accumulation of mtDNA deletions in normal aging (and the DNA mutator mouse). One of the models, the “twinkle mouse,” expresses a defective transgenic Twinkle, a putative helicase presumably involved in mtDNA replication and repair (Tyynismaa et al., 2005). The other model, the mito-Pst mouse, expresses mitochondrially-targeted restriction enzyme PstI, which generates double-stranded breaks in the mtDNA (Fukui & Moraes, 2008). Unlike in the mtDNA mutator mouse, increases in deletions in either twinkle or mito-Pst mice are not complicated by an increase of point mutations. These mouse models are promising with respect to exploring the role of mtDNA deletions in the aging process.
It has been proposed that premature aging of Polgamut/mut mice is caused by excessive cell death (Kujoth et al., 2005). Thus, one may suspect that the low fraction of deletions in premature aging mutator mice implies that cells bearing deletions have already died. While this scenario cannot be completely excluded, it is more important in the context of this paper to point out that there is no evidence to support it. Furthermore, such a scenario implies that deletions are much more efficient in causing cell death than the much more abundant point mutations found in cells that persist in the older polG mutator mice. In fact, at least some cell types as diverse as muscle fibers and kidney tubular epithelial cells appear capable of sustaining high levels of mtDNA deletions for a long time. In muscle, clonal expansions of deleted mtDNA may extend over a couple of millimeters of fiber length (Bua et al., 2006), which takes a long time to develop, so central parts of such fiber segments should remain viable for substantial time periods. In kidney, full turnover units of tubular epithelial cells appear to carry expanded mtDNA deletions (McKiernan et al., 2007), which implies that corresponding stem cells survive despite a high load of deletions.
The question whether mtDNA deletions cause premature aging in the mutator mouse is similar to the question whether mtDNA deletions cause normal aging in mice or in humans. Indeed, both in normal and premature aging, the fraction of deletions strongly correlates with the aging phenotype. Overall fractions of deletions in some cell types in elderly humans can reach levels that are similar to or even higher than those observed in the mtDNA mutator mouse. Unlike the mutator mouse, however, the distribution of deletions between tissues, cell types and individual cells in humans has been studied in substantial detail. In has been shown that deletions accumulate up to critical levels (i.e. levels that cause electron transport chain defects) in certain cells or cell types, for example, in pigmented neurons of the aged substantia nigra (Bender et al., 2006), (Kraytsberg et al., 2006)), or in segments of muscle fibers in both rodents (Herbst et al., 2007) and humans (Bua et al., 2006). Despite these advances, there is still no consensus on whether deletions drive normal aging (Khrapko & Vijg, 2008). Thus, it seems fair to postpone conclusions about the role of mtDNA deletions in mutator mice at least until more data are available about their detailed distribution. Interestingly, so far, our preliminary studies of mutator mice failed to discover any excess of mtDNA deletions in the putamen, an area that is particularly rich in cytochrome c oxidase-deficient cells in the mutator mice (M. Vermulst, pers. Comm.) and one that is traditionally considered enriched in mtDNA deletions in normal human aging. This observation may imply that the distribution of mtDNA deletions in mtDNA mutator mice is different from that in normal aging, which warrants even more caution in interpretation of the mutator mouse data.
In conclusion, though the possibility of a role for mtDNA deletions in premature aging of mtDNA mutator mice cannot be definitively excluded, an equally important point is that existing data are clearly not sufficient to infer that deletions drive premature aging. More work is needed to determine the contribution, if any, of mtDNA deletions to the phenotype in these mice. It remains possible that mtDNA deletions could play some role in normal aging or in degenerative diseases, but addressing that possibility will require detailed studies of the cellular distributions of mtDNA mutations and their physiological impact before drawing any conclusions about their significance.
Acknowledgments
We thank Drs. LA Loeb, TA Prolla, and M Vermulst for sharing brain tissue and DNA from the Polg mice, and M Vermulst and LA Loeb for sharing data, materials, and ideas, and for critical review of the manuscript. Funded in part by the NIA R01AG019787 (KK) and NINDS 1R01NS058988 (DKS).
References
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Bensch KG, Degraaf W, Hansen PA, Zassenhaus HP, Corbett JA. A transgenic model to study the pathogenesis of somatic mtDNA mutation accumulation in beta-cells. Diabetes Obes Metab. 2007;9(Suppl 2):74–80. doi: 10.1111/j.1463-1326.2007.00776.x. [DOI] [PubMed] [Google Scholar]
- Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, Aiken JM. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Grey ADNJ. Mitochondrial mutations in mammalian aging: an over-hasty about-turn? Rejuvenation Res. 2004;7:171–174. doi: 10.1089/rej.2004.7.171. [DOI] [PubMed] [Google Scholar]
- Fukui H, Moraes CT. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet. 2008;18:1028–1036. doi: 10.1093/hmg/ddn437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst A, Pak JW, McKenzie D, Bua E, Bassiouni M, Aiken JM. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J Gerontol A Biol Sci Med Sci. 2007;62:235–245. doi: 10.1093/gerona/62.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Nakada K, Ogura A, Isobe K, Goto Y, Nonaka I, Hayashi JI. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat Genet. 2000;26:176–181. doi: 10.1038/82826. [DOI] [PubMed] [Google Scholar]
- Khrapko K, Vijg J. Mitochondrial DNA mutations and aging: devils in the details? Trends Genet. 2008;25:91–98. doi: 10.1016/j.tig.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrapko K, Bodyak N, Thilly WG, van Orsouw NJ, Zhang X, Coller HA, Perls TT, Upton M, Vijg J, Wei JY. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999;27:2434–2441. doi: 10.1093/nar/27.11.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrapko K, Kraytsberg Y, de Grey ADNJ, Vijg J, Schon EA. Does premature aging of the mtDNA mutator mouse prove that mtDNA mutations are involved in natural aging? Aging Cell. 2006;5:279–282. doi: 10.1111/j.1474-9726.2006.00209.x. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Khrapko K. Single-molecule PCR: an artifact-free PCR approach for the analysis of somatic mutations. Expert Rev Mol Diagn. 2005;5:809–815. doi: 10.1586/14737159.5.5.809. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Kukat A, Trifunovic A. Somatic mtDNA mutations and aging–facts and fancies. Exp Gerontol. 2009;44:101–105. doi: 10.1016/j.exger.2008.05.006. [DOI] [PubMed] [Google Scholar]
- McKiernan SH, Tuen VC, Baldwin K, Wanagat J, Djamali A, Aiken JM. Adult-onset calorie restriction delays the accumulation of mitochondrial enzyme abnormalities in aging rat kidney tubular epithelial cells. Am J Physiol Renal Physiol. 2007;292:F1751–1760. doi: 10.1152/ajprenal.00307.2006. [DOI] [PubMed] [Google Scholar]
- Sato A, Kono T, Nakada K, Ishikawa K, Inoue S, Yonekawa H, Hayashi J. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc Natl Acad Sci USA. 2005;102:16765–16770. doi: 10.1073/pnas.0506197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, Spelbrink JN, Paetau A, Suomalainen A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci USA. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Mott JL, Chang SW, Denniger G, Feng Z, Zassenhaus HP. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics. 2000;69:151–161. doi: 10.1006/geno.2000.6333. [DOI] [PubMed] [Google Scholar]