

Abstract
The initial section deals with basic sciences; among the various topics briefly discussed are the anatomical features of ophthalmic, central retinal and cilioretinal arteries which may play a role in acute retinal arterial ischemic disorders. Crucial information required in the management of central retinal artery occlusion (CRAO) is the length of time the retina can survive following that. An experimental study shows that CRAO for 97 minutes produces no detectable permanent retinal damage but there is a progressive ischemic damage thereafter, and by 4 hours the retina has suffered irreversible damage. In the clinical section, I discuss at length various controversies on acute retinal arterial ischemic disorders.
Classification of acute retinal arterial ischemic disorders
These are of 4 types: CRAO, branch retinal artery occlusion (BRAO), cotton wools spots and amaurosis fugax. Both CRAO and BRAO further comprise multiple clinical entities. Contrary to the universal belief, pathogenetically, clinically and for management, CRAO is not one clinical entity but 4 distinct clinical entities – non-arteritic CRAO, non-arteritic CRAO with cilioretinal artery sparing, arteritic CRAO associated with giant cell arteritis (GCA) and transient non-arteritic CRAO. Similarly, BRAO comprises permanent BRAO, transient BRAO and cilioretinal artery occlusion (CLRAO), and the latter further consists of 3 distinct clinical entities - non-arteritic CLRAO alone, non-arteritic CLRAO associated with central retinal vein occlusion and arteritic CLRAO associated with GCA. Understanding these classifications is essential to comprehend fully various aspects of these disorders.
Central retinal artery occlusion
The pathogeneses, clinical features and management of the various types of CRAO are discussed in detail. Contrary to the prevalent belief, spontaneous improvement in both visual acuity and visual fields does occur, mainly during the first 7 days. The incidence of spontaneous visual acuity improvement during the first 7 days differs significantly (p<0.001) among the 4 types of CRAO; among them, in eyes with initial visual acuity of counting finger or worse, visual acuity improved, remained stable or deteriorated in nonarteritic CRAO in 22%, 66% and 12% respectively; in nonarteritic CRAO with cilioretinal artery sparing in 67%, 33% and none respectively; and in transient nonarteritic CRAO in 82%, 18% and none respectively. Arteritic CRAO shows no change. Recent studies have shown that administration of local intra-arterial thrombolytic agent not only has no beneficial effect but also can be harmful. Prevalent multiple misconceptions on CRAO are discussed.
Branch retinal artery occlusion
Pathogeneses, clinical features and management of various types of BRAO are discussed at length. The natural history of visual acuity outcome shows a final visual acuity of 20/40 or better in 89% of permanent BRAO cases, 100% of transient BRAO and 100% of nonarteritic CLRAO alone.
Cotton wools spots
These are common, non-specific acute focal retinal ischemic lesions, seen in many retinopathies. Their pathogenesis and clinical features are discussed in detail.
Amaurosis fugax
Its pathogenesis, clinical features and management are described.
1. INTRODUCTION
Central retinal artery occlusion (CRAO) results in sudden, catastrophic visual loss and is therefore one of the most important topics in ophthalmology. Similarly, branch retinal arteriolar occlusion (BRAO) causes sudden segmental visual loss and may recur to involve other branch retinal arterioles. Amaurosis fugax is a common transient acute retinal ischemic condition. Thus, acute retinal arterial occlusive disorders together comprise one of the major causes of acute visual loss. There is a voluminous literature on the subject, with conflicting findings. The subject has been and continues to be rife with misconceptions and mistaken theories. Recent studies have provided new data on various aspects of acute retinal arterial occlusive disorders.
Since 1955 I have investigated the subject comprehensively by doing basic, experimental and clinical studies. Those have revealed new information about the retinal arterial blood supply and its occlusive disorders, contradicting much of the conventional thinking. The objective of this review is to provide a comprehensive overview of this important subject, based on my studies combined with a review of the relevant literature.
The first essential for an in-depth understanding of the retinal arterial occlusive disorders is a good grasp of the relevant basic scientific facts about them; the basic sciences are the foundation of Medicine. Following is a brief discussion of some of those.
2. BLOOD SUPPLY OF THE RETINA
The retina is supplied by the central retinal artery (CRA) and in some eyes also by the cilioretinal artery. The primary source of blood supply to both the arteries is the ophthalmic artery. A brief account of the anatomy of these three arteries is essential to understanding the retinal arterial vascular disorders.
2.1 OPHTHALMIC ARTERY
The ophthalmic artery is the first major branch of the internal carotid artery. However, rarely the ophthalmic artery does not arise from the internal carotid artery. The most common abnormal origin is from the middle meningeal artery by an enlargement of an anastomosis between the recurrent branch of the lacrimal artery and the orbital branch of the middle meningeal artery through the superior orbital fissure or a foramen in the greater wing of the sphenoid (Fig. 1). This anastomosis is present during fetal life and becomes stronger when the ophthalmic artery is stenosed or not connected to the internal carotid artery. In a study of 170 specimens, the ophthalmic artery arose from the middle meningeal artery in 2 (Hayreh and Dass, 1962). In some cases the ophthalmic artery trunk arising from the internal carotid artery may be markedly stenosed, so that the major source of blood supply to the ophthalmic artery is then from the middle meningeal artery; in an anatomical study of 100 specimens (Singh (Hayreh) and Dass, 1960a), this was reported in 4% (Fig. 1). Other extremely rare abnormal modes of origin of the ophthalmic artery have been reported (Hayreh and Dass 1962, Hayreh, 2006). These variations in origin and blood supply of the ophthalmic artery may have clinical significance.
Figure 1.

Variations in origin and course of the ophthalmic artery.
A = Normal pattern; B,C,D, E = The ophthalmic artery arises firm the internal carotid artery as usual, but the major contribution comes from the middle meningeal artery. F,G = The only sources is the middle meningeal artery, as its connection with the internal carotid artery is either absent (F) or obliterated (G).
Abbreviations: ICA = Internal carotid artery; Lac. = Lacrimal artery; MMA = Middle meningeal artery; OA = Ophthalmic artery. (Reproduced from Singh (Hayreh) et al. 1960a.)
2.2 CENTRAL RETINAL ARTERY
This is the primary source of blood supply to the retina. A detailed discussion of it is essential to an understanding of the various aspects of CRAO. A detailed anatomical study of 100 human specimens described the various aspects of the CRA (Hayreh, 1958; Singh (Hayreh) and Dass, 1960a). Briefly, it showed the following:
2.2.1 Source of Origin
It arises from the ophthalmic artery, irrespective of whether the latter is a branch of the internal carotid artery or of the middle meningeal artery. In that study, it was the first branch of the ophthalmic artery in 77%, second in 19% and third in 4%. In two specimens the CRA had two trunks, arising independently from the ophthalmic artery (Fig. 2). Morandi et al. (1998) described a case where the ophthalmic artery arose from the middle meningeal artery and was associated with CRAO, and they considered that such an unusual origin could be a factor for CRAO. As discussed above, I have seen the ophthalmic artery arising from the middle meningeal artery in 6 cases, none with associated CRAO (Hayreh, 1958; Singh (Hayreh) and Dass, 1960a).
Figure 2.

Two trunks (1,2) of the central retinal artery, arising independently from the ophthalmic artery, as seen on splitting open the optic nerve (ON) behind the eyeball. (Reproduced from Hayreh 1958)
2.2.2 Mode of origin
The CRA arose independently in 37.5% from the ophthalmic artery, in 59.5% by a common trunk with one or another posterior ciliary artery (PCA) (Fig. 3) and extremely rarely with other branches of the ophthalmic artery. The fact that the CRA may arise by a common trunk with a PCA has great clinical significance, as discussed later.
Figure 3.

The central artery of the retina (CAR) arising by a common trunk with lateral posterior ciliary artery (LPCA) from the ophthalmic artery (OA). PPS = Point of penetration of the central artery of the retina into the optic nerve sheath. (Reproduced from Hayreh 1958)
2.2.3 Course
The course of the CRA is divided into 3 parts:
2.2.3.1 Intraorbital part
This is from its origin from the ophthalmic artery till it pierces the optic nerve sheath. This site of penetration in this study was found to vary from 5.0 mm to 15.5 mm (median 10 mm; mean 9.8 ± 1.8 mm) from the eyeball, and was located in the inferior medial (86%), inferior lateral (13%) or lateral (1%) aspect of the optic nerve.
2.2.3.2 Intravaginal part
This is situated in the subdural and subarachnoid spaces of the optic nerve sheath. The artery formed a tortuous loop in this part in 8%. The length of this section of the artery is 1.2 to 4.0 mm.
2.2.3.3 Intraneural part
This lies in the optic nerve. The artery enters the optic nerve in a well-defined fissure on the inferior surface of the optic nerve anterior to the site of penetration into the dural sheath and carries a fold of pia with it. The initial, vertical part of this section runs upwards and slightly forward to reach the center of the optic nerve, and then runs forwards horizontally in the center of the optic nerve to the optic disc, where it divides into its terminal branches. In the specimens with the two trunks of the CRA, the vertical part was missing and the two trunks ran straight to the optic disc from their point of penetration into the nerve (Fig. 2).
2.2.4 Branches
2.2.4.1 Incidence
In that study, when the artery had a satisfactory injection throughout its course (64 specimens), branches were present in 97% (Hayreh, 1958; Singh (Hayreh) and Dass, 1960a). They arose from the intraorbital part only in 1.6%, intravaginal part only in 9.4%, all three parts in 42.2% and from intravaginal and intraneural in 29.7%, intravaginal and intraorbital in 9.4% and intraorbital and intraneural parts in 4.7%. The size of the branches varied widely, from minute to as big as the CRA itself.
2.2.4.2 Distribution
Branches arising from the three parts of the CRA (Figs. 4–6) have variable supply.
Figure 4.

Two views of the same optic nerve in the retrobulbar regions – (A) from inferior aspect and (B) from the superior aspect after removal of dural sheath. They show pial branches of the central retinal artery (CRA) arising from the intravaginal part of the artery, and their anastomoses anteriorly with recurrent pial branches of the circle of Zinn and Haller (CZ) and posteriorly with collateral (Col.) branches from the ophthalmic artery (O.A.). (Reproduced from Hayreh 1958)
Figure 6.

Schematic representation, showing:
(A) The course of central retinal artery and its branches and anastomoses, central retinal vein and its tributaries, and blood supply of the optic nerve.
(B) Blood vessels on the optic disc and in the retina.
(Modified from Trans Am Acad Ophthalmol Otolaryngol 1974;78:OP240–OP254.).
Abbreviations: A = arachnoid; C = choroid; CRA = central retinal artery; Col. Br. = Collateral branches; CRV = central retinal vein; D = dura; LC = lamina cribrosa; OD = optic disc; ON = optic nerve; PCA = posterior ciliary artery; PR = prelaminar region; R = retina; S = sclera; SAS = subarachnoid space.
2.2.4.2.1
Intraorbital branches usually supply the dural sheath, about half of them penetrated the sheath to ramify on the pia of the optic nerve (Fig. 6) and a rare one entered the optic nerve.
2.2.4.2.2
Intravaginal branches ramified on the pia of the optic nerve in all specimens anterior to the site of penetration of the CRA and, in about half, also posterior to that, so that they supply the optic nerve from the eyeball to some distance posterior to the site of penetration of the dural sheath (Fig. 4). These branches ramified on the optic nerve all around the nerve in 15%, on the inferior aspect only in 36% and variably on other regions in the rest. Thus, intravaginal branches play an important role in the blood supply of the optic nerve. They are also important in establishing anastomoses in the event of CRA occlusion at its site of penetration into the dural sheath (see below),
2.2.4.2.3
Intraneural branches run radially as well as backwards and forward in the septa between the nerve fiber bundles to supply the optic nerve (Fig. 5,6). The CRA does not give out any branch in the region of the lamina cribrosa and the prelaminar region.
Figure 5.

This shows intravaginal (indicated by smaller arrow) and intraneural (anterior to the bigger arrow with asterisk where the optic nerve is split open to show the central region of the optic nerve) course of the central retinal artery (CRA) from below. It shows 2 pial branches (at and before the arrow with asterisk) and 6 intraneural branches anterior to that. (Reproduced from Hayreh 1958)
2.2.4.3 Anastomoses by the branches
This is an important subject when dealing with CRAO. Numerous anastomoses are established by the branches of the CRA with other branches of the ophthalmic artery, most on the pia of the optic nerve and between the pial branches of the CRA (mostly arising from the intravaginal part of the artery) and the recurrent pial branches of the circle of Haller and Zinn, and with the pial branches from the collateral branches of the ophthalmic artery (Figs. 4,6). These branches are most commonly situated on the inferior aspect of the optic nerve, less commonly on other aspects. The study showed that all these pial anastomoses were usually large enough to establish a variable amount of collateral circulation in the event of occlusion of the CRA at its site of penetration into the optic nerve sheath. This was also demonstrated by fluorescein fundus angiography in various studies dealing with experimental occlusion of the CRA at its site of penetration into the optic nerve in monkeys (Hayreh and Weingeist, 1980a; Petrig et al., 1999; Hayreh et al., 2004b).
2.2.5 Intraretinal Branches of the Central Retinal Artery
At the optic disc the CRA usually divides into two main branches (superior and inferior) and the two then further divide into temporal and nasal branches, which supply the four quadrants of the retina; however, there is marked variation in their vascular pattern and supply. These intraretinal arterial branches mainly lie in the nerve fiber and ganglion cell layer, usually under the internal limiting membrane; however, at the arteriovenous crossing they may extend down to the inner nuclear layer. The various branches, by multiple divisions, finally end in terminal or precapillary arterioles, which are usually not visible on ophthalmoscopy. Terminal arterioles play an important role in the regulation of retinal blood flow by constriction or dilation.
The so-called “branch retinal arteries” are in fact arterioles after the first branching in the retina, because their diameter near the optic disc is about 100 µm, which is typically the diameter of an arteriole, and, unlike arteries, they possess neither an internal elastic lamina nor a continuous muscular coat. This differentiation from arteries is important in understanding their pathological involvement in some diseases, e.g., giant cell arteritis (GCA). There are normally no interarterial or arteriovenous anastomoses in the retina, so that the retinal vascular bed is an end-arterial system.
2.3 CILIORETINAL ARTERY
These arteries belong to the PCA system. They usually arise from the peripapillary choroid or directly from one of the short PCAs. The cilioretinal artery has a characteristic hook-like appearance at its site of entry into the retina at the optic disc margin, usually on the temporal side and less frequently elsewhere. The size and area of the retina supplied by a cilioretinal artery vary widely (Figs. 7–10) - from a minute artery supplying a tiny area of the peripapillary retina (Fig. 7), to supplying half (Fig. 10A) or the entire retina (Fig. 10B). A histological study of two eyes of a rhesus monkey showed multiple cilioretinal arteries (8 in the right and 3 in the left) and the CRA was absent in the right eye and supplied only the upper part of the retina in the left (Hayreh, 1963). I have seen two cases where cilioretinal arteries supplied the entire retina. Similarly, Hegde et al. (2006) reported a case of a cilioretinal artery supplying the entire retina.
Figure 7.

Fundus photograph of the left eye with CRAO, with a tiny patent cilioretinal artery (Arrow). It shows the classical cherry-red spot in macular region and attenuated retinal vessels with “cattle-trucking” or “box-carring” in them.
Figure 10.

Two fundus photographs.
A = This shows a large cilioretinal artery (arrow with one asterisk) supplying the superior half of the retina, and the central retinal artery (arrow with two asterisks) supplying the lower half of the retina.
B = This shows two (1,2) large cilioretinal arteries supplying the entre retina and absent central retinal artery.
In 1963, I reviewed the literature on cilioretinal arteries since 1856 (Hayreh, 1963). The incidence of presence of a cilioretinal artery had been described variably by different studies, based on variable numbers of eyes, and it varied from none to 25%. Collier (1957) evaluated various aspects of the cilioretinal artery in 1,000 subjects and found an incidence of 22%, bilateral in 17% of cases, frequently associated with congenital optic disc abnormalities and most common in hypermetropic astigmatism. The incidence in all these studies was based on ophthalmoscopic evaluation, however, that can be deceptive, because what looks like a cilioretinal artery on ophthalmoscopy may actually be an early intraneural branch of the CRA, emerging at the optic disc as a separate artery. The most reliable way to ascertain the true incidence is by fluorescein fundus angiography. This is because the cilioretinal artery fills concurrently with the filling of the choroid and usually before the start of filling of the CRA. Justice and Lehmann (1976) evaluated the incidence of cilioretinal arteries in 2,000 eyes of 1,000 consecutive patients by reviewing stereoscopic color fundus photographs and fluorescein angiograms. One or more cilioretinal arteries were present in 49.5% of all patients or in 32% of the eyes. The arteries occurred bilaterally in 15% and contributed to some portion of the macular circulation in 19% of the patients. Great variation in size, number, and distribution of cilioretinal vessels was observed. The incidence reported by this study is much higher than reported previously based on ophthalmoscopic evaluation, because in this study early phase fluorescein angiography helped in the detection of these vessels that might otherwise have been missed.
2.4 RETINAL CAPILLARY BED
Each terminal arteriole gives out a plexus of 10–20 interconnected capillaries (Fig. 11). Capillaries lie between the feeding arterioles and venules. Around the retinal arterioles there is a capillary free zone. The retinal capillaries are arranged in two layers (Fig. 12); (i) superficial layer in the ganglion cell and nerve fiber layers, and (ii) deeper layer in the inner nuclear layer which is denser and more complex than the superficial layer. However, in the posterior retina, in the peripapillary region there may be three layers and in the perifoveal region there is only one layer. The capillaries are absent in the foveal avascular zone of about 400–500 µm in diameter. Also, in the peripheral retina the deep layer disappears and only the superficial layer is left, with a wider network. At the extreme periphery of the retina, there is an avascular zone of about 1.5 mm width.
Fig. 11.

Fluorescein fundus angiogram of a rhesus money eye showing retinal vessels and capillary network. A = Retinal arteriole; V = Retinal vein.
Figure 12.

Schematic representation of two layers of the retinal capillaries and radial peripapillary capillaries (RPC). (Reproduced from Henkind P: Trans Am Ophthalmol Otolaryng 1969;73:890–897.).
2.4.1 Radial peripapillary capillaries
The following special characteristics distinguish them from other retinal capillaries (Fig. 13):
Figure 13.

Schematic representation of radial peripapillary capillaries. X = Site of foveola (Reproduced from Henkind P: Br J Ophthalmol 1967;51:115–123.).
2.4.1.1
They are long, straight capillaries, measuring several hundred microns to several millimeters.
2.4.1.2
They form the most superficial layer, lying among the superficial nerve fibers, along the superior and inferior temporal arcades of the retinal vessels and the peripapillary region.
2.4.1.3
They rarely anastomose with one another.
2.4.1.4
They arise from the peripapillary retinal arterioles, and drain into retinal venules or veins on the optic disc.
Because of these characteristics, the radial peripapillary capillaries play an important role in the development of several lesions. For example, cotton wool spots are often located in their distribution, which indicates that they may play a role in their pathogenesis (see below). Also, in chronic optic disc edema these capillaries become dilated and develop microaneurysms and hemorrhages.
The wall of the retinal capillaries consists of endothelial cells, pericytes and basement membrane. Their diameter varies from 3.5 to 6 µm. The endothelial cells have tight cell junctions, which exercise a blood-retinal barrier. In addition to the endothelial cells, there are also pericytes which form a discontinuous layer within the basement membrane of the capillaries. They have a contractile property, by virtue of which they may play a role in regulating blood flow in the capillaries and autoregulation of blood flow (see below).
2.5 RETINAL VENOUS DRAINAGE
The postcapillary venules drain the blood from the capillaries, but occasionally capillaries may join a major vein directly. The terminal arterioles and postcapillary venules are situated in an alternating pattern, with the capillary bed in between the two (Fig. 11). The postcapillary venules drain into bigger venules and finally into the major branch retinal veins, which join at the optic disc to form the central retinal vein. In the central part of the retina, the branch retinal veins and arterioles usually run in close association and at places cross one another, but in the peripheral retina, the veins do not follow the course of the arterioles.
During the third month of intrauterine life, there are always two trunks of the central retinal vein in the optic nerve, one on either side of the CRA, one of which usually disappears before birth; however, in 20% of eyes a dual-trunked central retinal vein persists into adult life. There is usually one central retinal vein, but in 20% there are two trunks of the central retinal vein in the optic nerve (Chopdar, 1984); this represents a congenital anomaly (Fig. 14) (Hayreh and Hayreh, 1980). In such eyes, only one of the two trunks may develop occlusion in the optic nerve, resulting in development of the clinical entity called "hemi-central retinal vein occlusion” (Hayreh and Hayreh, 1980). The central retinal vein travels in the optic nerve temporal to the artery, where the central retinal vein and artery lie in the center of the optic nerve, surrounded by a fibrous tissue envelope (Fig. 15). During its intraneural course, the vein receives a large number of tributaries (Fig. 6). The central retinal vein exits the optic nerve and its sheath, and finally drains into either the superior ophthalmic vein or directly into the cavernous sinus.
Figure 14.

Schematic representation of two-trunked central retinal vein in the optic nerve. For abbreviations see Figure 6. (Reproduced from Hayreh and Hayreh 1980)
Figure 15.

Light micrograph showing the central retinal vessels and surrounding fibrous tissue envelope, as seen in a transverse section of the central part of the retrolaminar region of the optic nerve. (Masson’s trichrome staining) CRA = Central retinal artery; CRV = Central retinal vein. FTE = Fibrous tissue envelope, NASAL = Nasal side of the optic nerve. (Reproduced from Hayreh et al. 1999 J Glaucoma;8:56–71.)
3. NERVE SUPPLY TO RETINAL VESSELS
During its intraorbital and intraneural portion the CRA has adrenergic nerve supply by a sympathetic nerve called the nerve of Tiedemann (Hayreh and Vrabec, 1966); however, the retinal branches of the CRA have no adrenergic nerve supply. Therefore, there is no autonomic innervation of the retinal vascular bed.
4. BLOOD–RETINAL BARRIER
The blood-retinal barrier plays an important role in the regulation of the microenvironment in the retina. There are two types of blood-retinal barrier:
4.1 Inner blood-retinal barrier
This lies in the retinal vessels. It is produced by the tight cell junctions between the endothelial cells of the vessels (due to the presence of extensive zonulae occludentes). The tight interendothelial cell junctions block movement of macromolecules from the lumen toward the interstitial space. Pericytes, Müller cells and astrocytes also contribute to the proper functioning of this barrier.
4.2 Outer blood-retinal barrier
Tight cell junctions between the retinal pigment epithelial cells also produce a blood-retinal barrier, preventing the leakage of fluid from the choroid into the retina. This barrier breaks down when the retinal pigment epithelial cells are destroyed or subjected to ischemia, as in hypertensive choroidopathy (Hayreh et al., 1986). The retinal tissue itself has no barrier in its stroma, so fluid may diffuse from one part to the adjacent areas (Hayreh et al., 1986).
5. AUTOREGULATION OF RETINAL BLOOD FLOW
The object of blood flow autoregulation is to maintain the blood flow in a tissue relatively constant during changes in its perfusion pressure. This is an important mechanism to regulate blood flow. The retinal circulation has efficient autoregulation. The exact mechanism and site of autoregulation are still unclear, except that it most probably operates by altering the vascular resistance. It is generally considered a feature of the terminal arterioles; with the rise or fall of perfusion pressure beyond normal levels, the terminal arterioles constrict or dilate, respectively, to regulate the vascular resistance and thereby the blood flow. Studies have suggested that pericytes in the retinal capillaries play a role in autoregulation as well, because of their contractile property (Anderson, 1996; Anderson and Davis, 1996). The metabolic needs of the tissue also regulate the autoregulation. But autoregulation works only within a critical range of perfusion pressure, so that it breaks down with any rise or fall of the perfusion pressure beyond the critical autoregulatory range.
6. RETINAL TOLERANCE TIME TO ACUTE ISCHEMIA
Retinal tolerance to acute ischemia is key to understanding the retinal arterial occlusive disorders and their management. We investigated this experimentally in 38 elderly, atherosclerotic and hypertensive rhesus monkeys (mimicking patients with CRAO), producing transient occlusion of the CRA varying from 97 to 240 minutes, by temporarily clamping the CRA at its site of entry into the optic nerve (Hayreh et al., 2004b). The retinal circulation, function and changes were evaluated before and during CRA clamping, after unclamping, and serially thereafter by stereoscopic color fundus photography, fluorescein fundus angiography, electroretinography, and visual evoked potential. Finally, the eyes and optic nerves were examined histologically. These studies showed that the retina of old, atherosclerotic, hypertensive rhesus monkeys suffers no detectable damage with CRAO for 97 minutes, but above that level, the longer the CRAO, the more extensive the irreversible damage (Hayreh et al., 2004b). The study suggests that CRAO lasting for about 240 minutes results in massive, irreversible retinal damage. This was further confirmed by a study of retinal nerve fiber layer damage and optic disc changes in these monkey eyes; there was no apparent morphometric evidence of damage with CRAO of less than 97 minutes, but, occlusion of 105 minutes but less than 240 minutes produced a variable degree of damage, while occlusion for 240 minutes or more produced total optic nerve atrophy and nerve fiber damage (Hayreh and Jonas, 2000).
This study (Hayreh et al., 2004b) also showed that, surprisingly, contrary to the prevalent impression, the retina of old, atherosclerotic, hypertensive rhesus monkeys could tolerate ischemia for a much longer time than in younger, normal rhesus monkeys in an exactly identical study (Hayreh et al., 1980; Hayreh and Weingeist, 1980b); the reasons for that discrepancy are discussed at length elsewhere (Hayreh et al., 2004b). Those studies also showed that in eyes where the retinal circulation was restored to normal after CRAO of more than 2 but less than 4 hours’ duration, retinal function did not show signs of major improvement until many hours or even a day or more after restoration of circulation – the longer the ischemia, the longer the lag before any improvement of function started. This finding is important clinically; because the notion has unfortunately arisen that visual recovery can occur in eyes with CRAO lasting for much longer than 4 hours and that belief has fostered the use of various advocated treatments for CRAO of much longer than 4 hours (see below). In view of the above facts, treatment instituted at any time beyond 4 hours, after the onset of CRAO cannot have any scientific rationale for improvement of vision.
7. CAUSES OF RETINAL ARTERIAL OCCLUSION
In the management of a disease, the first essential requisite is to know what caused it; therefore the first necessary task is to find out the causes of retinal arterial occlusion. There is a colossal amount of literature about the association of retinal arterial occlusion with a large variety of systemic and hematological conditions. Most of that is based on anecdotal case reports, and, given that, it is not always possible to establish a true cause-and-effect relationship between the CRAO/BRAO and the reported disease. The following associations with retinal artery occlusion have been reported:
7.1 Both CRAO and BRAO
In the literature, the information for all types of retinal artery occlusion is often combined, instead of type of retinal artery occlusion being specified. CRAO and BRAO have been reported with a variety of conditions, including the following: embolism from atheromatous plaques in the carotid arteries, during carotid angiography and stenting or cardiac cauterization or coronary angiography, atrial fibrillation, mitral or aortic valve mass, bacterial endocarditis, atrial myxoma, patent foramen ovale, Takayasu’s arteritis, and migraine. Other reported rare causes are: after radial optic neurotomy or after retinal surgery, and herpes zoster ophthalmicus.
7.2 CRAO Alone
In addition many conditions have been reported as associated with CRAO alone. These include the following: polyarteritis nodosa, Wegener's granulomatosis, Churg-Strauss syndrome, Behcet's disease, sarcoidosis, sickle cell disease, carotid artery dissection, aneurysm of the internal carotid artery, plasma lipoprotein(a) levels, high homocysteine levels, higher lupus anticoagulant, acquired immunodeficiency syndrome, leukemia, non-Hodgkin lymphoma, T-cell lymphoma, oral contraceptives, incontinentia pigmenti, Fabry’s disease, cat scratch disease, following severe blow-out fracture, perioperative CRAO, peribulbar anesthesia or forehead injection with a corticosteroid suspension, and laser in situ keratomileusis. Transient CRAO has even been described following viperine snake bite (Singh et al., 2007; Hayreh, 2008c).
Combined CRAO and central retinal vein occlusion (CRVO) has often been reported. This is not a true CRAO but it is actually secondary to occlusion of the central retinal vein in the region of the lamina cribrosa; the mechanism of CRAO in this case is that blood cannot get out of the retinal vascular bed because of complete blockage in the central retinal vein – naturally if the blood cannot get out of the retina, it cannot get in, resulting in secondary CRAO (Hayreh, 2005). Therefore when CRAO is associated with CRVO, it is a hemodynamic blockage rather than actual block of the CRA, as discussed below in detail.
7.3 BRAO Alone
Apart from those already mentioned, association of a large number of conditions with BRAO alone has been reported. These include the following: retinal vasculitis, multifocal retinitis, toxoplasmic chorioretinitis, prepapillary loops, Crohn's disease, Whipple disease, Lyme disease, Viagra, or Meniere's disease. BRAO associated with GCA has been reported (Fineman et al., 1996), however, this is not credible because, as discussed above, the so-called “branch retinal arteries” in fact are arterioles. GCA is a disease of the medium-sized and large arteries and not of the arterioles. This “BRAO due to GCA” is actually cilioretinal artery occlusion due to involvement of the PCA by GCA, as discussed below.
There are large numbers of reports of BRAO in Susac's Syndrome, which consists of the clinical triad of encephalopathy, BRAO, and hearing loss. It is an autoimmune endotheliopathy affecting the precapillary arterioles of the brain, retina, and inner ear (cochlea and semicircular canals). The age range extends from 7 to 72 years, but young women (20–40) are most vulnerable (Rennebohm and Susac, 2007). I have, myself, seen a few patients with Susac's Syndrome and all were young women (Gordon et al., 1991). It tends to produce recurrent BRAO.
7.4 Cilioretinal Artery Occlusion
This has been reported in association with embolism, CRVO or GCA (see below), Viagra, systemic lupus erythematosus, antiphospholipid syndrome, migraine, and pregnancy.
In addition to these anecdotal reports of a few cases, the association of retinal artery occlusion with various systemic conditions has been reported by some large studies. Following are a few recent reports.
Schmidt et al. (2007) compiled cardiovascular risk factor findings (RFs) in a retrospective study of 134 patients with BRAO, 253 patients with CRAO, and 29 patients with hemi-CRAO. There were 66% males and 34% females, and the mean age was 66 years (range: 18–90). RFs were found in 58% - arterial hypertension in 76% (74 % among BRAO, 80% among CRAO, and 79% among hemi-CRAO). RFs such as arterial hypertension, carotid artery diseases, diabetes mellitus, hyperlipidemia, hyperuricemia, and chronic smoking did not differ statistically between patients with BRAO, CRAO or hemi--CRAO. But visible emboli in retinal arteries were observed in patients with BRAO (47 %,), or hemi-CRAO (41 %), much more often than in patients with CRAO (11 %). They concluded that every patient with retinal arterial obstruction should undergo extensive examination for RFs.
Rudkin et al. (2010) in a retrospective study of 33 consecutive patients with non-arteritic CRAO, found that 64% had at least one new vascular risk factor, with hyperlipidemia in 36% and hypertension in 27%. Nine patients had more than 50% of ipsilateral carotid stenosis; six of these proceeded with carotid endarterectomy or stenting. Systemic ischemic events occurred after CRAO in two patients (stroke and acute coronary syndrome).
Greven et al. (1995) reported 21 patients under 40 years old with retinal arterial occlusion. They found that cardiac valvular disease was the most commonly recognized etiologic agent (19%). Various associated factors leading to a hypercoagulable state or embolic condition were identified in 19 patients (91%). They concluded that retinal arterial occlusions in young adults occur via multiple mechanisms.
Hayreh et al. (2009b) in a prospective study of 439 consecutive untreated patients with retinal artery occlusion (249 CRAO, 190 BRAO) found that in both nonarteritic CRAO and BRAO the prevalence of diabetes mellitus, arterial hypertension, ischemic heart disease, and cerebrovascular accidents were significantly higher compared to the prevalence of these conditions in the matched US population (all p<0.0001). Smoking prevalence, compared to the US population, was significantly higher for males (p=0.001) with nonarteritic CRAO and for females with BRAO (p=0.02). The ipsilateral internal carotid artery had ≥50% stenosis in 31% of nonarteritic CRAO patients and 30% of BRAO, and plaques in 71% of nonarteritic CRAO and 66% of BRAO. An abnormal echocardiogram with embolic source was seen in 52% of nonarteritic CRAO and 42% of BRAO. Thus, this study showed that in CRAO as well as BRAO the prevalence of various cardiovascular diseases and smoking was significantly higher than the prevalence of these conditions in the matched US population. Embolism is the most common cause of CRAO and BRAO; plaque in the carotid artery is usually the source of embolism and less commonly the aortic and/or mitral valve. The presence of plaques in the carotid artery is generally of much greater importance than the degree of stenosis in the artery. This study showed that, contrary to a prevalent misconception (Duker et al., 1991), there is no cause-and-effect relationship between CRAO and neovascular glaucoma (see below) (Hayreh et al., 2009b).
Hematologic abnormalities
Information in the literature on the association of retinal artery occlusion with hematologic abnormalities is contradictory. Reported hematologic abnormalities include familial and acquired thrombophilia (low protein C, lupus anticoagulant) in patients with CRAO (Glueck et al., 2008), antiphospholipid antibodies (Palmowski-Wolfe et al., 2007) and homocysteinemia (Weger et al., 2002; Glueck et al., 2008); however, factor V Leiden, prothrombin 20210A and homozygosity for the MTHFR C677T have not been found to be associated with retinal artery occlusion (Weger et al., 2002). Salomon et al. (2001) in a study of 21 consecutive patients with retinal artery occlusion found at least one thrombophilic marker in 43%. Chua et al. (2006) in the Blue Mountains Eye cross-sectional population-based study of 3509, age ≥49 years concluded that elevated serum homocysteine is weakly associated with increased odds of retinal emboli in this age-group. Atchaneeyasakul et al. (2005) in a retrospective, case-control study found no association between plasma homocysteine level and retinal vascular occlusion, and also found that anticardiolipin IgG antibody was not a major cause of the development of retinal vascular occlusive disease.
7.6 Retinal emboli
Thus, in summary, embolism is by far the most common cause of nonarteritic CRAO and BRAO. The major source of emboli is carotid artery disease - most commonly plaques, and much less frequently stenosis. The heart is also an important source of these emboli, though less than the carotid artery.
7.6.1 Types of retinal emboli
As discussed above, the most common cause of retinal artery occlusion is embolism. Arruga and Sanders (1982) showed that 74% of retinal emboli are made of cholesterol (Hollenhorst plaque), 10.5% calcific material and only 15.5% platelet-fibrin. The carotid artery and the heart are the most common sources of embolism to the retinal arteries. In the carotid artery, plaque is the most common source. In the heart, sources of emboli are aortic and mitral valvular lesions, patent foramen ovale, and left atrial myxoma (Schmidt et al., 2005). The study by Hayreh et al. (2009b) showed that the most common abnormality and source of embolism in the carotid arteries is plaque(s) (66%); significant carotid stenosis (i.e. ≥50%) was seen in only 30%. In that study, relating carotid Doppler/angiography findings to echocardiography findings showed an abnormal echocardiogram with embolic source in 62% of those with plaque in the carotid artery in CRAO and in 44% with BRAO. This means that in such cases, the embolus could have come from either the carotid artery or the heart or possibly both. This indicates that one has to evaluate both sources for embolism in all patients with CRAO and BRAO.
7.7
Carotid artery disease, apart from embolism, can also produce CRAO by the following two other means:
7.7.1 Hemodynamic cause
A significant stenosis (usually about 70% or more), or complete occlusion of the internal carotid artery can produce hemodynamically induced retinal and/or ocular ischemia, by markedly reducing the ocular blood flow. A fall of blood pressure, particularly nocturnal arterial hypotension (Fig. 16), with a markedly stenosed or occluded internal carotid artery, can result in transient CRAO (see below) (Hayreh and Zimmerman, 2005). Anderson et al. (2002) found that the hemodynamic effects of carotid artery stenosis do not appear to be more important in the pathogenesis of retinal events than hemispheric ones; whereas severe stenosis of the extracranial internal carotid artery is the most common identified condition associated with retinal and ocular ischemia (Hayreh and Podhajsky, 1982; Mizener et al., 1997; Sharma et al., 1998; Babikian et al., 2001).
Figure 16.

A 24-hour ambulatory blood pressure recording, starting at about 11 AM and ending at about 10 AM next day. Note that during the waking hours the blood pressure is perfectly normal but shows a marked drop during the sleeping hours (nocturnal arterial hypotension). (Reproduced from Hayreh et al. 1999.)
Sharma et al. (1998) found hemodynamically significant carotid artery stenosis in only 19% of patients with acute retinal artery occlusion. In our study, ≥ 80% stenosis of the internal carotid artery was seen in 18% of CRAO cases and 14% of BRAO (Hayreh et al., 2009b). We have found that the presence of a retinal embolus is a poor predictor of hemodynamically significant carotid stenosis on carotid Doppler, as has also been pointed out by others (Sharma et al., 1998; Wakefield et al., 2003; McCullough et al., 2004).
7.7.1 Serotonin (5-hydroxytryptamine) induced arterial spasm
Serotonin is released by platelet aggregation on atherosclerotic plaques in the carotid artery. Serotonin is a vasoconstrictor. A study on atherosclerotic monkeys found that Serotonin, in the presence of atherosclerotic lesions, can cause transient or complete occlusion or impaired blood flow in the CRA, by producing a transient spasm (Hayreh et al., 1997) (Fig. 17). This may contribute to CRAO and retinal ischemic disorders,
Figure 17.

Fluorescein fundus angiograms of right eye of an atherosclerotic cynomolgus monkey.
(A) Fluorescein fundus angiogram about 4 minutes after the start of serotonin infusion, showing normal filling of the choroidal circulation but complete occlusion of the central retinal artery.
(B) An angiogram about 2 hours after stopping serotonin infusion, showing normal filling of the central retinal artery and choroidal circulations. (Reproduced from Hayreh et al. 1997.)
7.8 GIANT CELL ARTERITIS AND RETINAL ARTERIAL OCCLUSION
A study investigated this in 170 consecutive patients with temporal artery biopsy confirmed GCA (Hayreh et al., 1998b). In this study 50% (85 patients, 123 eyes) presented with ocular involvement. Of the 123 eyes, CRAO was present in 18%, cilioretinal artery occlusion in 25%, and ocular ischemia in 1%. Cilioretinal artery occlusion in GCA has been erroneously diagnosed as BRAO (Fineman et al., 1996); this is because the so-called branch retinal arteries are in fact arterioles and not arteries, and GCA is a disease of medium-sized and large arteries and not of arterioles. In almost every patient with GCA, fluorescein fundus angiography disclosed occlusion of the PCAs; these arteries supply the optic nerve head and cilioretinal arteries, and occlusion of them results in development of arteritic anterior ischemic optic neuropathy and cilioretinal artery occlusion.
8. RACIAL DIFFERENCES IN THE CAUSES OF RETINAL ARTERIAL OCCLUSION
Ahuja et al. (1999), in a retrospective study of consecutive 29 African American and 17 Caucasian patients with a diagnosis of amaurosis fugax, CRAO, BRAO, or intra-arterial retinal plaques, evaluated the causes of retinal artery occlusion. They found that their African American patients had a mean age of 61 years (range, 30 to 77 years) and the Caucasian patients a mean age of 73 years (range, 56 to 94 years) (P=0.003). There was no statistically significant difference between the 2 groups with respect to visible emboli on ophthalmoscopy, or with regard to risk factors for arterial occlusive disease such as arterial hypertension, coronary artery disease, hypercholesterolemia, tobacco use, and history of stroke or transient ischemic attacks. But Caucasian patients had a 41% incidence of high-grade ipsilateral internal carotid artery stenosis, compared with a 3.4% incidence in African American patients (P=0.002).
9. EVALUATION OF PATIENTS WITH ACUTE RETINAL ARTERIAL OCCLUSION
It is generally agreed that all patients with acute retinal arterial occlusion must be evaluated for the source of embolism, which is the commonest cause for its development.
A recent study evaluated the role of carotid Doppler/angiography and Echocardiography in a prospective study 249 CRAO and 190 BRAO patients (Hayreh et al., 2009b). It showed that the plaque in the carotid artery is the usual source of embolism and less commonly the aortic and/or mitral valve.
9.1 CAROTID DOPPLER/ANGIOGRAPHY
In general, when evaluating carotid arteries in these cases, there is a tendency to look only for the stenosis, but it is the presence of plaques in the carotid artery which is of prime interest in such cases; it is of much greater importance than the degree of stenosis in the artery. In this study, 34% had 50% or greater stenosis on the side of CRAO and 71% had plaque(s), and in BRAO that was 30% and 66% respectively (Hayreh et al., 2009b).
9.2 ECHOCARDIOGRAPHY
The transesophageal type of echocardiography is superior to the transthoracic type in detecting cardiac abnormalities. This study showed the following (Hayreh et al., 2009b):
9.2.1
In CRAO, abnormal echocardiograms of source from the mitral valve in 26%, from the aortic valve in 38%, and from both mitral and aortic valves in 36%. The mitral valve lesions comprised 57% calcified valve, 17% mitral valve prolapse, and 26% other types of lesions. The aortic valve lesions were 78% calcified valve and 22% of other types. Patent foramen ovale was detected in 2.4%.
9.2.2
In BRAO, abnormal echocardiograms of an embolic source showed an embolic source in 31% from the mitral valve, 28% from the aortic valve, and 41% from both valves. The mitral valve lesions comprised 70% calcified valve, 4% with mitral valve prolapse, and 26% with other types of lesions. The aortic valve lesions were 68% calcified valve and 32% of other types. Patent foramen ovale was detected in 2%.
9.3 SYSTEMIC EVALUATION
In CRAO as well as BRAO, the prevalence of diabetes mellitus, arterial hypertension, ischemic heart disease, and transient ischemic attacks / cerebrovascular accidents is significantly higher than the prevalence of these conditions in the matched U.S. population (all p<0.0001); so also is the prevalence of smoking, compared to the US population (p=0.001) (Hayreh et al., 2009b). Therefore these patients require a complete medical evaluation. Among the laboratory studies, the first step for all patients 50 years and older is to rule out GCA, which is an ophthalmic emergency, by doing instant evaluation of the erythrocyte sedimentation rate and C-reactive protein, the latter is more reliable. We do not have any definite evidence that hematologic abnormalities play a significant role. In my studies of about 450 patients with various types of retinal artery occlusion, I have found that usually ophthalmologists had already done a large number of hematologic studies to rule out coagulation, thrombotic and other hematologic disorders before referring them to my clinic, only very rarely was any abnormality detected. Therefore, I do not advocate doing all these studies as a routine, unless there is some other strong indication.
9.4 PREVALENT MISCONCEPTIONS IN EVALUATION OF PATIENTS WITH RETINAL ARTERY OCCLUSION
There are several misconceptions about this, as discussed in detail elsewhere (Hayreh, 2005). It is widely assumed that the absence of any abnormality on Doppler evaluation of the carotid artery or echography of the heart rules out those sites as the source of embolism. On the contrary, I have seen some patients with CRAO or emboli in their retinal arteries, without either of these tests showing any abnormality at all. Following are the explanations.
9.4.1 Doppler Evaluation of the Carotid Artery
This is the most common investigation done in retinal artery occlusion patients. I have found that, when evaluating the carotid artery on Doppler, vascular surgeons, neurologists and other physicians are almost invariably interested in the degree of stenosis of the carotid artery because their primary interest is hemodynamic insufficiency and carotid endarterectomy. However, retinal arterial occlusions are almost always due to microembolism, and the major source of microemboli is the plaque(s) which may be present with or without any significant carotid artery stenosis. Thus, absence of significant stenosis of the carotid artery does not necessarily rule out the carotid artery as the source of microembolism. Vascular insufficiency in the eye caused by a significant carotid artery stenosis is seen only rarely, and occurs primarily in ocular ischemic syndrome (Hayreh and Podhajsky, 1982; Mizener et al., 1997). Therefore, from the ophthalmic point of view, when evaluating the results of carotid Doppler study for ocular microembolism, the presence or absence of plaques is usually of much greater importance than the degree of stenosis.
The other reasons why an absence of abnormality on the carotid Doppler does not rules out the carotid artery as the source of the retinal microembolism are: (a) Carotid Doppler evaluates the artery only in the neck and not above and below that, in the skull and the thorax respectively – the source of microembolism may be at those locations. (b) The resolution of Doppler is not always sensitive enough to detect very small plaques which may be enough to produce microembolism to the retina.
9.4.2 Echocardiography
Once again, the absence of any abnormality on this test does not always rule out the heart as the source of microembolism, because its resolution may not be sensitive enough to detect very small valvular or other cardiac lesions. I have also found that when routine transthoracic cardiac echography shows no abnormality, transesophageal echography may reveal abnormalities.
9.4.3 Absence of an embolus
There is another serious misconception that the absence of an evident embolus in the retinal artery means the occlusion was not caused by an embolus. A clinical study on CRAO showed that migration and disappearance of retinal emboli is common (Hayreh and Zimmerman, 2007). Of the 42 eyes which were seen on multiple visits where an embolus was seen at least once, there were 69% in which the embolus was not consistently present at all of the visits. In other words, there was migration of retinal emboli in at least 69% – the actual incidence is likely to be much higher. Thus, the axiom is that if one sees an embolus, then that means it was responsible for the occlusion; but, if one does not see an embolus, that does not rule out that embolism was not responsible for the occlusion, because it might have migrated and disappeared by the time the eye is examined. This issue is further discussed below.
10. CENTRAL RETINAL ARTERY OCCLUSION
Von Graefe (1859) was the first to diagnose CRAO ophthalmoscopically, and he wrote a classical description of the appearance of the fundus which is still well worth reading. The sudden and catastrophic visual loss with CRAO is one of the most important topics in ophthalmology. That makes it an ophthalmic emergency. In spite of this clinical entity having been well known for 150 years, and a voluminous literature on its various aspects, there are still several controversial issues. My critical review of the available literature that has accumulated on the management of CRAO has revealed that the controversy has resulted from several misconceptions on some fundamental scientific aspects of CRAO (Hayreh, 2005). The objective of my studies on CRAO has been to investigate those systematically.
10.1 CLASSIFICATION OF CRAO
CRAO is universally considered as one clinical entity; however, from the point of view of pathogenesis, clinical characteristics and management, recent studies have shown that it actually consists of the following four distinct categories (Hayreh and Zimmerman, 2005, 2007).
10.1.1 Non-arteritic CRAO
This includes eyes with the classical clinical picture of permanent CRAO with retinal infarction, cherry-red spot (Fig. 7) and absent or poor residual retinal circulation on fluorescein angiography, and no evidence of GCA. This is the most common type. It is usually either embolic or thrombotic in origin - embolism is a far more common cause than thrombosis (Hayreh and Podhajsky, 1982). Very rarely, vasculitis or trauma can cause CRAO. The site of occlusion in CRAO is invariably thought to be at the level of the lamina cribrosa; however, a detailed anatomical study of 100 human central retinal arteries showed that the narrowest lumen of the artery is where it pieces the dura mater of the optic nerve sheath before entering the optic nerve (Fig. 18) (Hayreh 1958). Therefore, in cases of CRAO due to embolism, the chances of an embolus getting impacted at this site are much higher than at any other site in the artery. However, histopathological studies have shown that the lamina cribrosa is the site of occlusion by thrombosis. The site of occlusion is an important factor in determining the amount of residual retinal circulation seen on fluorescein angiography following CRAO (see below).
Figure 18.

A transverse section of the optic nerve showing the central retinal artery (CRA) inferiorly lying within the substance of dural sheath of the optic nerve (ON). (Reproduced from Hayreh 1958.)
10.1.2 Non-arteritic CRAO with cilioretinal artery sparing
This type of non-arteritic CRAO develops only in eyes with a cilioretinal artery. As discussed above, the cilioretinal artery may vary in size from minute (Fig. 7) to one supplying a large part of the retina (Figs. 9,10). The visual outcome (Hayreh and Zimmerman, 2005) and fundus findings (Hayreh and Zimmerman, 2007) in this type of CRAO are different from the classical non-arteritic CRAO. Since the presence and size of a patent cilioretinal artery in permanent non-arteritic CRAO can have a marked influence on the visual outcome and retinal circulation, it is imperative not to lump these eyes together with those having non-arteritic CRAO alone.
Figure 9.

Fundus photograph of the right eye with CRAO and patent cilioretinal artery (Arrow).
10.1.3 Arteritic CRAO
This is due to GCA, which has a special predilection for involvement of the PCAs and only very rarely involves the CRA directly. As discussed above, the CRA, instead of arising directly from the ophthalmic artery, can arise from the ophthalmic artery, not infrequently by a common trunk with the PCA (Fig. 3) (Hayreh 1958). When GCA involves that common trunk, it results in occlusion of both the CRA and PCAs, and consequently development of both arteritic CRAO and arteritic anterior ischemic optic neuropathy (Hayreh, 1974a, 1975; Hayreh et al., 1998b). Therefore, in these eyes, the massive visual loss is the result of acute ischemia, not only of the retina but also of the optic nerve head. Clinically, these eyes on ophthalmoscopy have the classical fundus findings of CRAO with or without optic disc edema, but, most importantly, on fluorescein angiography there is evidence of a PCA occlusion in addition to CRAO (Hayreh, 1974a, 1975; Hayreh et al., 1998b). Without fluorescein fundus angiography, the presence of a PCA occlusion and the diagnosis of GCA is usually missed completely, resulting in the tragedy of bilateral blindness that could have been prevented by doing fluorescein angiography in all CRAO patients 50 and older, and early institution of high-dose steroid therapy.
10.1.4 Transient NA-CRAO
This is due to the following 3 reasons:
10.1.4.1 Due to transient impaction of an embolus
An embolus may occlude the CRA temporarily and then dislodge, result in restoration of circulation. The extent of visual loss and clinical features depend upon the duration of the temporary occlusion of the artery by the embolus. The type of embolus which usually causes this is platelet-fibrin embolus.
10.1.4.2 Due to fall of perfusion pressure below the critical level in the retinal vascular bed
In addition to the above causes, our study has revealed that several factors which produce a fall of perfusion pressure below the critical level in the retinal vascular bed can cause CRAO - usually transient CRAO (Hayreh and Zimmerman, 2005). We reported several patients who, in the presence of other predisposing risk factors, developed CRAO because of a fall of perfusion pressure. To understand the mechanism for this, it is essential to know the factors that can cause such a marked. The perfusion pressure in the CRA is equal to the mean blood pressure in the artery minus the intraocular pressure. Therefore, a fall of perfusion pressure can be due to the following two factors:
10.1.4.2.1 Marked fall in mean arterial blood pressure
This can occur for several reasons, including nocturnal arterial hypotension (Fig. 16) (particularly in patients who take blood pressure lowering medicines in the evening or at bedtime) (Hayreh et al., 1994; Hayreh, 1996), severe shock, during hemodialysis, spasm of the CRA, marked stenosis or occlusion of the internal carotid or ophthalmic artery, or ocular ischemia ((Hayreh and Podhajsky, 1982; Mizener et al., 1997).
10.1.4.2.2 A rise of intraocular pressure
Causes may include ocular compression during certain surgical procedures or marked orbital swelling, acute angle closure glaucoma, or neovascular glaucoma in association with ocular ischemia ((Hayreh and Podhajsky, 1982; Mizener et al., 1997).
When the intraocular pressure goes above the mean blood pressure or the blood pressure falls below the intraocular pressure, there is no retinal blood flow. Eyes with already poor perfusion pressure in the CRA are especially vulnerable to CRAO with the fall in perfusion pressure, particularly due to a fall of mean blood pressure, which acts as “the straw that breaks the camel’s back” ((Hayreh and Podhajsky, 1982; Mizener et al., 1997). Since these patients suffer from transient CRAO lasting for several hours, usually during sleep, from marked nocturnal arterial hypotension (Fig. 16), many of them typically give a history of discovering visual loss on waking in the morning or after hemodialysis or surgery. Fluorescein angiography performed during waking hours, when the perfusion pressure has returned to normal, misleadingly reveals normal retinal blood flow in spite of severe visual loss and all the classical signs of CRAO (Fig. 19). I have found that this normal angiographic finding has frequently misled ophthalmologists about the reason for the development of CRAO or even the correct diagnosis.
Figure 19.

Fundus photograph (A) and fluorescein fundus angiogram during retinal arterial phase (B) of an eye with transient CRAO. Fundus photograph shows large number of cotton wool spots, maximum in the macular region. Fluorescein angiogram shows almost normal but slightly sluggish retinal circulation except for absence of filling in the foveal region, and cotton wool spots at places masking the background fluorescence.
10.1.4.3 Due to vasospasm of the CRA
This is a rare cause of transient CRAO. There is evidence to suggest that platelets stick and aggregate on atherosclerotic plaques in the carotid arteries. An experimental study in atherosclerotic monkeys showed that serotonin (5-hydroxytryptamine) released by platelet aggregation on atherosclerotic plaques, in the presence of atherosclerotic lesions, triggers transient, severe vasospasm of the CRA, resulting in transient CRAO (Fig. 17) (Hayreh et al., 1997).
Thus, transient CRAO can be produced not only by transient impaction of an embolus in the CRA but also by all these various factors. In transient CRAO, the clinical appearance and visual outcome naturally depend upon the duration of CRAO.
This classification is essential to characterize and differentiate the visual outcomes among these four types of CRAO.
10.2 DEMOGRAPHIC CHARACTERISTICS OF VARIOUS TYPES OF CRAO
A prospective study of 244 patients with CRAO showed the following demographic characteristics in the 4 types of CRAO (Hayreh and Zimmerman, 2005):
10.2.1. Non-arteritic CRAO
This was seen in 67% of cases. Age range in this type was 26.5 to 90.4 (mean ± SD = 67.7 ± 12.3). Of these patients 42% were female, and 5% had it in both eyes. There was a history of amaurosis fugax before development of CRAO in 12%. Visual loss was discovered on waking up in the morning by 35%.
10.2.2 Non-arteritic CRAO with cilioretinal artery sparing
This was seen in 14% of cases. Age range in this type was 39 to 87 (mean ± SD = 67.1 ± 11.6). Of these patients, 46% were female. There was a history of amaurosis fugax before development of CRAO in 20%. Visual loss was discovered on waking up in the morning by 29%.
10.2.3 Arteritic CRAO
This was seen in 4.5% of cases. Age range in this type was 62 to 87 (mean ± SD = 74.4 ± 7.2). Of these patients, 64% were female, and 18% had it in both eyes. There was a history of amaurosis fugax before development of CRAO in 9%. Visual loss was discovered on waking up in the morning by 30%.
10.2.4 Transient CRAO
This was seen in 16% of cases. Age range in this type was 20 to 89 (mean ± SD = 62.7 ± 17.1). Of these patients 54% were female, and 5% had it in both eyes. There was a history of amaurosis fugax before development of CRAO in 13%. Visual loss was discovered on waking up in the morning by 39%.
10.3 SYMPTOMS
Typically, there is a sudden, massive loss of vision in the involved eye. It can occur at any time of the day. As shown above, some discover the visual loss on waking up in the morning. In the latter case, it may be due to embolism, thrombosis or due to transient CRAO from a fall of perfusion pressure during sleep caused by nocturnal arterial hypotension (Fig. 16). The visual loss may be preceded by transient visual obscurations, which may be due to transient migrating embolism or GCA. Simultaneous onset of CRAO in both eyes does not normally occur; it happens rarely, when there is compression of both eyes during a prolonged surgical procedure, or during hemodialysis. Although CRAO is more common in the elderly, it does occur in other age groups, including the young and, rarely, even in infants.
10.4 VISUAL ACUITY
A prospective study of 260 consecutive eyes with CRAO showed that this can vary among different types of CRAO (Hayreh and Zimmerman, 2005). In this study, the time interval between the onset of CRAO and the first visit to the clinic varied greatly - 49% seen within 7 days, 26% in 8 to 30 days, 23% more than one month and in 2% it was unknown. This study showed that when CRAO was subdivided into its four types, the initial visual acuity of the eyes seen within 7 days of onset differed significantly between the four types (p<0.0001) (Hayreh and Zimmerman, 2005). Following was the initial visual acuity in various types of CRAO in that study:
10.4.1 Non-arteritic CRAO
In 171 eyes, overall the initial visual acuity was 20/200 in 3%, 20/400 in 8%, counting fingers in 39%, hand motion in 26%, light perception in 16% and no light perception in 7% of the eyes. Thus, 49% of the eyes had visual acuity of hand motion or worse. However, one eye had visual acuity of 20/30 and another one 20/70. Among 127 eyes seen within 7 days, initial visual acuity was 20/200 – 20/400 in 3% and counting fingers or worse in 93%.
10.4.2 Non-arteritic CRAO with cilioretinal artery sparing
In 35 eyes, the initial visual acuity overall was 20/30 or better in 29%, 20/60–20/100 in 14%, 20/200 in 6%, counting fingers in 20%, hand motion in 26% and light perception in 6%. Visual acuity in this group depends upon the size of the cilioretinal artery – the larger the artery, the better is the visual acuity. Among 20 patients seen within 7 days, visual acuity was 20/40 or better in 20%, 20/50 – 20/100 in 3%, 20/200 – 20/400 in 5% and counting fingers or less in 60%.
10.4.3 Arteritic CRAO
In 13 eyes, all had visual acuity 20/400 or worse, with 54% having no light perception. This type has the worst visual acuity of all types of CRAO. Among 4 patients seen within 7 days, it was 20/200 – 20/400 in 1 and counting fingers or worse in 3 eyes.
10.4.4 Transient non-arteritic CRAO
There were 41 eyes overall in this category, of which the initial visual acuity was 20/20 or better in 27%, 20/25–20/30 in 15%, 20/60–20/70 in 10%, 20/100 in 2%, 20/200 in 7%, 20/400 in 5%, counting fingers in 24% and hand motion in 10%. This type has much better visual acuity than other types of CRAO. Among 29 patients seen within 7 days, initial visual acuity was 20/40 or better in 38%, 20/50–20/100 in 7%, 20/200 – 20/400 in 17% and counting fingers or worse in 38%.
This shows that, of the eyes seen within 7 days of onset, initial visual acuity differed significantly among the 4 CRAO types (p<0.0001), 38% of transient nonarteritic CRAO patients having counting fingers or worse compared to 93% of those with nonarteritic CRAO. There is a widespread impression that all eyes with CRAO initially have very poor visual acuity, e.g., hand motion or worse. As is evident from the above, an eye with CRAO may, rarely, present with visual acuity as good as 20/30 or better. This has often resulted in the diagnosis of CRAO being missed because of the conventional thinking. In the literature, visual acuity has unfortunately always been lumped together as one group; that does not provide valid information.
10.5 VISUAL FIELDS DEFECTS
Visual acuity essentially represents the function of the foveal region and not of the rest of the retina, while visual fields plotted with a Goldmann perimeter provide information about the entire retina and its function – after all, CRAO involves the entire retina. Unfortunately, there has been little information on the visual field defects in CRAO in the literature, except for one recent prospective study of 145 eyes (Hayreh and Zimmerman, 2005). Visual field defects in that study were divided into two categories: (i) scotomas seen in the central 30°, and (ii) general visual field defects.
10.5.1 Scotomas in the central 30°
Central scotoma was the most common type of all types of CRAO; its pathogenesis is discussed below. Paracentral scotoma was the next most common. There was no scotoma in 37% of the eyes with transient NA-CRAO.
10.5.2 General visual field defects
Eyes with nonarteritic CRAO with cilioretinal artery sparing showed an intact central island field corresponding to the area of the retina supplied by the patent cilioretinal artery. Generalized constriction of the peripheral fields was more common than other types of field defect in eyes with nonarteritic CRAO with cilioretinal artery sparing (32%) and also in those with transient nonarteritic CRAO (17%). By contrast, 52% of nonarteritic CRAO eyes had only a peripheral island residual field, most frequently located in the temporal region (42%). Most interestingly, the peripheral visual field was normal in 63% of the eyes with transient nonarteritic CRAO and even in 22% with nonarteritic CRAO; the clinical importance of intact peripheral visual field is discussed below.
10.6 NATURAL HISTORY OF VISUAL OUTCOME IN CRAO
Information on the natural history of a disease is vital, so that claims of “success” for various advocated treatments can be measured against the natural history. A recent large, planned, systematic study on the natural history of visual outcome in CRAO has given useful information (Hayreh and Zimmerman, 2005). This study had 168 eyes with a follow-up of 1.1 years (interquartile range: 0.2–3.5 years). It revealed the following:
10.6.1 Visual acuity
This study revealed that visual acuity improvement essentially occurs during the first 7 days, with minimal chance of any appreciable improvement thereafter. Also, the incidence of visual acuity improvement during the first 7 days differed significantly (p<0.001) among the 4 types of CRAO. Of the eyes that presented with counting fingers or worse initial visual acuity, 37% of those seen within 7 days of onset showed an improvement in visual acuity. In contrast, only 5% of those seen 8 to 30 days after onset and 9% of those seen more than 30 days after onset had any improvement in visual acuity (p<0.0001). Among the patients seen within 7 days with initial visual acuity of counting finger or worse, visual acuity improved, remained stable or deteriorated in nonarteritic CRAO in 22%, 66% and 12% respectively; in nonarteritic CRAO with cilioretinal artery sparing in 67%, 33% and none respectively; and in transient nonarteritic CRAO in 82%, 18% and none respectively. This shows that the popular concept of the dismal outlook in CRAO is not correct, and that it varies markedly with the type of CRAO. It is possible that many of the claims of visual acuity improvement following various treatments may be no more than the natural history of the disease.
10.6.2 Visual fields
In all types of CRAO, both central and peripheral visual fields showed improvement. In eyes with a follow-up of at least 30 days, the study found that in all types of CRAO, both central and peripheral visual fields showed improvement in a variable number of eyes.
10.6.2.1 Central visual fields
These improved or remained stable in nonarteritic CRAO in 21% and 73% respectively; in nonarteritic CRAO with cilioretinal artery sparing, in 25% and 69% respectively; and in transient nonarteritic CRAO in 39% and 30% respectively.
10.6.2.2 Peripheral visual fields
These improved, remained stable or deteriorated in nonarteritic CRAO in 39% 39% and 15% respectively; in nonarteritic CRAO with cilioretinal artery sparing in 19%, 75% and 6% respectively; and in transient nonarteritic CRAO in 39%,30% and none respectively.
Most importantly, in transient nonarteritic CRAO with field defects initially, the central and peripheral visual fields recovered to normal in 26% and 30% respectively.
Thus, this natural history study (Hayreh and Zimmerman, 2005) showed:
Contrary to the prevalent impression, spontaneous improvement in both visual acuity and visual fields does occur in the first few days after onset of CRAO, and the extent of improvement depends very much upon the type of CRAO. It is worst in arteritic CRAO.
Visual acuity improvement essentially occurs during the first 7 days, and differs significantly (p<0.001) among the 4 types of CRAO. There is minimal chance of any appreciable visual acuity improvement after 7 days.
It is essential to classify CRAO into 4 types to evaluate the visual outcome realistically – this has never been done in previous studies.
10.6.2.3 Pathogenesis of central scotoma in CRAO
There is an common impression that CRAO causes generalized loss of vision. However, it is not widely realized that CRAO can produce only central scotoma (the most common visual field defect in CRAO) with fairly normal peripheral visual fields (Fig. 20-C). The mechanism of selective development of central scotoma without any peripheral visual field defect in CRAO is as follows. It is well known that the macular region has more than one layer of retinal ganglion cells (unlike the rest of the retina), and it is the thickest part of the retina – maximal thickness being close to the foveola. Experimental (Hayreh and Weingeist, 1980a; Hayreh et al., 2004b) and clinical (Hayreh, 1976; Hayreh and Zimmerman, 2005) CRAO studies have shown that the ischemic retinal whitish opacity and swelling of CRAO is essentially located in the macular region – maximal in the perifoveolar region (Figs. 7,20-A). If there is restoration of circulation in the CRA, the retinal capillaries in the central, thickest part of the macular region do not re-fill (Fig.20-B) because of compression by the surrounding swollen superficial retinal tissue, resulting in the “no re-flow phenomenon” (Hayreh and Weingeist, 1980b), and consequently in permanent ganglion cell death in the non-perfused retina; the area of central retinal capillary non-filling may vary from eye to eye, depending upon the severity of retinal swelling in the macula region. This results in the variable size of the permanent central scotoma. Oxygen supply and nutrition from the choroidal vascular bed to the thinner peripheral retina help in its much longer survival, and the maintenance of peripheral visual fields.
Figure 20.

Fundus photograph (A) and fluorescein angiogram (B) of right eye at initial visit in an eye with transient non-arteritic CRAO 10 days earlier.
A. Fundus photograph shows cherry-red spot, retinal opacity of posterior fundus – most marked in the macular region, and a small area of normal retina temporal to the optic disc corresponding to a patent cilioretinal retinal artery.
B. Angiogram during the retinal arteriovenous phase shows normal filling of the retinal vascular bed with complete absence of filling in the macular region, corresponding to the area with most marked retinal swelling.
C. Visual fields of a left eye, plotted with a Goldmann perimeter 2 months after the development of transient non-arteritic CRAO. It shows an absolute central scotoma, and slightly constricted peripheral visual field with I-4e but normal with V-4e, and visual acuity of 20/200. (Reproduced from Hayreh and Zimmerman 2005.)
Patients with a central scotoma often learn with experience to fixate eccentrically, resulting in an apparent visual acuity improvement that does not represent a genuine improvement in the retinal function and may erroneously be attributed to a therapy. Natural history studies of various types of ocular vascular occlusive disorders have shown that for genuine visual acuity improvement, there must be simultaneous improvement in both the central scotoma and visual acuity (Hayreh et al., 2002; Hayreh and Zimmerman, 2005; Hayreh et al., 2011).
10.6.2.4 Role of Visual Field Findings in Judging Functional Visual Disability in CRAO
This has important clinical implications. It is well established that the constant tracking provided by the peripheral visual fields is essential for sensory input in our day-to-day activity, for example for driving and “navigating” generally (Hayreh, 2005). In view of that, to assess the visual function disability produced by CRAO, it is important to realize that persisting peripheral fields even in the presence of central scotoma (see above) do provide the patient fairly useful functional vision for day to day living, in spite of the fact that he/she cannot see well enough to read, write, drive or do any fine work with that eye. This is particularly important if the patient loses the fellow eye. Unfortunately, this important fact is not fully realized by much of the ophthalmic community.
10.6.3 Factors Influencing the Visual Outcome in CRAO
This is an important subject, because an in-depth understanding of the fundamental facts behind the factors that play a crucial role in determining the visual outcome in CRAO is essential. Experimental (Hayreh, 1971; Hayreh et al., 1980; Hayreh and Weingeist, 1980a, b; Hayreh et al., 2004b; Kwon et al., 2005) and clinical (Hayreh, 1971, 1975, 1976; (Hayreh and Podhajsky, 1982; Mizener et al., 1997) studies on CRAO have provided important information on these factors. The factors influencing the visual outcome in CRAO are discussed at length elsewhere (Hayreh and Zimmerman, 2005) and following is a brief summary.
10.6.3.1 Duration of CRAO
This is by far the most important factor. As discussed above, CRAO lasting for 97 minutes does not produce any detectable visual loss; but after that there is a progressive loss, and when ischemia lasts for 240 minutes or more that results in massive, irreversible retinal damage (Hayreh and Jonas, 2000; Hayreh et al., 2004b). Also, studies have shown that the longer the ischemia, the longer the time-lag before the start of any visual improvement (Hayreh and Weingeist, 1980b; Hayreh et al., 2004b).
10.6.3.2 Residual retinal circulation in CRAO
This has caused a good deal of controversy as to whether there is complete or incomplete CRAO. This is discussed below, in the section dealing with fluorescein fundus angiography soon after the onset of CRAO.
10.6.3.3 Site of occlusion in the central retinal artery
It is generally believed that the site of occlusion in CRAO is at the level of the lamina cribrosa (as revealed by histopathologic studies of enucleated eyes). As discussed above, the narrowest lumen of the artery is where it pierces the dura mater of the optic nerve sheath (Fig.18) (Hayreh 1958). Therefore, in cases of CRAO due to embolism, the chances of an embolus getting impacted at this site are much higher than at any other site in the artery. In contrast to that, the site of occlusion in thrombosis of the CRA is at the lamina cribrosa, as shown by histopathological studies. The site of occlusion is an important factor in determining the amount of residual retinal circulation in CRAO, as discussed below.
10.6.3.4 Presence and area of supply by a patent cilioretinal artery
This can have a major impact on the visual outcome in CRAO, as shown above. The incidence of presence of cilioretinal arteries is discussed above. In CRAO, if the patent cilioretinal artery supplies the foveal region, the visual acuity is almost always normal, with an intact central island visual field corresponding to the distribution of the artery (Coverdale, 1929; Karjalainen, 1971; Brown and Shields, 1979; (Hayreh and Podhajsky, 1982; Maharajan et al., 2002). Importantly, in CRAO when the supply by the cilioretinal artery just touches the foveola (Fig. 9), initially there is a marked decline in visual acuity, but within 2–3 weeks the visual acuity improves markedly, spontaneously (to almost 20/20) which, again, may erroneously be attributed to whatever treatment is being administered (Hayreh, 2005). However, if it supplies only a small peripapillary region or retina nasal to the disc, a small island visual field may be present, with poor visual acuity (Fig. 7).
10.6.3.5 Causes of occlusion in CRAO
These are discussed above. It is generally believed that the CRAO is always either embolic or thrombotic in origin. Our previous studies on CRAO have shown that embolism is far more common than thrombosis ((Hayreh and Podhajsky, 1982), as was pointed out almost a century ago by Coats (1905, 1913).
GCA is an important and a well-known cause of CRAO, and it is an ophthalmic emergency because of the high risk of bilateral visual loss, which is preventable. Detailed studies have revealed that when these patients develop CRAO, although ophthalmoscopy reveals a classic picture of CRAO with or without optic disc edema, fluorescein fundus angiography discloses combined occlusion of the CRA and one of the PCAs (Hayreh, 1974a; Hayreh et al., 1998b). This is because the two arteries often arise by a common trunk from the ophthalmic artery (Fig. 3) (Hayreh, 1958). GCA has a special predilection for involving the PCA. When GCA causes occlusion of the common trunk of the PCA and the CRA, both arteries are occluded. Therefore, these eyes actually have combined arteritic CRAO and arteritic anterior ischemic optic neuropathy, resulting in a much worse visual loss than in NA-CRAO (see above). Fluorescein angiography is the only way to diagnose this. Therefore, in all patients aged 50 years and older, it is essential to rule out GCA by doing fluorescein angiography, to prevent devastating visual loss due to GCA, which is preventable by early detection and treatment.
10.6.3.6 Fall of perfusion pressure below the critical level in the retinal vascular bed causing CRAO
In addition to the above causes, a fall of perfusion pressure below the critical level in the retinal vascular bed can cause CRAO, in the presence of other predisposing risk factors - usually transient CRAO. The mechanism for development of CRAO in this case is discussed above.
In conclusion, all these factors play important roles in the visual outcome of CRAO and require evaluation. Therefore, it is essential to classify CRAO into its different types to make scientifically accurate observations about the visual outcome, which, as shown above, can be very different in the various types.
10.7 ANTERIOR SEGMENT OF THE EYE
This shows no abnormality specifically related to the CRAO. In these eyes, often the intraocular pressure tends to be slightly lower than in the fellow normal eye, similar to that seen with retinal vein occlusion (Hayreh et al., 2004a), but, unlike in the latter the lower pressure does not last long.
10.8 OPHTHALMOSCOPIC FINDINGS IN CRAO
Nettleship (1891) published a detailed and comprehensive description of the ophthalmoscopic appearance of acute CRAO in the English literature. He stated: “The classical dense, white haze of the central region of the retina with a well-marked clear patch (“cherry-red spot”) at the yellow-spot was very well shown; there were no haemorrhages; the arteries and veins were of about the normal size, but no pulsation could be produced in any of them by pressure with the finger upon the globe.” He went on to describe the appearance of the blood vessels: “The second feature of interest ․… is the stagnation of the arterial blood stream without diminution in the size of the arterial column. ‥… the large size of the vessels appeared to be inconsistent with the conclusion that their supply was cut off. This phenomenon is by no means uncommon in recent cases of embolism or thrombosis of the arteria centralis, and, … is quite compatible with almost, if not quite complete extinction of sight; on the other hand in some cases the arteries are, as is well known, found to be extremely shrunken from the first.” He also gave an excellent diagram of cilioretinal collaterals on the surface of the optic disc, seen in a case 12 months after development of CRAO.
The late fundus findings of CRAO were described by Lorentzen (1969) and Karjalainen (1971) in a retrospective review of the records of 37 and 50 CRAO patients respectively – the patients, however, were not seen by the authors during the acute phase.
While the classical ophthalmoscopic findings of acute and late CRAO are well known, there is limited information concerning the prevalence and temporal development of the fundus findings, drawn from large studies, in part due to the predictability of the clinical picture and the rarity of CRAO. A recent prospective study (Hayreh and Zimmerman, 2007), based on 248 eyes with CRAO, comprehensively described the pattern and evolution of fundus findings in the four types of CRAO. The main findings during the initial examination for permanent CRAO were: retinal opacity in the posterior pole (58%), cherry-red spot (90%), box-carring (19%), retinal arterial attenuation (32%), and optic disc edema (22%) and pallor (39%). The most frequent findings identified at the late stage, based on survivorship curves, were: optic atrophy (91%), retinal arterial attenuation (58%), cilioretinal collaterals (18%), and macular retinal pigment epithelial changes (11%). Compared to the above findings, permanent CRAO with cilioretinal artery sparing had a lower incidence of all macular and optic disc abnormalities. For transient CRAO, the incidence of initial findings varied greatly compared to the other types. Intra-arterial emboli were observed in 20% of patients. Four percent of CRAOs presented with simultaneous bilateral onset.
The study showed that clinicians should be cautioned: reliance upon only one single feature or subgroup of CRAO-associated findings does not provide robust or sensitive diagnosis of this serious, vision threatening condition (Hayreh and Zimmerman, 2007). The type and incidence of fundus findings at the initial visit and in the late phase of CRAO vary by its type. Combined information from this clinical study (Hayreh and Zimmerman, 2007) and experimental studies (Hayreh and Weingeist, 1980a; Hayreh et al., 2004b) on CRAO also showed the following:
10.8.1 Retinal Opacity
The earliest characteristic change in CRAO is the development of retinal opacity, located essentially in the posterior pole, mostly within the temporal arcades, with normal-looking peripheral fundus (Fig. 7,20-A). There is a common misconception that in CRAO retinal whitening must involve the entire retina to justify the diagnosis of CRAO. The opacity is essentially due to opacification of the retinal ganglion cells, caused by acute ischemia, and is seen ophthalmoscopically mainly where there is more than one layer of ganglion cells, that is, mainly in the macular region, extending up to the retinal vascular arcade above and below. Since the number of ganglion cells progressively increases towards the center of the macula, the retinal opacity and associated swelling become progressively worse towards the center of the macula (Figs. 7, 20A) (Hayreh and Zimmerman, 2005, 2007). However, since there are no ganglion cells in the foveola and its avascular region, it is not affected by the retinal opacity. Consequently the normal color of the fundus is still seen through the normal transparent foveolar retina, giving rise to the “cherry-red spot” (Figs. 7, 20A). The absence of retinal opacity in the peripheral retina is due to thinner retina with a single layer of ganglion cells, so that nutrition of the inner layers of the retina, along with that of the outer layers, can be maintained by the choroidal vascular supply. That explains why either normal or varying amounts of peripheral visual field are still present in many of these eyes (see above).
Important information about the time it takes for the retinal opacity to develop following the onset of CRAO was provided by our experimental study in primates with occlusion of the CRA (Hayreh and Weingeist, 1980a). That showed that at the earliest post-occlusion examination of the fundus, i.e. 7 minutes after CRAO, the retina in the posterior pole was already showing a mild degree of opacity. The severity of retinal opacity progressively increased, so that after CRAO of 60 minutes it was marked and after 70 minutes or longer it was of severe degree. The cherry-red spot was evident with CRAO of about 20 minutes or longer. In eyes with short term transient CRAO, multiple retinal opacities, similar to cotton wool spots, are the only findings at the posterior pole (Fig. 19A); this finding has resulted in missing the diagnosis of CRAO because of the common impression that in CRAO the entire retina shows uniform opacity.
Once the retinal opacity resolves, the retina regains its transparency, but histopathology of eyes with long term CRAO showed permanent damage to the inner retina (Hayreh et al., 2004b). When the CRAO lasts for several hours or is permanent, the macular retina looks atrophic. Some eyes develop macular retinal pigment epithelial granularity.
10.8.2 Retinal Vascular Changes
An experimental CRAO study showed the appearance of the retinal vessels that soon after the onset of CRAO varied markedly, irrespective of the duration of CRAO, from being extremely attenuated to normal-looking (Hayreh and Weingeist, 1980a). Box-carring was usually seen in eyes with severe retinal opacity although it was sometimes seen even with less marked opacity; box-carring was usually found in both arteries and veins (Fig. 7,21). On long-term follow-up, in eyes with transient CRAO of less than 105 minutes, the retinal vessels remained normal-looking at all times; however, in those with CRAO for longer than that, the retinal vessels became attenuated. Both experimental (Hayreh and Weingeist, 1980a) and clinical (Hayreh and Zimmerman, 2007) studies showed that normal looking retinal vessels do not rule out complete CRAO, as Nettleship also pointed out as long ago as 1891 (Nettleship, 1891) (see above).
Figure 21.

Fluorescein fundus angiogram of right eye of a patient with CRAO, 29 seconds after injection of the dye, showing normal filling of the choroid and optic disc vessels (supplied by the posterior ciliary artery circulation), but no filling of the retinal vasculature at all as yet. The retinal vessels, particularly the veins, show typical “cattle-trucking” or “box-carring”. (Reproduced from Hayreh 2005.)
10.8.3 Optic Disc Changes
Experimental CRAO study showed that, although the optic disc usually looked pale initially, it was sometimes normal-looking or even hyperemic or edematous (Hayreh and Weingeist, 1980a). The late optic disc findings depend upon the duration of CRAO. If CRAO lasted for about 97 minutes, the disc looks normal; however, with CRAO for more than 97 minutes, the longer the CRAO, the more marked the pallor.
10.8.4 Cilioretinal Optic Disc Collaterals
Since Nettleship (Nettleship, 1891) first reported these vessels in 1891, there have been many such reports. Cilioretinal collaterals represent a compensatory enlargement of capillary anastomoses between the retinal capillaries on the surface of the disc and ciliary capillaries (supplied by the PCAs) in the deeper parts of the optic nerve head (Figs. 6,22) (Hayreh, 1969, 1995, 2001). Since the collaterals do not develop for months, they are not effective in protecting the retina from acute ischemic damage. Cilioretinal collaterals have sometimes been misdiagnosed as disc neovascularization and, under the mistaken notion that CRAO can cause optic disc neovascularization (which is never the case), even treated with panretinal photocoagulation.
Figure 22.
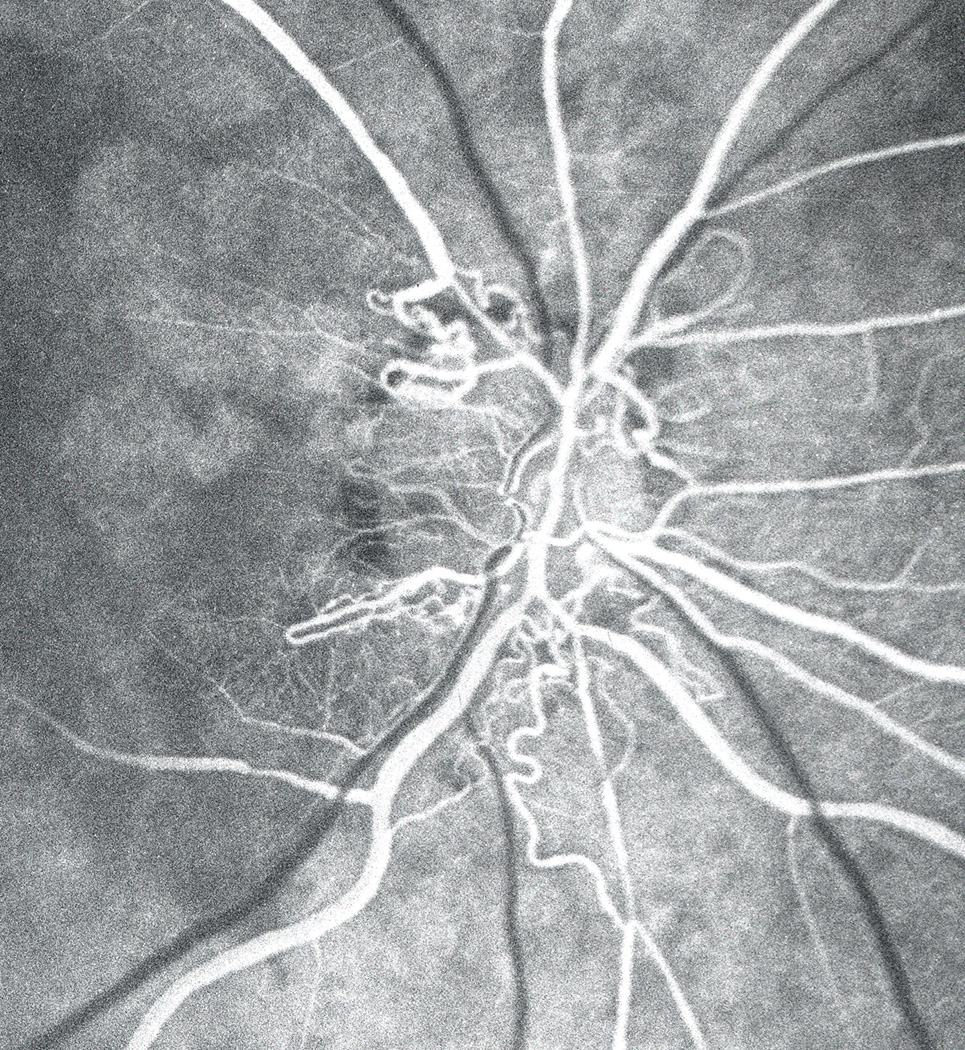
Fluorescein angiogram of an eye with old CRAO, showing multiple retinociliary collaterals. (Reproduced from Hayreh 2005.)
10.8.5 Emboli Seen in the Retinal Arteries
A clinical study on CRAO showed that migration and disappearance of retinal emboli is a common finding (Hayreh and Zimmerman, 2007). In this study, there was migration of retinal emboli in at least 69% of eyes – the actual incidence is likely to be much higher. I have highly instructive and unique fundus photography videos of 3 eyes, given to me by Dr. William Haining from Dundy, Scotland (one during carotid endarterectomy and two without any manipulation). The videos show two fascinating phenomena: (a) during carotid endarterectomy, showers of multiple, moving emboli kept appearing and disappearing without any permanent residual embolus seen later on in the retinal arteries, or visual loss, and (b) the recurrent moving emboli in the retinal arteries followed exactly the same path again and again, as if a hemodynamic process directs the emboli to the same retinal arteriole each time. This latter phenomenon explains why the location of visual loss is usually identical in recurrent episodes of embolic amaurosis fugax. This phenomenon of migratory emboli has practical implications in determining the role of embolism causing CRAO and BRAO. Obviously, if one finds an embolus that is most likely to be responsible for retinal arterial occlusion. However, if one does not find an embolus in CRAO, there may be two reasons. (a) If the embolus is impacted in the proximal part of the CRA, it will not be seen. (b) The embolus may have caused CRAO but migrated and disappeared by the time the eye is examined, so that fluorescein angiography shows normal retinal circulation in spite of classical fundus findings of CRAO (Figs. 19,20). Thus, one cannot always conclude that because no embolism was seen, the occlusion was not caused by an embolus. Calcific emboli usually get impacted and do not migrate, being rough in texture, whereas Hollenhorst emboli, and especially platelet-fibrin emboli, do migrate frequently.
10.9 FLUORESCEIN FUNDUS ANGIOGRAPHY IN CRAO
Fluorescein angiography yields important information in several ways, and it must be performed in all patients who present with fresh visual loss due to CRAO.
It provides definite information to differentiate the various types of CRAO, as is evident from the following:
10.9.1 Arteritic CRAO
Fluorescein angiography is virtually the only way to diagnose this type of CRAO for certain. As discussed above, in these eyes there is also associated PCA occlusion resulting in absent filling of the choroid in the distribution of the occluded PCA – that information is provided only by fluorescein angiography. Missing a diagnosis of arteritic CRAO and consequently of GCA can result in disastrous visual loss in both eyes. Thus, fluorescein angiography plays a crucial role in the diagnosis of arteritic CRAO and GCA – it should be performed in all CRAO patients 50 years and older.
10.9.2
In transient CRAO: it shows normal or almost normal retinal circulation (Fig. 19B, 20B), in spite of the presence of the classical ophthalmoscopic fundus findings of CRAO, i.e. cherry-red spot (Fig. 20A) or a large number of cotton wool spots (Fig. 19A). Thus, without fluorescein angiography the diagnosis may be missed.
10.9.3 In CRAO with cilioretinal artery sparing
Fluorescein angiography is the only way to definitely outline the region supplied by the cilioretinal artery and that is important for determining the visual loss.
10.9.4 Non-arteritic CRAO
These eyes almost always show a variable amount of retinal circulation, with sluggish filling of the retinal vasculature (Fig. 23), and it is rare to see complete absence of retinal vascular filling in these eyes (Fig. 21). Presence of variable amount of retinal circulation has resulted in a firm belief by the vast majority of ophthalmologists that in CRAO the CRA is not completely occluded (Brown and Magargal, 1982; David et al., 1967; Karjalainen, 1971). This in turn, is used as a justification for claims of visual improvement from many advocated therapies, even when instituted many hours or even days after the onset of CRAO. For example, Watson (1969) very well reflected the prevailing view that if there is even minimal retinal circulation after CRAO, it is possible to obtain a considerable recovery of vision in these eyes. Therefore, this topic - the presence of some retinal circulation in eyes with CRAO - requires detailed discussion, because it has important practical implications for management.
Figure 23.

Fluorescein fundus angiogram (14 seconds after injection of the dye) of the left eye of a rhesus monkey, immediately after experimentally cutting of the central retinal artery at its site of entry into the optic nerve. It shows slow filling of the retinal arterioles in the posterior part of the fundus in spite of cutting of the central retinal artery. (Reproduced from Hayreh 2005.)
10.9.5 In Eyes With CRAO, Is The Artery Usually Only Partially or Completely Occluded?
Experimental studies provide vital clue to this. In our experimental studies in rhesus monkeys, where CRAO was produced by either clamping or cutting the CRA, fluorescein fundus angiography showed some slow filling of the retinal circulation in the vast majority of eyes (Fig. 23), in spite of the presence of a cherry-red spot and marked retinal infarction clearly indicating massive retinal ischemia (Hayreh, 1969, 1971; Hayreh et al., 1978; Hayreh and Weingeist, 1980a; Hayreh et al., 2004b; Juarez et al., 1986a, b; Kwon et al., 2005; Petrig et al., 1999). Similarly, in our fluorescein angiographic clinical studies of patients with CRAO, we have found identical slow filling of the retinal circulation in the vast majority of eyes (Hayreh, 1971, 1974b; (Hayreh and Podhajsky, 1982; Hayreh and Zimmerman, 2005; Mizener et al., 1997). This definitely proves, that in spite of fluorescein fundus angiography indicating a highly variable amount of retinal circulation, the CRA is completely occluded in CRAO. The following are the mechanisms responsible for the filling of the retinal circulation despite complete occlusion of the CRA.
10.9.5.1 Collateral circulation via cilioretinal capillary anastomoses within the optic nerve head
The optic nerve head has two main sources of blood supply: (a) the superficial layers of the optic nerve head (i.e., the surface of the optic disc) by the CRA circulation, and (b) the deeper layers (i.e. prelaminar, lamina cribrosa and retrolaminar regions) by the PCA circulation (Hayreh, 1969, 1995, 2001) (Fig. 6). The capillary bed in the entire optic nerve is continuous. In CRAO, this permits blood to flow from the normally filling deeper capillaries of the optic nerve head (supplied by the PCAs) to the adjacent empty capillaries of the superficial layers of the optic disc (supplied by the occluded CRA) (Fig. 21). This blood flow in the capillaries of the surface of the optic disc derived from the deeper ones is one of the sources for the retinal circulation seen on fluorescein angiography in these eyes (Hayreh, 1969; Hayreh and Weingeist, 1980a; Petrig et al., 1999; Hayreh et al., 2004b).
10.9.5.2 Collateral circulation via pial and intraneural anastomoses of the CRA
As discussed above, the CRA, at its site of entry into the optic nerve, gives out prominent pial branches in 95% of eyes (Fig. 4), and up to 8 branches while traveling in the optic nerve in 75% (Fig. 5). The great majority of pial branches of the CRA establish very conspicuous anastomoses with pial branches from circle of Haller and Zinn or other orbital arteries (Figs. 4,6) [described and illustrated in detail elsewhere (Hayreh, 1958; Singh (Hayreh) and Dass, 1960b)] and labeled as “collateral branches” in Figs. 4 and 6. My anatomical studies on 100 CRAs in the human showed that, contrary to the prevalent impression, the narrowest lumen of the CRA is where the artery pierces the dural sheath of the optic nerve (Fig. 18) and NOT at the lamina cribrosa (Hayreh, 1958; Singh (Hayreh) and Dass, 1960a). Clinical studies in CRAO suggest that embolism is the most common cause of CRAO ((Hayreh and Podhajsky, 1982). Therefore, the most likely site for embolic occlusion of the CRA is where it enters the sheath of the optic nerve, not, as commonly believed, at the lamina cribrosa (Hayreh, 1971). In spite of complete occlusion of the CRA at its site of entry into the optic nerve dural sheath, all the downstream pial anastomoses of the CRA (Figs. 4,6) are still functioning, and fill the intraneural part of the CRA to a variable extent. This is quite evident on fluorescein angiography because in such eyes the CRA gradually fills from its intraneural part (Fig. 23). In our experimental and clinical studies, the speed and extent of filling of the residual retinal circulation varied markedly from eye to eye, depending upon the size of the anastomoses.
When the site of occlusion is at the lamina cribrosa, no collaterals are available to establish retinal circulation (Singh (Hayreh) and Dass, 1960b); in these eyes, angiography shows only filling of capillaries on the surface layer of the optic disc from deeper PCA circulation (Fig. 21), without any filling of the CRA trunk itself. Thus, angiography provides very useful information about the site of occlusion.
10.9.5.3 Presence of a patent cilioretinal artery of medium or larger size
My angiographic studies have shown that that appreciably improves the slow filling of the retinal vascular bed.
10.9.5.4 When CRAO Eyes Are Seen Many Days or Weeks after the Visual Loss
Our serial angiographic studies in both experimental and clinical CRAO eyes have always shown marked improvement in retinal circulation filling in these eyes (Hayreh, 1971, 1974b; Hayreh et al., 1978; Hayreh and Weingeist, 1980a; Hayreh and Zimmerman, 2008; Hayreh et al., 2004b; Juarez et al., 1986b). The time taken and the extent of this restoration of retinal circulation vary markedly from eye to eye, depending upon the site of occlusion and availability and number of anastomoses. The mechanism of this residual retinal circulation with complete occlusion of the CRA is discussed above.
10.9.5.5 Is It Possible To Obtain Substantial Recovery Of Vision In Eyes With Even A Minimal Residual Retinal Circulation After CRAO?
The prevailing view is that if there is even minimal retinal circulation after CRAO, considerable recovery of vision is possible in these eyes (Watson, 1969). This issue was especially investigated by us in 101 eyes of rhesus monkeys, in two experimental CRAO studies (Hayreh et al., 1980; Hayreh and Weingeist, 1980a, b; Hayreh et al., 2004b). Both clearly showed that there was no correlation between the residual retinal circulation and recovery of visual function, as judged by electrophysiologic and morphologic studies. This is presumably because the very low oxygen tension in the blood of the residual retinal circulation is not adequate to maintain retinal viability (Hayreh and Weingeist, 1980a, 1980b; Hayreh et al., 2004b). Both studies showed that it is almost always the duration of CRAO which is the principal determinant in the production of irreversible retinal damage, i.e. the longer the ischemia, the more marked the retinal damage. The only exception was when a large patent cilioretinal artery was present; the relatively quicker and better filling of the retinal circulation seems to have some protective effect, particularly in the macular retina.
The rationale for various treatments for CRAO and claims of visual improvement have been based on the presence of this residual retinal circulation and the misconception that CRAO is always “incomplete”.
10.10 CRAO AND NEOVASCULAR GLAUCOMA
This is an important and controversial subject and requires discussion. There is a prevalent idea that CRAO can cause anterior segment neovascularization and neovascular glaucoma (Hayreh, 2005), similar to that seen following ischemic CRVO. For example, Duker et al. (1991), based on a study of 33 eyes with CRAO, attributed the development of neovascular glaucoma in 18% to CRAO per se. They claimed that in the majority of cases, carotid artery disease was not responsible for ocular neovascularization in their series because in 5 of the 7 (71%) patients there was no “ipsilateral hemodynamically significant carotid artery disease”. There are several problems with that conclusion.
10.10.1
The common argument put forward has been that the carotid arteries show no severe stenosis in CRAO to cause ocular ischemic and consequently anterior segment neovascularization. However, studies show that ocular ischemia as well as neovascular glaucoma can be present when the internal carotid artery on that side does not show “hemodynamically significant” stenosis or complete occlusion on Doppler evaluation ((Hayreh and Podhajsky, 1982; Mizener et al., 1997). For example, in our study on ocular ischemia (Mizener et al., 1997), 26% of the cases showed only mild stenosis of the internal carotid artery in spite of definite ocular ischemia, and 77% with ocular ischemia had 70% or less stenosis. As discussed above, this reflects the limitations of carotid Doppler evaluation.
10.10.2
In a comprehensive anatomical study on the ophthalmic artery by Hayreh and Dass (1962), the ophthalmic artery was markedly stenosed at its origin from the internal carotid artery in some cases, without any stenosis of the internal carotid artery at all. Similarly, the ophthalmic artery or its ocular branches themselves may be markedly stenosed to produce ocular ischemia and ocular neovascularization.
10.10.3
In the study by Duker et al. (1991) the incidence of development of neovascular glaucoma was 18% among 33 patients with CRAO, while in our study of 232 eyes with complete CRAO, it was only 2.5%. That implies that the sample of neovascular glaucoma patients in Duker et al.’s series must have been biased (Duker et al., 1991). Other factors must have been responsible for that high incidence of neovascular glaucoma in their series, e.g., diabetes mellitus.
10.10.4
As far as the development of neovascular glaucoma is concerned, ischemic CRVO and CRAO cannot be considered as similar in nature. In CRVO, there is chronic retinal hypoxia, while in CRAO there is acute, severe retinal ischemia and infarction. It is the chronically hypoxic retina that is thought to liberate vasoproliferative factor to cause ocular neovascularization, which is totally missing in CRAO. Thus, there is little scientific rationale for CRAO per se to cause ocular neovascularization. Moreover, if CRAO were causing ocular neovascular glaucoma, it would logically cause that in far more than only 2.5% of eyes. In ischemic CRVO, for example, neovascular glaucoma develops in about 45% (Hayreh et al., 1983).
10.11 MANAGEMENT OF CRAO
CRAO is an ophthalmic emergency associated with a catastrophic visual loss. Therefore, there has been tremendous interest in its management since von Graefe described it in 1859 (Von Graefe, 1859). A host of measures have been advocated to improve visual outcome; these include the following:
10.11.1 No treatment
Currently we have no treatment with definitely proven beneficial effect.
10.11. 2 Conventional advocated treatments
These include (i) ocular massage in an effort to dislodge the embolus in the CRA; (ii) a reduction of intraocular pressure by various medical and surgical means to increase retinal perfusion pressure; (iii) vasodilation of the CRA by sublingual isosorbide dinitrate, rebreathing of expired CO2 in a bag, or breathing Carbogen or retrobulbar vasodilators; (iv) antiplatelet therapy; and (v) heparin therapy.
10.11.3 Other miscellaneous advocated treatments
These include: (i) thrombolysis by administering a thrombolytic agent intravenously; (ii) isovolumic hemodilution; (iii) hyperbaric oxygen; (iv) reduction of red blood cell rigidity by giving Pentoxifylline; (v) systemic steroids intravenously to reduce vascular endothelial edema following CRAO; (vi) neodymium:yttrium-aluminum-garnet laser arteriotomy and embolectomy; and (vii) cannulation of the supraorbital artery and retrograde injection of antispasmodic papaverine.
Margo and Mack (1996) stated that if any new therapy for CRAO were to have a major impact on managing this disease, it must at least double or triple the success rate of conventional therapy. I feel that an even more important benchmark would be whether the visual outcome of any advocated therapy is better than the natural history. Atebara et al. (1995), based on 89 patients, concluded that there was no significant difference between patients who underwent conventional treatment and untreated groups (P=0.87). Similarly, Mueller et al. (2003), based on 102 patients, concluded that commonly used minimally invasive treatments for CRAO do not improve the natural course of the disease.
10.11.4 Local Intra-arterial Thrombolysis
In this mode of treatment, intra-arterial fibrinolysis is delivered directly into the ophthalmic artery by super-selective administration of thrombolytic agent. This is currently the most widely advocated therapy and success has been enthusiastically claimed for it. I discussed this in detail elsewhere (Hayreh, 2008a).
10.11.5 Therapeutic considerations for CRAO
When discussing any therapy for CRAO, one has to consider the following issues:
10.11.5.1
The first essential: does a therapy have a scientific rationale? Without it, any treatment must eventually prove useless or even harmful.
10.11.5.2
The gold standard is to compare the outcome of treatment with the natural history of the disease.
10.11.5.3
There are the other important considerations when evaluating any treatment for CRAO. These include the following:
10.11.5.3.1 Retinal tolerance time to acute retinal ischemia
For the management of CRAO, this is the most crucial information, because the chance of recovery of vision only exists as long as the retina has reversible ischemic damage. As discussed above, the retina suffers no detectable damage with CRAO of up to 97 minutes, but after that, the longer the CRAO, the more extensive the irreversible ischemic retinal damage. CRAO lasting for about 4 hours results in massive and irreversible ischemic retinal damage. A review of the various reported studies on thrombolysis in CRAO shows that the time interval between the onset of CRAO and start of thrombolytic therapy has never been less than 4 hours, varying between 6 and 18 hours or even longer; by that time the retina is already dead. Claims of visual improvement in such cases have no scientific merit.
10.11.5.3.2 Proof of improvement of retinal circulation following thrombolysis
In CRAO, this is the most crucial piece of information required, and can only be provided by doing fluorescein fundus angiography before (showing occlusion of the CRA) and immediately after the treatment (showing improved retinal circulation). No other method can demonstrate that thrombolysis is indeed restoring the retinal circulation and retinal function. That information is absent from all the studies claiming a benefit from thrombolysis.
10.11.5.3.3 Nature of the embolus causing CRAO
This is also important, because fibrinolytic agents can dissolve only platelet-fibrin emboli. Arruga and Sanders (1982) showed that 74% of retinal emboli are made of cholesterol, 10.5% of calcific material and only 15.5% of platelet-fibrin. Fibrinolytic agents cannot dissolve cholesterol or calcified material. Therefore, there is no scientific rationale for the use of fibrinolytic agents in at least 85% of CRAO cases. Moreover, there is the important but often overlooked fact that when the CRA is completely occluded, there is no blood flow in the artery from its site of origin from the ophthalmic artery to the site of occlusion; consequently, there is a little chance of a fibrinolytic agent getting to the site of the thrombus in an adequate concentration to dissolve the thrombus.
10.11.5.3.4 Proof of genuine visual improvement
An effective therapy must result in improvement in both visual acuity and central visual fields. As discussed above, central scotoma is invariably present in CRAO (Hayreh and Zimmerman, 2005). Visual acuity improvement without a corresponding improvement in central visual fields implies that the patient has learned to fixate eccentrically, rather than that a genuine improvement has occurred (Hayreh et al., 2002, 2011; Hayreh and Zimmerman, 2005, 2008).
Success with thrombolysis in CRAO has been enthusiastically claimed by many studies (Weber et al., 1998; Weill et al., 1998; Padolecchia et al., 1999; Richard et al., 1999; Fernandez et al., 2002; Kattah et al., 2002; Schmidt et al., 2002; Butz et al., 2003; Mueller et al., 2003; Arnold et al., 2005; Pettersen et al., 2005;). However, a critical review of the published studies shows several fundamental problems, including the following. (a) Almost all studies are retrospective. (b) There is almost no randomized, controlled, masked study. (c) The fundamental flaw in most studies claiming visual improvement is the one stated above: they contain no fluorescein fundus angiographic evidence to document improved blood flow immediately after the therapy, compared to that beforehand. (d) Practically all the studies lack comparison with a satisfactory natural history control. (e) Almost all studies have lumped CRAO into one category and not classified it into its various types to determine the visual outcome; as shown above; a recent study has shown that visual outcome varies greatly among the different types (Hayreh and Zimmerman, 2005). (f) This therapy lacks a scientific rationale in the vast majority, because, as mentioned above, only 15% of the emboli consist of platelet-fibrin, which fibrinolytic therapy could possibly dissolve – the remaining 85% of emboli are made of cholesterol or calcified material, which the fibrinolytic agents cannot dissolve. (g) Most importantly, thrombolytic therapy has invariably been administered 6 to 18 hours or even longer after onset of CRAO, by which time the retina is already dead (see above). Other serious problems have also been pointed out (Hayreh, 1999).
Beatty and Au Eong (2000) performed a meta-analysis of all the published literature germane to local intra-arterial fibrinolysis in CRAO. They concluded that all studies were retrospective and non-randomized, their methodology was often unsatisfactory, and outside of a randomized clinical trial, the use of superselective fibrinolytic therapy for CRAO cannot be recommended based on current evidence. Moreover, Framme et al. (2001), on comparison of visual recovery after intra-arterial fibrinolysis with conventional treatment, found no statistically significant difference between patients treated with the two types of therapies with regard to improvement of visual acuity, and noted, additionally, that thrombolytic therapy carries an increased risk of a stroke. Fraser and Siriwardena (2002) searched the Cochrane Controlled Trials Register, which included only randomized controlled trials in which one treatment for CRAO was compared to another, but found no such trials on CRAO that met their inclusion criteria. They found that the treatments reported in the literature are of unproven efficacy, and concluded that there is currently not enough evidence to decide which, if any, interventions for non-arteritic CRAO have any beneficial effect. Most recently, a prospective, randomized, multicenter clinical trial on thrombolysis was launched by the “Multicenter study of the European Assessment Group for Lysis in the Eye (EAGLE) for the treatment of CRA occlusion” to evaluate the role of local intra-arterial fibrinolysis in CRAO (Feltgen et al., 2006). Schumacher et al., (2010), in that study, found no beneficial effect on visual outcome on the treated versus the untreated groups; however, there was a significantly higher rate of adverse reactions in the treated group (37%) than the control group (4%). This is not surprising, because, as discussed above and elsewhere, thrombolysis therapy has little scientific rationale in CRAO and is not entirely safe (Hayreh, 1999, 2005; Hayreh and Zimmerman, 2005).
Thus, in conclusion CRAO is a classic case of “a disease without any treatment has many treatments”. None of the proposed treatments has so far stood the test of time, in spite of enthusiastic claims. As regards intra-arterial thrombolytic therapy, a beneficial effect is possible only if this treatment is started aggressively in much less than 4 hours from the onset of CRAO, and the occlusion is caused by a fibrin embolus and not cholesterol or calcific embolus.
10.12 PREVALENT MISCONCEPTIONS ON CRAO
The subject of CRAO is plagued with misconceptions, discussed at length elsewhere (Hayreh, 2005). These are responsible for much confusion and controversy on its management. The following is list of some of the major misconceptions (many of them are discussed above).
10.12.1
That in CRAO the CRA is usually only partially and not completely occluded (perhaps the most prevalent misconception)
10.12.2
That the site of occlusion is always at the level of lamina cribrosa.
10.12.3
That retinal function can improve even when acute retinal ischemia due to CRAO has lasted for 20 hours or even longer.
10.12.4
That an eye with CRAO has no chance of spontaneous visual improvement.
10.12.5
That CRAO cannot produce only central scotoma, with intact peripheral visual fields.
10.12.6
That eyes with CRAO, like ischemic CRVO, are at risk of developing neovascular glaucoma.
10.12.7
That CRAO is one clinical entity. As discussed above, CRAO actually consists of four distinct clinical entities, with different visual outcome and fundus findings (Hayreh and Zimmerman, 2005, 2007).
10.12.8
That CRAO is always embolic or thrombotic. As discussed above, it may also be hemodynamic in nature (Hayreh and Zimmerman, 2005, 2007).
10.12.9
That asymptomatic plaque(s) in retinal arteries do not require a detailed evaluation: This misconception may result in a patient later developing retinal arterial occlusion and visual loss, or even a cerebrovascular accident. It is prudent to evaluate the source of the asymptomatic retinal plaque(s) in all cases to prevent these tragedies.
10.12.10
That a treatment which is effective in myocardial infarction and stroke is equally effective in CRAO. This assumption has invariably been put forward for the use of thrombolysis in CRAO, implying that the pathogenesis and management of CRAO are similar to those disorders. However, the morphology, physiology and responses to acute ischemia of the retina are very different from those in the brain and heart. Moreover, CRAO is usually embolic in nature, whereas myocardial infarction is thrombotic - which is why thrombolytic agents work in myocardial infarction. The response of the brain to acute ischemia and its tolerance to ischemia are very different from those of the retina (Hayreh et al., 2004b). Thus, it is an error of judgment to equate CRAO, myocardial infarction and stroke.
11. HEMI-CENTRAL RETINAL ARTERY OCCLUSION
This has recently been reported by Karagoz et al. (2009) in one patient due to thromboembolism and by Rishi et al. (2010) in 4 young adults aged between 22 and 36. Color Doppler examination of the CRA confirmed branching of the artery behind the lamina cribrosa. My anatomical studies of CRA showed two examples of two CRAs in the optic nerve (Fig. 2) (Hayreh 1958). Thus, occlusion of one of those can cause hemi-CRAO. This clinical entity should not be confused with occlusion of one of the two branches of the CRA at the optic disc, which can cause similar ophthalmoscopic findings.
12. BRANCH RETINAL ARTERIOLAR OCCLUSION
As discussed above, the so-called “branch retinal arteries” are in fact arterioles (Hogan et al., 1971). Therefore the universally used term of “branch retinal artery occlusion” is incorrect.
12.1 CLASSIFICATION OF BRANCH RETINAL ARTERIOLAR OCCLUSION
12.1.1 Branch Retinal Arteriolar Occlusion (BRAO)
This may be permanent or transient. It is usually due to embolism and occasionally due to vasculitis.
12.1.2 Cilioretinal Arteriolar Occlusion (CLRAO)
This is a distinct clinical entity, because the cilioretinal arterioles arise from the PCA, instead of the CRA. Furthermore, etiologically CLRAO is of three distinct types: (i) nonarteritic CLRAO alone, (ii) arteritic CLRAO associated with GCA (Hayreh, 1974a, 1990; Hayreh et al., 1998b), and (iii) nonarteritic CLRAO associated with CRVO (Hayreh et al., 2008). Of the three types of CLRAO, those associated with GCA and those with CRVO require detailed discussion about their pathogeneses, which are controversial.
12.1.2.1 Pathogenesis of Arteritic CLRAO Associated with Giant Cell Arteritis
This is an extremely important type of CLRAO. As Kearns (1975) rightly stressed rightly stressed, GCA "ranks as the prime medical emergency in ophthalmology, there being no other disease in which the prevention of blindness depends so much on prompt recognition and early treatment." It can result in massive bilateral visual loss if not detected and treated urgently – the sooner high dose steroid therapy is instituted, the smaller is the risk of any further visual loss (Hayreh and Zimmerman, 2003a, b).
The PCA supplies the optic nerve head (Hayreh, 1969, 1995, 2001) as well as the cilioretinal artery (Fig. 6). GCA has a selective tendency to involve the PCA (Hayreh, 1974a; Hayreh et al., 1998b; Hayreh and Zimmerman, 2003a), resulting in occlusion of it, which in turn results in simultaneous development of both arteritic anterior ischemic optic neuropathy and CLRAO (Fig. 24) (Hayreh, 1974a, 1990; Hayreh et al., 1998b; Hayreh and Zimmerman, 2003a). The arteritic anterior ischemic optic neuropathy causes massive visual loss in these eyes.
Figure 24.

Fundus photograph (A) and fluorescein angiogram (B) of left eye with arteritic anterior ischemic optic neuropathy and associated cilioretinal artery occlusion (arrow), in a patient with giant cell arteritis. Fluorescein angiogram (B) shows normal filling of the area supplied by the lateral PCA, but no filling of the choroid and entire optic disc supplied by the medial PCA or of the cilioretinal artery (Arrow). (Reproduced from Hayreh, S.S., 1978. Int. Ophthalmol. 1, 9–18.)
CLRAO is sometime erroneously diagnosed as “branch retinal artery occlusion” (Fineman et al., 1996), but the so-called “branch retinal arteries”, as discussed above, are in fact arterioles, and GCA is a disease of the medium-sized and large arteries and not of the arterioles (Hayreh et al., 1998b; Hayreh and Zimmerman, 2003a). I have seen patients with CLRAO occlusion diagnosed by ophthalmologists as ordinary BRAO and left untreated, resulting in catastrophic and unnecessary visual loss in both eyes. These eyes present with the following classical, diagnostic clinical picture. A combination of chalky white optic disc edema, retinal infarct in the region of the occluded cilioretinal artery and the presence of PCA occlusion on fluorescein angiography is diagnostic of GCA (Fig. 24) (Hayreh, 1974a, 1990; Hayreh et al., 1998b; Hayreh and Zimmerman, 2003a). In all patients ≥50 years who have cilioretinal artery occlusion, it is essential to rule out GCA and associated PCA occlusion by fluorescein angiography.
12.1.2.2 Nonarteritic CLRAO associated with central retinal vein occlusion
This clinical entity was first described by Oosterhuis (1968) and Hayreh (1971). Since then a large number of reports have been published (McLeod, 2009), mostly anecdotal in nature or on small series of 10 or 11 eyes, except for one recent report based on 38 eyes (Hayreh et al. 2008). In this condition, there is cilioretinal artery occlusion secondary to central or hemi-CRVO.
12.1.2.2.1 Pathogenesis of CLRAO Associated With CRVO/Hemi-CRVO
Several hypotheses have been put forward to explain the simultaneous development of CLRAO and CRVO; that has resulted in a controversy on the subject, which requires detailed discussion. McLeod and Ring (1976), based on a study of 11 eyes, initially postulated that in these patients, “partial obstruction of their PCAs may be the explanation for at least some of our cases”. They went on: “This series may represent a spectrum of ocular vascular lesions intermediate between acute central retinal vein occlusion and acute ischaemic optic neuropathy”. McLeod (2009) most recently, based on a series of presumptions and speculations has postulated the following confusing hypothesis to explain development of CLRAO with CRVO. He concluded: “In eyes with a cilioretinal supply, the probability that cilioretinal infarction will complicate retinal vein occlusion increases with increasing severity of venous obstruction and the more distally the cilioretinal artery arises from the posterior ciliary arterial tree. A distal branch point also facilitates observation of dye front reciprocation within the artery. Indicators of the degree of venous obstruction that may be necessary to instigate cilioretinal infarction include very prolonged dye transit times in the central retinal circulation, exaggerated venous cyanosis and tortuosity, perivenous cotton-wool sentinels, and macular perivenular whitening.” He goes on to state: “In eyes with CRVO and a cilioretinal supply, the degree of venous outflow obstruction that will be sufficient to trigger branch flow exclusion and diversion depends primarily on the site of cilioretinal branching from the PCA circulation. Cilioretinal territories supplied by arteries that arise from the proximal part of the PCA tree are unlikely to be selectively hypoperfused after CRVO, especially if they are hemodynamically isolated from the choroid. Finally, in theory, choroidal arterial steal might also operate by diverting blood from the CRA after CRVO in some of the 60% of individuals in whom the CRA arises from the ophthalmic artery in company with one or more PCAs.”
I have myself performed fluorescein angiography in many such cases, and looking into the fundus camera while the dye is injected, I evaluated the actual dynamics of blood flow in the retinal vasculature and cilioretinal arteries. This provided information that is not always available from the pictures taken during angiography. I discuss below the fluorescein angiographic findings in such eyes, based on my observations. From these angiographic studies in these eyes and my extensive basic, experimental and clinical studies on the PCA circulation, CRVO and CRAO, I find the hypothesis postulated by McLeod inadequate to explain the pathogenesis of development of CLRAO with CRVO and hemi-CRVO. There is no hemodynamic logic in the speculation by McLeod that “choroidal arterial steal might also operate by diverting blood from the CRA after CRVO in some of the 60% of individuals in whom the CRA arises from the ophthalmic artery in company with one or more PCAs.”
Glacet-Bernard et al. (1987) stated that the possibility of primary occlusion of the cilioretinal artery must be considered in these eyes. It has also been proposed that optic disc edema caused by CRVO can cause CLRAO, but this postulate is not valid because optic disc edema cannot cause CLRAO. Some have even attributed CLRAO with CRVO to embolism. Schatz et al. (1991), based on a study of 10 cases, discussed the various possible mechanisms.
To comprehend the pathogenesis of this clinical entity logically, one has to understand the factors that influence the ocular blood flow. Blood flow in general depends upon the intraluminal perfusion pressure (Perfusion pressure = arterial pressure minus venous pressure). Therefore, any factors that either reduce the arterial pressure or increase the venous pressure, or a combination of both, result in reduced perfusion pressure, and consequently decreased blood flow or even no circulation.
Eyes with an additional cilioretinal artery differ from eyes with the CRA as the only source of blood supply, in their retinal arterial and venous drainage systems. The venous drainage from the entire retina is by the central retinal vein, irrespective of the numbers and sources of the arteries that supply the retina. In eyes with a cilioretinal artery, the arterial supply to the retina is obviously from two independent sources - the CRA is the major source, and the cilioretinal artery usually supplies only a small part of the retina, though its distribution can vary widely (Figs.7–10,20) (Hayreh, 1963). The CRA and cilioretinal artery belong to two types of arterial systems with different physiological properties. The CRA has efficient blood flow autoregulation, so that when there is fall in perfusion pressure in the retinal arterial bed caused by a rise in the retinal venous pressure, the autoregulatory mechanism in the retinal arterial bed causes a rise in its pressure, to try to maintain retinal circulation. By contrast, since the cilioretinal artery belongs to the choroidal vascular system, the following two entirely different mechanisms are working in the cilioretinal artery circulation in eyes with CRVO: (a) the choroidal vascular bed has no autoregulation, and (b) in eyes with CRVO, there is no vortex venous obstruction. Therefore, with a sudden onset of CRVO, the decrease in perfusion pressure in the CRA kicks in the autoregulatory mechanism to maintain its blood flow; by contrast, no such compensatory mechanism exists in the choroid or the short PCAs supplying the choroid. Moreover, studies have shown that the perfusion pressure in the choroidal vascular bed is normally lower than that in the CRA (Hayreh, 1970; Hayreh et al., 1970; Blumenthal et al., 1971). Thus, the theory that in these eyes there is partial obstruction of their PCAs, and that this represents “a spectrum of ocular vascular lesions intermediate between acute central retinal vein occlusion and acute ischaemic optic neuropathy” (McLeod and Ring, 1976) cannot be accepted; moreover, contrary to what they stated, there is no evidence of anterior ischemic optic neuropathy in these eyes.
In the light of the above facts, let us consider then the hemodynamic situation in eyes that have a cilioretinal artery and develop CRVO. Sudden occlusion of the central retinal vein results in a marked rise of intraluminal pressure in the entire retinal capillary bed; when that intraluminal pressure rises above that in the cilioretinal artery, the result is a hemodynamic stasis in the cilioretinal artery. In many of these eyes, angiography performed during the early acute phase provided information about the in vivo dynamics of blood flow in the eye. During the early stages of the transit of the dye, the cilioretinal artery in these eyes usually fills up to the optic disc, since the optic nerve head is supplied mainly by the PCA circulation (Fig. 6) (Hayreh, 1969, 1995, 2001). During systole, the cilioretinal artery often fills for a variable length from the optic disc into the retina, but during diastole, the filling retracts to the optic disc, resulting in an oscillating blood column in the cilioretinal artery, moving back and forth from the optic disc for a variable distance into the retina; I have often observed this while observing the fundus during angiography - it presents a fascinating phenomenon. Thus, the CLRAO in these eyes is simply a hemodynamic stasis and is not due to embolism or thrombosis. The hemodynamic stasis is invariably transient, lasting from a few hours to several days, depending upon how severe the retinal venous stasis and how rapidly the collateral circulation is established by the central retinal vein through its multiple tributaries in the optic nerve (Fig. 6) (Hayreh et al., 2011). Therefore, in the optic nerve, the further anterior the site of occlusion in the central retinal vein, the fewer tributaries are available (Fig. 6), and the longer it takes to re-establish the circulation - and vice versa. As soon as those tributaries establish collateral circulation, there is a fall of intraluminal pressure in the retinal capillary bed to below that of the blood pressure in the cilioretinal artery, resulting in restoration of retinal circulation in the distribution of the cilioretinal artery. Hence, if a patient with CRVO is seen for the first time many days after the onset of CLRAO, a retinal infarct is present ophthalmoscopically, but on angiography the cilioretinal artery fills normally; that can result in confusion and mistaken diagnosis of the cause of retinal infarct – this may be why occlusion of the cilioretinal artery has mistakenly been attributed to embolism. In hemi-CRVO, when one of the two trunks of the central retinal vein is occluded (Fig. 14), the above mechanism applies only to the segment of the retina drained by the occluded trunk (Hayreh and Hayreh, 1980).
Patients with CRVO associated with CLRAO often have two complaints:
-
1
Visual loss is often first discovered on waking up from sleep, or in the morning on first opportunity to use fine central vision. To comprehend the reason for that, one has to consider the important phenomenon of nocturnal arterial hypotension (Fig. 16).
Nocturnal arterial hypotension
A fall of blood pressure during sleep is a well-established physiological phenomenon. We have investigated that by doing 24-hour ambulatory blood pressure monitoring in more than 700 patients, whose blood pressure was recorded every 10 minutes during waking hours and every 20 minutes during sleep. In our studies (Hayreh et al., 1999; Hayreh et al., 1994), we found that during sleep systolic blood pressure fall by 34.8±1.2% and diastolic by 44.0±1.3% from the daytime pressures (Fig. 16) (Hayreh et al., 1999). This fall is aggravated by overmedication with blood pressure lowering medication, particularly when given in the evening or at bedtime. There were two other important relevant findings of our study.
The most impressive finding in our 24-hour ambulatory blood pressure monitoring study was that systemic arterial blood pressure is the most volatile parameter in the human body, and is greatly influenced instantaneously by emotional factors. This is particularly true of a patient who has just suffered visual loss and is emotionally upset. In view of that, arterial hypertension discovered in CRVO patients at the time of their diagnosis may be of one of the three types: (a) genuine, (b) temporary arterial hypertension due to emotional upset at sudden visual loss, or (c) “white coat hypertension”. Unfortunately, it is not unusual that a patient newly diagnosed with CRVO and found to have transient arterial hypertension in his ophthalmologist’s office may be treated aggressively by physicians without realizing that that may not be genuine arterial hypertension at all. That action has the potential of precipitating or aggravating the visual loss in CRVO eyes with cilioretinal artery (see below), and also can convert non-ischemic CRVO to ischemic CRVO.
Daytime blood pressure usually has no relationship to the nighttime blood pressure (Fig. 16). Since blood pressure is always evaluated during the daytime, and that almost invariably gives no information about the blood pressure during sleep.
Thus, an understanding of nocturnal arterial hypotension has important implication both for comprehension of the mechanism of development of CLRAO with CRVO and for management of these patients (see below).
Therefore, in eyes with CRVO and cilioretinal artery, the following sequence of events takes place: a fall of systemic blood pressure during the night → secondary falls in the cilioretinal artery blood pressure (without any appreciable change in the intraluminal pressure in the retinal capillary bed caused by CRVO) → hemodynamic block in the cilioretinal artery during sleep → no retinal circulation in its distribution for several sleeping hours → retinal infarct in the distribution of the cilioretinal artery. Thus, a marked fall of blood pressure during sleep (Fig. 16) may play an important role in the development of CLRAO in these patients.
-
2
In our study (Hayreh et al., 2008) at least one third of patients gave a definite history of episode(s) of transient visual blurring before the onset of constant blurred vision. This is most probably also due to a transient fall in systemic blood pressure during waking hours, for whatever reason (e.g., orthostatic hypotension), resulting in a transient hemodynamic stasis in the cilioretinal artery. Naturally the question arises – why did only one third of our patients experience that, and not all? Whether a patient gets these episodes of transient blurring before developing CLRAO depends upon the difference between the intraluminal pressures in the retinal capillary bed and in the cilioretinal artery (see above). There can be two scenarios. (i) If the difference between the two pressures is very small, even a transient, mild fall of systemic blood pressure is enough to precipitate such an episode, e.g., with orthostatic hypotension. (ii) But, if that difference is substantial, it would require, proportionately, a much greater fall of systemic blood pressure (e.g. with marked nocturnal arterial hypotension); in the latter case, these episodes may be occurring during sleep but the patient is not aware of them.
As discussed above, there are eyes with a cilioretinal artery, which do not develop CLRAO with CRVO. We have seen the following two types of cases (Hayreh et al., 2008).
Many patients, when first seen many days or weeks after the onset of visual blurring, showed no evidence of infarction or any significant delay in filling of the cilioretinal artery. In these eyes, obviously, the intraluminal pressure in the retinal capillary bed was never high enough to interfere with cilioretinal artery filling. This may be due to slow development of CRVO, which allows time for collaterals to develop in the optic nerve, so that the intraluminal pressure in the retinal capillaries never goes high enough to cause hemodynamic stasis in the cilioretinal artery. The other possible explanation is that the cilioretinal artery is a direct branch of the PCA and not a part of the choroidal vascular bed, so that it has the same intraluminal pressure as the CRA – both arising from the ophthalmic artery (Fig. 3).
We have also seen an occasional patient who presented soon after having developed transient visual obscuration, with markedly engorged retinal veins and none or a rare retinal hemorrhage. In these eyes, angiography revealed markedly delayed filling of the cilioretinal artery but no occlusion. This indicates that in these eyes, although the intraluminal pressure in the retinal capillary bed is high enough to cause delayed filling of the cilioretinal artery, it is not high enough as yet to produce a complete hemodynamic block and infarction, although enough to produce transient visual obscuration. This would indicate that delayed filling of the cilioretinal artery occurs much earlier than development of complete hemodynamic block and retinal infarction. Most likely, the retinal infarction develops in these eyes when nocturnal arterial hypotension during sleep causes intraluminal pressure in the cilioretinal artery to fall below the critical level, resulting in a hemodynamic block in the cilioretinal artery lasting many hours.
To understand why some eyes with CRVO associated with CLRAO develop permanent visual loss in the distribution of cilioretinal artery while others have only temporary loss, it is essential to consider retinal tolerance time to acute ischemia (Hayreh and Jonas, 2000; Hayreh et al., 2004b) (see above). The severity of visual loss and the recovery of retinal function depend upon the duration and severity of retinal ischemia in the area of retina supplied by the cilioretinal artery. If in these eyes acute retinal ischemia lasts for ≤ 97 minutes, that causes no irreversible retinal damage and the retina recovers its function fully, but after that, the longer the acute ischemia, the greater the irreversible ischemic damage. If the acute retinal ischemia lasts for about 4 hours, they show no recovery of visual function.
12.2 RECURRENT BRAO
This has been reported by several authors as distinct clinical entity (Barak et al., 1997; Beiran et al., 1995; Beversdorf et al., 1997; Johnson et al., 1994), and I have seen such patients. Most of these patients probably have Susac’s syndrome or recurrent embolization from lesions in the carotid arteries or the heart.
12.3 DEMOGRAPHIC CHARACTERISTICS OF VARIOUS TYPES OF BRAO
Following were the findings in various types of BRAO reported in a recent study (Hayreh et al., 2009a), based on 199 consecutive untreated patients (212 eyes).
12.3.1 Permanent BRAO
This was seen in 61% of the patients with BRAO; 39% were female and 61% male. In this group, 9% had bilateral BRAO. The mean age at first onset was 61±14(SD) years, with a range of 13 to 83 years. The two most common types were superior temporal BRAO (31%) and inferior temporal BRAO (28%), and less common were those involving the entire superior (11%) or inferior (8%) halves, those involving only the macular region (10%), combined superior and inferior temporal (5%) and other miscellaneous combinations in the rest. Of the 133 eyes in this group, 46% were first seen within 7 days of onset. 58% had follow-up of at least 6 months, with median follow-up of 2 years.
12.3.2 Transient BRAO
This was seen in 6% of the patients (18 eyes). This type of BRAO involved the superior temporal in 7 eyes, inferior temporal in 6, superior temporal + inferior temporal in 1, peripheral superior temporal in 1, foveolar arteriolar 1, and unknown in 2.
12.3.3 Cilioretinal Artery Occlusion
As discussed above, CLRAO is of three types. In the recent study on 199 patients with BRAO, there was non-arteritic CLRAO alone in 6%, non-arteritic CLRAO associated with CRVO/hemi-CRVO in 19% and CLRAO associated with GCA in 6% (Hayreh et al., 2009a).
12.3.3.1 Non-arteritic CLRAO alone
The location of the CLRAO in 11 eyes in this group was superior in 5, inferior in 2, and macular in 4.
12.3.3.2 Non-arteritic CLRAO associated with CRVO/hemi-CRVO
CLRAO in this group was associated with nonischemic CRVO in 80%, ischemic CRVO in 13% and nonischemic hemi-CRVO in 8%. Patients with nonischemic CRVO were significantly younger (mean 45.3±16.0 years) than those with ischemic CRVO (72.3 ± 9.2 years; P=0.001) and nonischemic hemi- CRVO (64.7 ± 7.5 years; P=0.018). At least one third of the patients gave a definite history of episode(s) of transient visual blurring before the onset of constant blurred vision. McLeod (2009) recently stated that CRVO with cilioretinal artery occlusion “is arguably the most common cause of retinal infarction in young individuals.” I have studied about 700 CRVO, 200 hemi-CRVO, 300 CRAO and 200 BRAO cases; my findings do not support the statement by McLeod (2009).
12.3.3.3 Arteritic CLRAO associated with GCA
This was present in 11 patients (12 eyes) with temporal artery biopsy confirmed GCA. There were 7 females and 4 males, with an age range of 57 to 79 (mean 69.4 ± 6.8 SD) years. Three eyes had episode/s of amaurosis before permanent visual loss. Of the 12 eyes, 10 had associated arteritic anterior ischemic optic neuropathy, one arteritic posterior ischemic optic neuropathy and in only one eye CLRAO was not associated with optic neuropathy.
12.4 VISUAL OUTCOME IN BRAO
A recent study of 199 consecutive untreated patients (212 eyes) with BRAO provides detailed information on the visual acuity, visual fields and natural history of visual outcome in the various types of BRAO (Hayreh et al., 2009a). Following is a brief account based on the findings of that study.
12.4.1 VISUAL ACUITY
This varies in different types of BRAO.
12.4.1.1 Permanent BRAO
At the first clinic visit
A visual acuity of 20/40 or better was recorded in 74% of those first seen within 7 days of onset (64% in those with superior temporal BRAO and 82% in those with inferior temporal BRAO), in 78% of those seen between >1 weeks and one month, and in 84% of those seen more a month after onset. Visual acuity of 20/200 or worse was seen in 11%, 9% and 9% respectively.
Visual acuity on follow-up
In eyes with initial visual acuity of worse than 20/40, it improved by at least 3 lines in 79% among those seen within one week, in 25% among those seen >1 week and 1 month, and in 67% among those seen one month after onset.
12.4.1.2 Transient BRAO
There were 18 patients (18 eyes) with transient BRAO and only one presented with worse than 20/40 initial visual acuity, which, on follow-up, improved to 20/30, with the rest maintaining vision of 20/40 or better.
12.4.1.3 Cilioretinal Artery Occlusion
CLRAO is of three types:
12.4.1.3.1 Non-arteritic CLRAO alone
Of 11 eyes in this category, 3 presented with worse than 20/40 visual acuity, and all 3 improved during follow-up.
12.4.1.3.2 Arteritic CLRAO associated with GCA
There were 11 patients (12 eyes) in this group. Initial deterioration of visual acuity was primarily due to associated arteritic anterior/posterior ischemic optic neuropathy in all but one, where it was 20/70. Visual acuity depends upon the type of visual field defect caused by the ischemic optic neuropathy. In the remaining 11 eyes, initial visual acuity was 20/20 in 1, 20/25 in 2, 20/40 in 1, counting fingers in 2, hand motion in 1, light perception in 2 and no light perception in 2. All were treated with systemic corticosteroids without any visual change on follow-up.
12.4.1.3.3 CLRAO associated with CRVO/hemi-CRVO
There were 38 eyes of this type. A detailed account of visual acuity in this is given elsewhere (Hayreh et al., 2008). Initial deterioration of visual acuity was due to either the CLRAO involving the foveal region or macular edema caused by CRVO or hemi-CRVO; therefore, visual acuity data are not necessarily related to CLRAO in this group. During follow-up, visual acuity improved markedly in eyes with associated nonischemic CRVO (P<0.001) and nonischemic hemi-CRVO but deteriorated in those with associated ischemic CRVO (primarily due to ischemic CRVO).
12.4.1.4 Conclusion
Visual acuity of 20/40 or better is seen on final follow-up in 89% of permanent BRAO cases, 100% of transient BRAO and 100% of nonarteritic CLRAO alone. The effectiveness of various treatment modalities for visual outcome has to be judged against this background. Similarly, Yuzurihara and Iijima (Yuzurihara and Iijima, 2004), in a retrospective study of 30 eyes with BRAO concluded, “visual acuity in patients with BRAO is far better both at presentation and at the final visit”. Mason et al. (Mason et al., 2008), based on a retrospective study of 52 eyes with BRAO concluded: “Visual prognosis after BRAO seems to be correlated to presenting VA (visual acuity)” but the findings of the study by Hayreh et al. (2009a) in 212 eyes with BRAO does not support the conclusion of Mason et al. (2008).
12.4.1.5 Factors influencing the visual acuity improvement in BRAO and CLRAO
12.4.1.5.1
Most importantly, when the junction between the normal and infarcted retina in BRAO (as is the case in the majority of temporal BRAO eyes – Fig. 25) and CLRAO (Fig. 8) passes through the fovea, the visual acuity may suddenly deteriorate initially, but marked spontaneous visual acuity improvement can occur within several days or weeks, even from 20/200 or worse to normal (Hayreh, 2005). This is an important fact which most ophthalmologists are unaware of. Persons advocating various treatments often see this spontaneous improvement (or improvement due to the patient learning to fixate eccentrically) as the beneficial result of their therapy (Garcia-Arumi et al., 2006; Opremcak et al., 2008).
Figure 25.

Fundus photograph of the left eye with inferior branch retinal artery occlusion, with an embolus (white) impacted at its origin on the optic disc. Note the junction of the normal (upper half) and infarcted (lower half) parts of the retina, passing through the fovea. (Reproduced from Hayreh 2005.)
Figure 8.

Two fundus photographs (A = right eye, B = left eye) of eyes with non-ischemic central retinal vein occlusion associated with occlusion of cilioretinal arteries (arrows). Note the difference in size of the cilioretinal arteries in the two eyes - a large one in A and a small one in B.
12.4.1.5.2
The retina can recover/improve function only so long it is not irreversibly damaged by acute ischemia. As discussed above, the retina suffers no detectable damage with CRAO of ≤ 97 minutes but above that level, the longer the CRAO, the more extensive and irreversible the damage, so that CRAO lasting for about 4 hours results in massive irreversible retinal damage. That must apply to BRAO and CLRAO too. Almost all the advocated modes of treatments claiming visual acuity improvement have been performed far longer than 4 hours after the onset, which indicates that their results simply reflected natural history.
12.4.2 VISUAL FIELD DEFECTS
There is practically no information in the literature on the visual field defects in BRAO, except for a recent detailed study (Hayreh et al., 2009a), which showed that visual field defects differ among the various types of BRAO. The location of BRAO obviously determines the location and type of visual field defect.
12.4.2.1 Permanent BRAO
At the first clinic visit
Of those seen within 7 days of onset, 7% had no scotoma, 20% had central scotoma, and 13% had inferior central altitudinal defect. There was no peripheral defect in 15% of eyes seen within 1 week of onset; there was a peripheral defect in the inferior nasal sector in 29% and in the superior nasal sector in 24%.
Visual field on follow-up
Of those seen within 7 days of onset, abnormal central visual field defect improved in 47%, while only 6% got worse. The abnormal peripheral visual field defect improved in 52% and got worse in 3%.
12.4.2.2 Transient BRAO
Central and peripheral visual fields remained normal during the entire follow-up period.
12.4.2.3 Cilioretinal Artery Occlusion
12.4.2.3.1 Non-arteritic CLRAO alone
All the 11 eyes in this group had a central visual field defect at the initial visit (6 eyes with centrocecal scotoma, 3 with central scotoma, 1 with superior central altitudinal defect, and 1 with inferior central altitudinal defect). A peripheral visual field defect was seen in 3 eyes with cilioretinal arteries of large size only (Fig. 8A), corresponding to their location. Of the 9 eyes with follow-up, the central field improved in 4 eyes, worsened in 1, and remained stable in 4. The 8 eyes with normal peripheral fields all remained normal during the follow-up period.
12.4.2.3.2 Arteritic CLRAO associated with GCA
In this type, associated arteritic ischemic optic neuropathy is the primary cause of visual field loss (Fig. 24). All were treated with systemic corticosteroids without any visual change on follow-up.
12.4.2.3.3 CLRAO associated with CRVO/hemi-CRVO
In this type, central visual field defects were usually due to CLRAO, and the most common (42%) and typical defect was centrocecal scotoma. As with the visual acuity, visual field improvement was common in the nonischemic CRVO group.
12.5 OPHTHALMOSCOPIC FINDINGS IN BRAO
The following information was provided by a recent study (Hayreh et al., 2009a):
12.5.1 Permanent BRAO
Initially there is segmental retinal infarction in the region supplied by the occluded branch retinal arteriole (Fig. 25); there is frequently attenuation and box-carring of the blood column in the vessels in the involved region, with or without the presence of an embolus, which when present is almost invariably at the site of bifurcation of the arteriole (Fig. 25). When BRAO is due to vasculitis, there is evidence of that. When there is superior or inferior temporal BRAO, the junction between the normal and infarcted retina usually passes through the foveal region (Fig. 25) - the importance of this fact is discussed above, regarding spontaneous visual acuity improvement. On follow-up, there is resolution of the retinal infarct; the median time to resolution was 4 to 4.4 weeks, with 20% resolving at 2.1 weeks and 80% resolved by 5.6 weeks. Median time to disc pallor in the corresponding sector was 6.7 to 6.9 weeks, with pallor present in 25% at 4 to 4.6 weeks, and 67% at 12.7 to 13.6 weeks.
12.5.2 Transient BRAO
Ophthalmoscopic findings in this type depend upon the duration of transient BRAO. Quite often there is patchy retinal opacity and that has been misdiagnosed as cotton-wool spots so that the diagnosis of transient BRAO is missed. In some eyes with prolonged occlusion, a cherry-red spot may be seen in some eyes. The retinal lesions resolve faster than in permanent BRAO.
12.5.3 Cilioretinal Artery Occlusion
The findings depend upon the type of CLARO. In non-arteritic CLRAO alone, the findings are similar those described above in permanent BRAO. In arteritic CLRAO, apart from retinal infarction in the involved region, there is also optic disc edema which is mostly chalky white swelling – a diagnostic appearance (Fig. 24). In non-arteritic CLRAO associated with CRVO, there are fundus findings of the latter with retinal infarct in the region of the involved cilioretinal arteriole (Fig. 8).
12.6 FLUORESCEIN FUNDUS ANGIOGRAPHIC FINDINGS IN BRAO
12.6.1 Permanent BRAO
When angiography is performed soon after the onset of visual loss, the involved retinal arteriole does not fill. Later on, retrograde circulation in the involved region starts to fill from the adjacent normal retinal vessels. When BRAO is caused by an embolus and angiography is done within the first 2–3 days of onset, this usually shows staining of the vessels wall at the site of embolus, even if it has moved away.
12.6.2 Transient BRAO
By the time a patient is seen, the retinal circulation is normal. This usually results in missing the diagnosis.
12.6.3 Cilioretinal Artery Occlusion
The findings depend upon the type of CLRAO.
12.6.3.1 Non-arteritic CLRAO alone
The findings are similar to those described above in permanent BRAO.
12.6.3.2 Arteritic CLRAO associated with GCA
Apart from non-filling of the involved cilioretinal arteriole, typically there is also absent filling of the choroid in the distribution of the occluded PCA; that is the diagnostic feature of this type of CLRAO (Fig. 24).
12.6.3.3 CLRAO associated with CRVO/Hemi-CRVO
Fluorescein fundus angiography provides useful information in these eyes. Normally, the cilioretinal artery starts to fill just before the CRA at the optic disc, though in some eyes the cilioretinal and central retinal arteries start to fill at the same time. However, in eyes with CRVO associated with CLRAO, the filling pattern seen in the cilioretinal artery depends upon the time lapse between the onset of visual symptoms and fluorescein angiography. (i) When the eyes are seen shortly after the onset, they show the classical oscillating blood column in the cilioretinal artery described above, i.e. the artery fills for a variable distance from the optic disc during systole but the filling is retracted to the optic disc during diastole. (ii) However, when the eyes are seen a few days after the onset of symptoms, the cilioretinal artery starts to fill – the shorter the time interval, the longer it takes the artery to fill; this filling can cause confusion about the cause of the retinal infarct in these eyes. The time the cilioretinal arteriole takes to fill on angiography depends upon: (a) the severity of retinal venous stasis caused by CRVO – the more marked the stasis, the longer it takes to fill, (b) the speed with which the venous collaterals developed in the optic nerve, and (c) the time lapse between the onset of visual symptoms and the first clinic visit (and angiography), which can vary widely among patients.
12.7 MANAGEMENT OF BRAO
This depends upon the type of BRAO.
12.7.1 Permanent BRAO
Surgical embolectomy has recently been advocated by some in this type of BRAO. Garcia-Arumi and colleagues (Garcia-Arumi et al., 2006) claimed that “Surgical removal of retinal arterial emboli seems to be an effective and safe treatment for RAO”. However, as discussed elsewhere (Hayreh, 2007), the visual acuity improvement attributed by Garcia-Arumi and colleagues to embolectomy simply represents natural history (Hayreh et al., 2009a). Opremcak et al. (2008) advocated doing translumenal Nd:YAG embolysis/embolectomy. As discussed elsewhere (Hayreh, 2008b), the claims made were unfortunately undermined by a series of flaws in this study. The only possible role of translumenal Nd:YAG embolysis/embolectomy for CRAO and BRAO is if the procedure is done within two or three hours of the onset of visual loss, while the retina is still not irreversibly damaged by acute ischemia. Thus, like CRAO, permanent BRAO lacks any proven treatment for so far.
12.7.2 Transient BRAO
Because of the transient nature of BRAO, these eyes do not require any treatment when first seen.
12.7.3 Cilioretinal Artery Occlusion
12.7.3.1 Non-arteritic CLRAO alone
There is no proven treatment for this type of CLRAO.
12.7.3.2 Arteritic CLRAO associated with GCA
In these cases, the treatment is basically the management of GCA. These patients require urgent high-dose steroid therapy to control GCA, to prevent any further visual loss.
12.7.3.3 CLRAO associated with CRVO/Hemi-CRVO
There is no proven treatment for this type of CLRAO. While we do not have any established treatment for BRAO, the most important consideration is the prevention of development of another BRAO, CRAO or even stroke; this is achieved by a detailed evaluation of the cause of embolization and careful management of it. Most importantly, as discussed above, the natural history of visual outcome for these conditions is excellent, with visual acuity of 20/40 or better is seen finally on follow-up in 89% of permanent BRAO cases, 100% of transient BRAO and 100% of nonarteritic CLRAO alone.
13. COTTON WOOL SPOTS
Cotton wool spots (CWSs) are common, acute, non-specific retinal lesions, seen in retinopathies due to a whole host of conditions, including diabetic retinopathy, hypertensive retinopathy, retinal vein occlusion, GCA, systemic lupus erythematosus, Wegener's granulomatosis, dermatomyositis, human immunodeficiency virus retinopathy, cytomegalovirus retinitis, Purtscher's retinopathy, Bartonella henselae neuroretinitis, radiation retinopathy, bone marrow transplantation retinopathy, Interferon-associated retinopathy, sarcoidosis, acute leukemia, Dengue fever, Takayasu's arteritis, malaria retinopathy and retinopathy of pancreatitis.
The presence of CWSs in GCA requires special mention since, as discussed above, GCA is an ophthalmic emergency where early detection and immediate management with high-dose steroid therapy can prevent catastrophic visual loss. There are several reports of the presence of CWSs in GCA. In our study of ocular involvement by GCA, of the 123 eyes with visual loss, one third had CWSs during the early stages of the disease (Hayreh et al., 1998b). So in all patients aged 50 years and over, the presence of CWSs is a signal to rule out GCA to prevent visual loss.
13.1 CLINICAL FEATURES OF CWSs
To understand the true nature and pathogenesis of CWSs, one has to have an in-depth understanding of the retinal capillary bed; this is discussed above in some detail. The retinal capillaries are arranged in two layers (Fig. 12); (i) a superficial layer in the ganglion cell and nerve fiber layers, and (ii) a deeper layer in the inner nuclear layer, which is denser and more complex than the superficial layer. Among the superficial layer of retinal capillaries in the posterior part of the fundus, there is a special type of retinal capillaries called “radial peripapillary capillaries” (Fig. 13). Their characteristic features are discussed above. Radial peripapillary capillaries, because of their distinctive features, play an important role in the development of several lesions. For example, CWSs are often located in their distribution, which indicates that they may play a role in their pathogenesis
I have conducted detailed studies of CWSs in 60 eyes with hypertensive retinopathy (Hayreh et al., 1989) and in more than a thousand eyes with various types of retinal vein occlusion. These have revealed the following:
13.1.1 Ophthalmoscopic findings
Typically, CWS are initially fluffy white focal areas of retinal opacity, having irregular polymorphous shapes, frequently with somewhat feathery margins; this appearance is due to axoplasmic flow accumulation caused by focal ischemia of the nerve fiber layer. CWSs are located in the retinal nerve fiber layer, usually located in the distribution of the radial peripapillary capillaries of the retina (Fig. 13), and more closely related to the arterioles than to the venules. During the acute phase they usually bulge slightly forward on the surface of the retina. The characteristic life cycle of a CWS is that it resolves into a dull,fragmenting white patch before disappearing completely, leaving a normal-looking transparent retina on ophthalmoscopy. According to McLeod et al. (1977) the opaque lesion of CWSs represents accumulated axoplasm and cytoplasmic debris in the nerve fiber layer due to obstruction of orthograde or retrograde axoplasmic transport.
13.1.2 Fluorescein fundus angiographic findings
During the acute phase it shows non-filling of the retinal capillaries in the region of the CWSs, which could be partly due to the overlying opaque retina. Once the retinal opacity associated with CWSs has cleared and the retina looks normal, fluorescein fundus angiography reveals permanent focal capillary nonperfusion in its location (as a lasting foot-print of the lesion). The extent of retinal capillary obliteration in an eye depends essentially upon the extent and distribution of the CWSs (Fig. 26). A terminal retinal arteriole, corresponding to a large patch of capillary obliteration, is occluded and frequently develops sheathing. At the junction between obliterated capillaries and the surrounding normal filling capillaries stumps of the remaining capillaries are seen which have erroneously been diagnosed as microaneurysms.
Figure 26.

Fluorescein fundus angiogram of an eye of a rhesus monkey with experimental malignant arterial hypertension, after resolution of multiple large cotton wool spots, showing patches of retinal capillary obliteration corresponding to the old cotton wool spots. Fluorescent leaking spots represent focal intraretinal periarteriolar transudates.
13.1.3
Retinal nerve fiber loss: has been reported corresponding to the location of the CWSs (Fig. 27).
Figure 27.

Fundus photographs of right eye of a rhesus monkey with experimental malignant arterial hypertension. It shows resolution of old cotton wool spots and appearance of new ones, with development of multiple areas of nerve fiber bundle loss in the location of resolved cotton wool spots (seen as dark semicircular bands in the macular region). (Reproduced from Hayreh et al. 1989)
13.1.4 Histopathological studies
There are many such studies of CWSs. For example, Leishman (1957) found the CWSs related to a small thrombotic arteriole. Ashton (1959), in his India ink injected specimens, found no filling of the capillaries in the CWSs. Ashton and Harry (1963) in flat preparation of the retina, found the terminal or precapillary arterioles supplying the CWSs obstructed. Thus, they concluded that CWSs in hypertensive retinopathy are a result of sudden occlusion of a small arteriole and that CWSs represent infarcts. The cause of terminal retinal arteriolar occlusion in malignant hypertension is most probably fibrinoid necrosis, a basic pathologic lesion of malignant hypertension. Pathological studies have shown the presence of cytoid bodies in the location of the CWSs. Electron microscopy has shown that the cytoid bodies are distended ends of disrupted nerve axons.
13.1.5 Experimental studies
CWSs have been produced by experimental occlusion of precapillary retinal arterioles (Gay et al., 1964; Ashton and Henkind, 1965; Dollery et al., 1966). McLeod et al. (1977) produced experimental cotton wools spots by occlusion of small retinal arterioles in the pig retina, and on axoplasmic flow studies found that the axonal swelling and collection of cytoplasmic organelles on their peripheral border and on the border nearest to the optic disc are caused by obstruction of orthograde and retrograde axoplasmic flows respectively, by ischemia. They stated that: “We concluded that cotton-wood spots should be redefined as accumulations of cytoplasmic debris in the retinal nerve-fibre layer caused by obstruction of orthograde or retrograde axoplasmic transport in ganglion cell axons.”
13.2 PATHOGENESIS OF CWSs
From the above evidence and discussion, it is very evident that CWSs are due to occlusion of the terminal retinal arterioles in the nerve fiber and ganglion cell layer, with focal nonperfusion of the retinal capillaries in their distribution, resulting in acute focal inner retinal ischemia and infarction. This is further supported by the following: (i) Experimental occlusion of precapillary retinal arterioles produce CWSs (Gay et al., 1964; Ashton and Henkind, 1965; Dollery et al., 1966). (ii) They are commonly seen in many systemic thromboembolic disorders (see above). (iii) In our studies discussed above, there is definite fluorescein fundus angiographic evidence of occlusion of terminal retinal arterioles and non-perfusion of the retinal capillaries in the area of the CWSs (Fig. 26), as well as retinal nerve fiber loss corresponding to the distribution and development of the CWSs (Fig. 27) (Hayreh et al., 1989). These findings contradict the claim by McLeod (2005) that “cotton wool spots should not be regarded as retinal nerve fibre layer infarcts”.
13.3 TERMINOLOGY
Over the years several different eponyms have been used for these lesions, including pathologically "cytoid bodies", and clinically "soft exudates" and "cotton-wool spots" (or patches) (Wagener et al., 1947; Leishman, 1957). The term "soft exudates" is wrong and unjustified since these are not exudates at all (as is evident from the discussion above). "Cotton wool spot" simply describes the ophthalmoscopic appearance during the acute phase, without giving any information about pathogenesis or any other feature. Based on the available evidence on the pathogenesis and other features of this lesion, I recommend the term "inner retinal ischemic spot" because this is a truly descriptive and pathogenetic term.
14. AMAUROSIS FUGAX
Amaurosis fugax (AF), or transient visual loss in an eye, is conventionally described as due to transient acute retinal ischemia. While that is true in most cases, it is important to remember that it does also occur due to conditions not involving the retina. An important and well documented example of the latter is the AF seen in GCA, which is due to transient acute ischemia of the optic nerve head only. I have seen patients who experienced episodes of transient blurring of vision due to acute transient anterior chamber hemorrhage from a vascularized cataract scar or anterior chamber lens implant, or a large central vitreous floater. Before starting extensive and expensive evaluations of patients complaining of AF, to determine the cause of retinal ischemia, it is important to rule out other possible causes.
There is a voluminous literature on the causes of AF. The most common is embolism into the retinal arterial circulation. By far the most common cause of embolism is carotid artery disease (both common and internal carotid arteries), particularly atherosclerotic plaques throwing emboli, and less commonly during angioplasty and stenting of the carotid artery, or hemodynamically significant internal carotid artery stenosis. The next most common cause is embolism from a cardiac source - from the aortic or mitral valves, prosthetic heart valves, and much less commonly patent foramen ovale, subacute bacterial endocarditis, atrial fibrillation or myxoma of the heart. The many other causes reported in the literature include orthostatic hypotension, vasovagal syncope, ulcerated aortic atheromatous plaque, familial thrombophilia, hypercoagulable states, antiphospholipid syndrome, ophthalmic artery stenosis, retinal migraine, ocular ischemic syndrome, Takayasu arteritis, Wegener's granulomatosis, exercise-induced vasospastic AF, dural arteriovenous fistula, angle-closure glaucoma, gaze-evoked AF, or atherosclerotic plaques in the external carotid artery in eyes where the ophthalmic artery arises from the middle meningeal artery (Figs. 1B–G). Episodes of AF also occur in eyes with marked optic disc edema from any cause, e.g., in idiopathic intracranial hypertension. I have seen many patients presenting with AF as the primary complaint in eyes with sudden onset of marked non-ischemic CRVO (Hayreh, 1998) or of cilioretinal artery occlusion associated with non-ischemic CRVO (Hayreh et al., 2008). As discussed above, AF has been reported in various types of BRAO before a permanent visual loss.
It is universally believed that in atherosclerotic plaques in the carotid arteries, it is embolism which causes AF. However, our experimental study in atherosclerotic monkeys showed that that is not always the case (Hayreh et al., 1997). This is because our study showed that serotonin (5-hydroxytryptamine), released by platelet aggregation on atherosclerotic plaques in the carotid artery, produces transient vasospasm and occlusion of the CRA in atherosclerotics (Fig. 17), and that may produce AF.
14.1 AF IN THE MIDDLE AGED AND ELDERLY POPULATION
AF is extremely important in these patients. In our prospective study of 85 consecutive patients with GCA with visual loss, at least 31% gave a history of AF preceding the development of visual loss (Hayreh et al., 1998b). Thus, development of AF in these patients is an ominous sign of impending blindness in one or both eyes. In view of that, when a patient 50 years or older complains of AF, it is absolutely critical first of all and immediately to rule out GCA by doing erythrocyte sedimentation rate and C-reactive protein estimations – the latter is most reliable. Most physician rely entirely on the systemic symptoms of GCA, but our study showed that 21% of those with visual loss due to GCA, report no systemic symptoms of GCA at all in spite of a positive temporal artery biopsy, i.e. they have occult GCA (Hayreh et al., 1998a). Thus, blood tests are far more reliable than systemic symptoms in the diagnosis of GCA. I have found that in patients with AF due to GCA, fluorescein fundus angiography shows evidence of PCA occlusion or stasis and that is important in the diagnosis of GCA.
AF is also produced by hypoperfusion of the retinal or optic nerve head vascular bed. Blood flow in the retinal and optic nerve head vascular bed depends upon the perfusion pressure in them. Perfusion pressure is equal to the mean blood pressure minus the intraocular pressure (or arterial minus venous blood pressures). The marked fall of blood pressure in these vascular beds seen in complete or marked stenosis of the internal carotid or ophthalmic artery, or in eyes with ocular ischemic syndrome, results in hypoperfusion in the retinal or optic nerve head vascular bed. That is also seen in some eyes at the onset of non-ischemic CRVO or in CLRAO associated with CRVO. Any sudden fall of perfusion pressure in the retina or optic nerve head vascular beds in such eyes due to either a further fall of blood pressure (e.g., in orthostatic hypotension) or rise in intraocular pressure due to compression of the eyeball or stooping down can precipitate development of AF.
14.2 EVALUATION OF PATIENTS WITH AF
This is identical to that discussed above regarding retinal arterial occlusion.
14.3 MANAGEMENT
If one can find a cause for it, then obviously one has to try to manage that. As discussed above, in persons aged 50 years and older, it is essential to rule out first GCA immediately and if there is any suspicion of that, they should be immediately started on high-dose steroid therapy to prevent catastrophic visual loss. In the vast majority of patient it is due to embolism from the atheromatous plaques in the carotid arteries or from the heart. For them, unfortunately, there are limited options. On a risk:benefit ratio, unless there is 70% or more carotid artery stenosis, carotid endarterectomy is not advisable even if there are atheromatous plaques throwing emboli. The same applies when the heart is the source of emboli. In such cases I seek the advice of the vascular surgeons and cardiologists. My routine usually is to put the patients on aspirin; I have found that effective in many cases. As regards anticoagulant therapy, it has its own serious systemic risks and one should be aware of those. I personally feel uncomfortable using this compared to the use of aspirin.
15. CONCLUSIONS AND FUTURE DIRECTIONS
There has been great confusion and controversy on the various types of acute retinal arterial occlusive disorders. For example, there is a very common impression that CRAO has a dismal visual outcome and no successful treatment. The primary reason for all this has been describing CRAO as all one clinical entity; the same applies to BRAO. Recent studies have proven otherwise. They have shown that, based on their pathogeneses and clinical features, CRAO and BRAO each consist of multiple distinct clinical entities. For example, CRAO actually comprises 4 distinct clinical entities – non-arteritic CRAO, non-arteritic CRAO with cilioretinal artery sparing, arteritic CRAO associated with GCA and transient non-arteritic CRAO. Their clinical features and visual outcomes are distinctly different. Similarly, BRAO consists of permanent BRAO, transient BRAO and cilioretinal artery occlusion (CLRAO), and the latter further consists of 3 distinct clinical entities - non-arteritic CLRAO alone, non-arteritic CLRAO associated with central retinal vein occlusion and arteritic CLRAO associated with GCA. Each of these has a different pathogenesis and clinical features. Understanding these classifications and pathogeneses is essential for a full comprehension of the various aspects of these disorders, and their management. Since we now understand their pathogeneses, we should be better able to explore the management of the different clinical entities individually rather that lumping diverse conditions into one group.
ACKNOWLEDGEMENTS
I owe a very great debt of gratitude to Mrs. Patricia Duffel, librarian of our department, for her invaluable help in researching the literature on the subject, formatting the references to the style of Journal and inserting them in the text. I am also grateful to my wife Shelagh for her editorial assistance.
Supported by grants EY-1151 and 1576 from the National Institutes of Health, and in part by unrestricted grant from Research to Prevent Blindness, Inc., New York.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The author has no conflict of interest.
REFERENCES
- Ahuja RM, Chaturvedi S, Eliott D, Joshi N, Puklin JE, Abrams GW. Mechanisms of retinal arterial occlusive disease in African American and Caucasian patients. Stroke. 1999;30:1506–1509. doi: 10.1161/01.str.30.8.1506. [DOI] [PubMed] [Google Scholar]
- Anderson DC, Kappelle LJ, Eliasziw M, Babikian VL, Pearce LA, Barnett HJ. Occurrence of hemispheric and retinal ischemia in atrial fibrillation compared with carotid stenosis. Stroke. 2002;33:1963–1967. doi: 10.1161/01.str.0000023445.20454.a8. [DOI] [PubMed] [Google Scholar]
- Anderson DR. Glaucoma, capillaries and pericytes. 1. Blood flow regulation. Ophthalmologica. 1996;210:257–262. doi: 10.1159/000310722. [DOI] [PubMed] [Google Scholar]
- Anderson DR, Davis EB. Glaucoma, capillaries and pericytes. 2. Identification and characterization of retinal pericytes in culture. Ophthalmologica. 1996;210:263–268. doi: 10.1159/000310723. [DOI] [PubMed] [Google Scholar]
- Arnold M, Koerner U, Remonda L, Nedeltchev K, Mattle HP, Schroth G, Sturzenegger M, Weber J, Koerner F. Comparison of intra-arterial thrombolysis with conventional treatment in patients with acute central retinal artery occlusion. J Neurol Neurosurg Psychiatry. 2005;76:196–199. doi: 10.1136/jnnp.2004.037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruga J, Sanders MD. Ophthalmologic findings in 70 patients with evidence of retinal embolism. Ophthalmology. 1982;89:1336–1347. doi: 10.1016/s0161-6420(82)34626-6. [DOI] [PubMed] [Google Scholar]
- Ashton N. Diabetic retinopathy: a new approach. Lancet. 1959;2:625–630. doi: 10.1016/s0140-6736(59)91407-2. [DOI] [PubMed] [Google Scholar]
- Ashton N, Harry J. The Pathology of Cotton Wool Spots and Cytoid Bodies in Hypertensive Retinopathy and Other Diseases. Trans Ophthalmol Soc U K. 1963;83:91–114. [PubMed] [Google Scholar]
- Ashton N, Henkind P. Experimental Occlusion of Retinal Arterioles: Using Graded Glass Ballotini. British journal of ophthalmology. 1965;49:225–234. doi: 10.1136/bjo.49.5.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchaneeyasakul LO, Trinavarat A, Bumrungsuk P, Wongsawad W. Anticardiolipin IgG antibody and homocysteine as possible risk factors for retinal vascular occlusive disease in thai patients. Jpn J Ophthalmol. 2005;49:211–215. doi: 10.1007/s10384-005-0190-3. [DOI] [PubMed] [Google Scholar]
- Atebara NH, Brown GC, Cater J. Efficacy of anterior chamber paracentesis and Carbogen in treating acute nonarteritic central retinal artery occlusion. Ophthalmology. 1995;102:2029–2035. doi: 10.1016/s0161-6420(95)30758-0. [DOI] [PubMed] [Google Scholar]
- Babikian V, Wijman CA, Koleini B, Malik SN, Goyal N, Matjucha IC. Retinal ischemia and embolism. Etiologies and outcomes based on a prospective study. Cerebrovasc Dis. 2001;12:108–113. doi: 10.1159/000047689. [DOI] [PubMed] [Google Scholar]
- Barak N, Ferencz JR, Freund M, Mekori Y. Urticaria in idiopathic bilateral recurrent branch retinal arterial occlusion. Acta Ophthalmol Scand. 1997;75:107–108. doi: 10.1111/j.1600-0420.1997.tb00264.x. [DOI] [PubMed] [Google Scholar]
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. British journal of ophthalmology. 2000;84:914–916. doi: 10.1136/bjo.84.8.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiran I, Dori D, Pikkel J, Goldsher D, Miller B. Recurrent retinal artery obstruction as a presenting symptom of ophthalmic artery aneurysm: a case report. Graefes Arch Clin Exp Ophthalmol. 1995;233:444–447. doi: 10.1007/BF00180950. [DOI] [PubMed] [Google Scholar]
- Beversdorf D, Stommel E, Allen C, Stevens R, Lessell S. Recurrent branch retinal infarcts in association with migraine. Headache. 1997;37:396–399. doi: 10.1046/j.1526-4610.1997.3706396.x. [DOI] [PubMed] [Google Scholar]
- Blumenthal M, Best M, Galin MA, Gitter KA. Ocular circulation: analysis of the effect of induced ocular hypertension on reginal and choroidal blood flow in man. Am J Ophthalmol. 1971;71:819–825. doi: 10.1016/0002-9394(71)90247-9. [DOI] [PubMed] [Google Scholar]
- Brown GC, Magargal LE. Central retinal artery obstruction and visual acuity. Ophthalmology. 1982;89:14–19. doi: 10.1016/s0161-6420(82)34853-8. [DOI] [PubMed] [Google Scholar]
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol. 1979;97:84–92. doi: 10.1001/archopht.1979.01020010024006. [DOI] [PubMed] [Google Scholar]
- Butz B, Strotzer M, Manke C, Roider J, Link J, Lenhart M. Selective intraarterial fibrinolysis of acute central retinal artery occlusion. Acta Radiol. 2003;44:680–684. doi: 10.1080/02841850312331287829. [DOI] [PubMed] [Google Scholar]
- Chopdar A. Dual trunk central retinal vein incidence in clinical practice. Arch Ophthalmol. 1984;102:85–87. doi: 10.1001/archopht.1984.01040030069038. [DOI] [PubMed] [Google Scholar]
- Chua B, Kifley A, Wong TY, Mitchell P. Homocysteine and retinal emboli: the Blue Mountains Eye Study. Am J Ophthalmol. 2006;142:322–324. doi: 10.1016/j.ajo.2006.03.039. [DOI] [PubMed] [Google Scholar]
- Coats G. Obstruction of the central artery of the retina. Royal London Ophthalmic Hospital Reports. 1905;16:262–306. [Google Scholar]
- Coats G. Pathology of obstruction of the central artery of the retina. Royal London Ophthalmic Hospital Reports. 1913;19:45–70. [Google Scholar]
- Collier M. Frequence des vaisseaux cilioretiniens, leur rapport avec les ametropies, leur association avec d’autres anomalies du fond de l’oeil [Incidence of cilioretinal vessels; their relation with ametropia; their association with other anomalies of the eye fundus] Bull Soc Fr Dermatol Syphiligr. 1957;64:598–601. [PubMed] [Google Scholar]
- Coverdale HV. The Cause and Results of Obstruction of the Central Artery of the Retina: A Study of Eleven Cases. Br J Ophthalmol. 1929;13:529–571. doi: 10.1136/bjo.13.11.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David NJ, Norton EW, Gass JD, Beauchamp J. Fluorescein angiography in central retinal artery occlusion. Arch Ophthalmol. 1967;77:619–629. doi: 10.1001/archopht.1967.00980020621010. [DOI] [PubMed] [Google Scholar]
- Dollery CT, Henkind P, Paterson JW, Ramalho PS, Hill DW. I. Ophthalmoscopic and circulatory changes in focal retinal ischaemia. Br J Ophthalmol. 1966;50:285–324. doi: 10.1136/bjo.50.6.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duker JS, Sivalingam A, Brown GC, Reber R. A prospective study of acute central retinal artery obstruction. The incidence of secondary ocular neovascularization. Arch Ophthalmol. 1991;109:339–342. doi: 10.1001/archopht.1991.01080030041034. [DOI] [PubMed] [Google Scholar]
- Feltgen N, Neubauer A, Jurklies B, Schmoor C, Schmidt D, Wanke J, Maier-Lenz H, Schumacher M. Multicenter study of the European Assessment Group for Lysis in the Eye (EAGLE) for the treatment of central retinal artery occlusion: design issues and implications. EAGLE Study report no. 1 : EAGLE Study report no. 1. Graefes Arch Clin Exp Ophthalmol. 2006;244:950–956. doi: 10.1007/s00417-005-0140-2. [DOI] [PubMed] [Google Scholar]
- Fernandez FJ, Guelbenzu S, Barrena C, Larrosa JM, Gonzalvo FJ, Melcon B, Honrubia FM. Fibrinolisis selectiva de arteria oftálmica en la oclusión aguda de la arteria central de la retina [Selective ophthalmic artery fibrinolysis in acute central retinal artery occlusion] Arch Soc Esp Oftalmol. 2002;77:81–86. [PubMed] [Google Scholar]
- Fineman MS, Savino PJ, Federman JL, Eagle RC., Jr Branch retinal artery occlusion as the initial sign of giant cell arteritis. Am J Ophthalmol. 1996;122:428–430. doi: 10.1016/s0002-9394(14)72073-2. [DOI] [PubMed] [Google Scholar]
- Framme C, Spiegel D, Roider J, Sachs HG, Lohmann CP, Butz B, Link J, Gabel VP. Zentralarterienverschluss. Stellenwert der selektiven intraarteriellen Fibrinolyse [Central retinal artery occlusion. Importance of selective intra-arterial fibrinolysis] Ophthalmologe. 2001;98:725–730. doi: 10.1007/s003470170079. [DOI] [PubMed] [Google Scholar]
- Fraser S, Siriwardena D. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev (1) 2002 doi: 10.1002/14651858.CD001989. CD001989. [DOI] [PubMed] [Google Scholar]
- Garcia-Arumi J, Martinez-Castillo V, Boixadera A, Fonollosa A, Corcostegui B. Surgical embolus removal in retinal artery occlusion. Br J Ophthalmol. 2006;90:1252–1255. doi: 10.1136/bjo.2006.097642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay AJ, Goldor H, Smith M. Chorioretinal Vascular Occlusions with Latex Spheres. Invest Ophthalmol. 1964;3:647–656. [PubMed] [Google Scholar]
- Glacet-Bernard A, Gaudric A, Touboul C, Coscas G. Occlusion de la veine centrale de la rétine avec occlusion d'une artère cilio-rétinienne: à propos de 7 cas [Occlusion of the central retinal vein with occlusion of a cilioretinal artery: apropos of 7 cases] J Fr Ophtalmol. 1987;10:269–277. [PubMed] [Google Scholar]
- Glueck CJ, Ping W, Hutchins R, Petersen MR, Golnik K. Ocular vascular thrombotic events: central retinal vein and central retinal artery occlusions. Clin Appl Thromb Hemost. 2008;14:286–294. doi: 10.1177/1076029607304726. [DOI] [PubMed] [Google Scholar]
- Gordon DL, Hayreh SS, Adams HP., Jr Microangiopathy of the brain, retina, and ear: improvement without immunosuppressive therapy. Stroke. 1991;22:933–937. doi: 10.1161/01.str.22.7.933. [DOI] [PubMed] [Google Scholar]
- Greven CM, Slusher MM, Weaver RG. Retinal arterial occlusions in young adults. Am J Ophthalmol. 1995;120:776–783. doi: 10.1016/s0002-9394(14)72731-x. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Thesis for the Master of Surgery. India: Panjab University; 1958. A study of the central artery of the retina in human beings. [Google Scholar]
- Hayreh SS. The Ophthalmic Artery: III. Branches. Br J Ophthalmol. 1962;46:212–247. doi: 10.1136/bjo.46.4.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS. The Cilio-Retinal Arteries. Br J Ophthalmol. 1963;47:71–89. doi: 10.1136/bjo.47.2.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS. Blood supply of the optic nerve head and its role in optic atrophy, glaucoma, and oedema of the optic disc. Br J Ophthalmol. 1969;53:721–748. doi: 10.1136/bjo.53.11.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS. Pathogenesis of visual field defects. Role of the ciliary circulation. Br J Ophthalmol. 1970;54:289–311. doi: 10.1136/bjo.54.5.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS. Pathogenesis of occlusion of the central retinal vessels. Am J Ophthalmol. 1971;72:998–1011. doi: 10.1016/0002-9394(71)91706-5. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Anterior ischaemic optic neuropathy. II. Fundus on ophthalmoscopy and fluorescein angiography. Br J Ophthalmol. 1974a;58:964–980. doi: 10.1136/bjo.58.12.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS. Central retinal vascular occlusion: morphology and pathogenesis. In: Brancato R, Pratesi F, editors. Angiology. Rome: Centro Minerva Medica; 1974b. pp. 15–40. [Google Scholar]
- Hayreh SS. Anterior Ischemic Optic Neuropathy. Heidelberg: Springer-Verlag; 1975. [Google Scholar]
- Hayreh SS. So-called "central retinal vein occlusion". I. Pathogenesis, terminology, clinical features. Ophthalmologica. 1976;172:1–13. doi: 10.1159/000307579. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Anterior ischaemic optic neuropathy. Differentiation of arteritic from non-arteritic type and its management. Eye (Lond) 1990;4(Pt. 1):25–41. doi: 10.1038/eye.1990.4. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. The 1994 Von Sallman Lecture. The optic nerve head circulation in health and disease. Exp Eye Res. 1995;61:259–272. doi: 10.1016/s0014-4835(05)80121-6. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Duke-elder lecture: Systemic arterial blood pressure and the eye. Eye (Lond) 1996;10(Pt. 1):5–28. doi: 10.1038/eye.1996.3. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Central retinal vein occlusion. Ophthalmology Clinics of North America. 1998;11:559–590. [Google Scholar]
- Hayreh SS. Retinal arterial occlusion with LIF using rTPA. Ophthalmology. 1999;106:1236–1239. doi: 10.1016/S0161-6420(99)10101-5. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. The blood supply of the optic nerve head and the evaluation of it - myth and reality. Prog Retin Eye Res. 2001;20:563–593. doi: 10.1016/s1350-9462(01)00004-0. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Prevalent misconceptions about acute retinal vascular occlusive disorders. Prog Retin Eye Res. 2005;24:493–519. doi: 10.1016/j.preteyeres.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Orbital vascular anatomy. Eye (Lond) 2006;20:1130–1144. doi: 10.1038/sj.eye.6702377. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Surgical embolus removal in retinal artery occlusion. Br J Ophthalmol. 2007;91:1096–1097. [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS. Intra-arterial thrombolysis for central retinal artery occlusion. Br J Ophthalmol. 2008a;92:585–587. doi: 10.1136/bjo.2007.136481. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Restoration of retinal blood flow via translumenal Nd:YAG embolysis/embolectomy (TYL/E) for central and branch retinal artery occlusion. Retina. 2008b;28:1369–1372. doi: 10.1097/IAE.0b013e3181846640. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Transient central retinal artery occlusion following viperine snake bite. Arch Ophthalmol. 2008c;126:870–871. doi: 10.1001/archopht.126.6.870-a. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Dass R. The Ophthalmic Artery: I. Origin and Intra-Cranial and Intra-Canalicular Course. Br J Ophthalmol. 1962;46:65–98. doi: 10.1136/bjo.46.2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS, Fraterrigo L, Jonas J. Central retinal vein occlusion associated with cilioretinal artery occlusion. Retina. 2008;28:581–594. doi: 10.1097/IAE.0b013e31815ec29b. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Hayreh MS. Hemi-central retinal vein occulsion. Pathogenesis, clinical features, and natural history. Arch Ophthalmol. 1980;98:1600–1609. doi: 10.1001/archopht.1980.01020040452011. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Jonas JB. Optic disk and retinal nerve fiber layer damage after transient central retinal artery occlusion: an experimental study in rhesus monkeys. Am J Ophthalmol. 2000;129:786–795. doi: 10.1016/s0002-9394(00)00384-6. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Kolder HE, Weingeist TA. Central retinal artery occlusion and retinal tolerance time. Ophthalmology. 1980;87:75–78. doi: 10.1016/s0161-6420(80)35283-4. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Piegors DJ, Heistad DD. Serotonin-induced constriction of ocular arteries in atherosclerotic monkeys. Implications for ischemic disorders of the retina and optic nerve head. Arch Ophthalmol. 1997;115:220–228. doi: 10.1001/archopht.1997.01100150222012. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Podhajsky P. Ocular neovascularization with retinal vascular occlusion. II. Occurrence in central and branch retinal artery occlusion. Arch Ophthalmol. 1982;100:1585–1596. doi: 10.1001/archopht.1982.01030040563002. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Podhajsky P, Zimmerman MB. Role of nocturnal arterial hypotension in optic nerve head ischemic disorders. Ophthalmologica. 1999;213:76–96. doi: 10.1159/000027399. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Podhajsky PA, Zimmerman B. Occult giant cell arteritis: ocular manifestations. Am J Ophthalmol. 1998a;125:521–526. 893. doi: 10.1016/s0002-9394(99)80193-7. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Podhajsky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol. 1998b;125:509–520. doi: 10.1016/s0002-9394(99)80192-5. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Podhajsky PA, Zimmerman MB. Branch retinal artery occlusion: natural history of visual outcome. Ophthalmology. 2009a;116:1188–1194. e1181–e1184. doi: 10.1016/j.ophtha.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology. 2009b;116:1928–1936. doi: 10.1016/j.ophtha.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS, Podhajsky PA, Zimmerman MB. Natural history of visual outcome in central retinal vein occlusion. Ophthalmology. 2011;118:119–133. e111–e112. doi: 10.1016/j.ophtha.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS, Revie IH, Edwards J. Vasogenic origin of visual field defects and optic nerve changes in glaucoma. Br J Ophthalmol. 1970;54:461–472. doi: 10.1136/bjo.54.7.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS, Rojas P, Podhajsky P, Montague P, Woolson RF. Ocular neovascularization with retinal vascular occlusion-III. Incidence of ocular neovascularization with retinal vein occlusion. Ophthalmology. 1983;90:488–506. doi: 10.1016/s0161-6420(83)34542-5. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Servais GE, Virdi PS. Fundus lesions in malignant hypertension. VI. Hypertensive choroidopathy. Ophthalmology. 1986;93:1383–1400. doi: 10.1016/s0161-6420(86)33554-1. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Servais GE, Virdi PS. Cotton-wool spots (inner retinal ischemic spots) in malignant arterial hypertension. Ophthalmologica. 1989;198:197–215. doi: 10.1159/000309999. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, van Heuven WA, Hayreh MS. Experimental retinal vascular occlusion. I. Pathogenesis of central retinal vein occlusion. Arch Ophthalmol. 1978;96:311–323. doi: 10.1001/archopht.1978.03910050179015. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Vrabec F. The structure of the head of the optic nerve in rhesus monkey. Am J Ophthalmol. 1966;61:136–150. doi: 10.1016/0002-9394(66)90758-6. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Weingeist TA. Experimental occlusion of the central artery of the retina. I. Ophthalmoscopic and fluorescein fundus angiographic studies. Br J Ophthalmol. 1980a;64:896–912. doi: 10.1136/bjo.64.12.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS, Weingeist TA. Experimental occlusion of the central artery of the retina. IV: Retinal tolerance time to acute ischaemia. Br J Ophthalmol. 1980b;64:818–825. doi: 10.1136/bjo.64.11.818. (also see corrected bibliography and text in Br J Ophthalmol 1981) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS, Zimmerman B. Management of giant cell arteritis. Our 27-year clinical study: new light on old controversies. Ophthalmologica. 2003a;217:239–259. doi: 10.1159/000070631. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Zimmerman B. Visual deterioration in giant cell arteritis patients while on high doses of corticosteroid therapy. Ophthalmology. 2003b;110:1204–1215. doi: 10.1016/S0161-6420(03)00228-8. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Zimmerman B, Kardon RH. Visual improvement with corticosteroid therapy in giant cell arteritis. Report of a large study and review of literature. Acta Ophthalmol Scand. 2002;80:355–367. doi: 10.1034/j.1600-0420.2002.800403.x. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol. 2005;140:376–391. doi: 10.1016/j.ajo.2005.03.038. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Zimmerman MB. Fundus changes in central retinal artery occlusion. Retina. 2007;27:276–289. doi: 10.1097/01.iae.0000238095.97104.9b. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Zimmerman MB. Nonarteritic anterior ischemic optic neuropathy: natural history of visual outcome. Ophthalmology. 2008;115:298–305. e292. doi: 10.1016/j.ophtha.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS, Zimmerman MB, Beri M, Podhajsky P. Intraocular pressure abnormalities associated with central and hemicentral retinal vein occlusion. Ophthalmology. 2004a;111:133–141. doi: 10.1016/j.ophtha.2003.03.002. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Zimmerman MB, Kimura A, Sanon A. Central retinal artery occlusion. Retinal survival time. Exp Eye Res. 2004b;78:723–736. doi: 10.1016/s0014-4835(03)00214-8. [DOI] [PubMed] [Google Scholar]
- Hayreh SS, Zimmerman MB, Podhajsky P, Alward WL. Nocturnal arterial hypotension and its role in optic nerve head and ocular ischemic disorders. Am J Ophthalmol. 1994;117:603–624. doi: 10.1016/s0002-9394(14)70067-4. [DOI] [PubMed] [Google Scholar]
- Hegde V, Deokule S, Matthews T. A case of a cilioretinal artery supplying the entire retina. Clin Anat. 2006;19:645–647. doi: 10.1002/ca.20362. [DOI] [PubMed] [Google Scholar]
- Hogan MJ, Alvarado JA, Wedell JE. Histology of the human eye: an atlas and textbook. Philadelphia: Saunders; 1971. pp. 508–513. [Google Scholar]
- Johnson MW, Thomley ML, Huang SS, Gass JD. Idiopathic recurrent branch retinal arterial occlusion. Natural history and laboratory evaluation. Ophthalmology. 1994;101:480–489. doi: 10.1016/s0161-6420(94)31309-1. [DOI] [PubMed] [Google Scholar]
- Juarez CP, Tso MO, van Heuven WA, Hayreh MS, Hayreh SS. Experimental retinal vascular occlusion. II. A clinico-pathologic correlative study of simultaneous occlusion of central retinal vein and artery. Int Ophthalmol. 1986a;9:77–87. doi: 10.1007/BF00159836. [DOI] [PubMed] [Google Scholar]
- Juarez CP, Tso MO, van Heuven WA, Hayreh MS, Hayreh SS. Experimental retinal vascular occlusion. III. An ultrastructural study of simultaneous occlusion of central retinal vein and artery. Int Ophthalmol. 1986b;9:89–101. doi: 10.1007/BF00159837. [DOI] [PubMed] [Google Scholar]
- Justice J, Jr, Lehmann RP. Cilioretinal arteries. A study based on review of stereo fundus photographs and fluorescein angiographic findings. Arch Ophthalmol. 1976;94:1355–1358. doi: 10.1001/archopht.1976.03910040227015. [DOI] [PubMed] [Google Scholar]
- Karagoz B, Ayata A, Bilgi O, Uzun G, Unal M, Kandemir EG, Ozgun A, Turken O. Hemicentral retinal artery occlusion in a breast cancer patient using anastrozole. Onkologie. 2009;32:421–423. doi: 10.1159/000218369. [DOI] [PubMed] [Google Scholar]
- Karjalainen K. Occlusion of the central retinal artery and retinal branch arterioles. A clinical, tonographic and fluorescein angiographic study of 175 patients. Acta Ophthalmol Suppl. 1971;109:1–95. [PubMed] [Google Scholar]
- Kattah JC, Wang DZ, Reddy C. Intravenous recombinant tissue-type plasminogen activator thrombolysis in treatment of central retinal artery occlusion. Arch Ophthalmol. 2002;120:1234–1236. [PubMed] [Google Scholar]
- Kearns TP. Collagen and rheumatic diseases: ophthalmic aspects. In: Mausolf FA, editor. The eye and systemic disease. Mosby: St. Louis; 1975. pp. 105–118. [Google Scholar]
- Kwon YH, Rickman DW, Baruah S, Zimmerman MB, Kim CS, Boldt HC, Russell SR, Hayreh SS. Vitreous and retinal amino acid concentrations in experimental central retinal artery occlusion in the primate. Eye (Lond) 2005;19:455–463. doi: 10.1038/sj.eye.6701546. [DOI] [PubMed] [Google Scholar]
- Leishman R. The eye in general vascular disease: hypertension and arteriosclerosis. Br J Ophthalmol. 1957;41:641–701. doi: 10.1136/bjo.41.11.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentzen SE. Occlusion of the central retinal artery. A follow-up. Acta Ophthalmol (Copenh) 1969;47:690–703. doi: 10.1111/j.1755-3768.1969.tb08157.x. [DOI] [PubMed] [Google Scholar]
- Maharajan VS, Dua HS, Scott R, Maharajan P. Unexpected visual improvement with a pinhole in central retinal artery occlusion and cilioretinal artery sparing fovea. Eye (Lond) 2002;16:194–196. doi: 10.1038/sj.eye.6700076. [DOI] [PubMed] [Google Scholar]
- Margo CE, Mack WP. Therapeutic decisions involving disparate clinical outcomes: patient preference survey for treatment of central retinal artery occlusion. Ophthalmology. 1996;103:691–696. doi: 10.1016/s0161-6420(96)30631-3. [DOI] [PubMed] [Google Scholar]
- Mason JO, 3rd, Shah AA, Vail RS, Nixon PA, Ready EL, Kimble JA. Branch retinal artery occlusion: visual prognosis. Am J Ophthalmol. 2008;146:455–457. doi: 10.1016/j.ajo.2008.05.009. [DOI] [PubMed] [Google Scholar]
- McCullough HK, Reinert CG, Hynan LS, Albiston CL, Inman MH, Boyd PI, Welborn MB, 3rd, Clagett GP, Modrall JG. Ocular findings as predictors of carotid artery occlusive disease: is carotid imaging justified? J Vasc Surg. 2004;40:279–286. doi: 10.1016/j.jvs.2004.05.004. [DOI] [PubMed] [Google Scholar]
- McLeod D. Why cotton wool spots should not be regarded as retinal nerve fibre layer infarcts. Br J Ophthalmol. 2005;89:229–237. doi: 10.1136/bjo.2004.058347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod D. Central retinal vein occlusion with cilioretinal infarction from branch flow exclusion and choroidal arterial steal. Retina. 2009;29:1381–1395. doi: 10.1097/IAE.0b013e3181b85f41. [DOI] [PubMed] [Google Scholar]
- McLeod D, Marshall J, Kohner EM, Bird AC. The role of axoplasmic transport in the pathogenesis of retinal cotton-wool spots. Br J Ophthalmol. 1977;61:177–191. doi: 10.1136/bjo.61.3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod D, Ring CP. Cilio-retinal infarction after retinal vein occlusion. British Journal of Ophthalmology. 1976;60:419–427. doi: 10.1136/bjo.60.6.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizener JB, Podhajsky P, Hayreh SS. Ocular ischemic syndrome. Ophthalmology. 1997;104:859–864. doi: 10.1016/s0161-6420(97)30221-8. [DOI] [PubMed] [Google Scholar]
- Morandi X, Le Bourdon E, Darnault P, Brassier G, Duval JM. Unusual origin of the ophthalmic artery and occlusion of the central retinal artery. Surg Radiol Anat. 1998;20:69–71. doi: 10.1007/BF01628119. [DOI] [PubMed] [Google Scholar]
- Mueller AJ, Neubauer AS, Schaller U, Kampik A. Evaluation of minimally invasive therapies and rationale for a prospective randomized trial to evaluate selective intra-arterial lysis for clinically complete central retinal artery occlusion. Arch Ophthalmol. 2003;121:1377–1381. doi: 10.1001/archopht.121.10.1377. [DOI] [PubMed] [Google Scholar]
- Nettleship E. Unusual appearance in a case of retinal embolism about 30 hours after its occurrence. Festschrift zur Feier des Siebzigsten Geburtstages von Herman von Helmholtz. 1891;7:7–8. [Google Scholar]
- Oosterhuis JA. Perspectives in ophthalmology. Amsterdam: Excerpta Medica; 1968. Fluorescein fundus angiography in retinal venous occlusion; pp. 29–47. [Google Scholar]
- Opremcak E, Rehmar AJ, Ridenour CD, Borkowski LM, Kelley JK. Restoration of retinal blood flow via translumenal Nd:YAG embolysis/embolectomy (TYL/E) for central and branch retinal artery occlusion. Retina. 2008;28:226–235. doi: 10.1097/IAE.0b013e31814b1d6e. [DOI] [PubMed] [Google Scholar]
- Padolecchia R, Puglioli M, Ragone MC, Romani A, Collavoli PL. Superselective intraarterial fibrinolysis in central retinal artery occlusion. AJNR Am J Neuroradiol. 1999;20:565–567. [PMC free article] [PubMed] [Google Scholar]
- Palmowski-Wolfe AM, Denninger E, Geisel J, Pindur G, Ruprecht KW. Antiphospholipid antibodies in ocular arterial and venous occlusive disease. Ophthalmologica. 2007;221:41–46. doi: 10.1159/000096521. [DOI] [PubMed] [Google Scholar]
- Petrig BL, Riva CE, Hayreh SS. Laser Doppler flowmetry and optic nerve head blood flow. Am J Ophthalmol. 1999;127:413–425. doi: 10.1016/s0002-9394(98)00437-1. [DOI] [PubMed] [Google Scholar]
- Pettersen JA, Hill MD, Demchuk AM, Morrish W, Hudon ME, Hu W, Wong J, Barber PA, Buchan AM. Intra-arterial thrombolysis for retinal artery occlusion: the Calgary experience. Can J Neurol Sci. 2005;32:507–511. [PubMed] [Google Scholar]
- Rennebohm RM, Susac JO. Treatment of Susac's syndrome. J Neurol Sci. 2007;257:215–220. doi: 10.1016/j.jns.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Richard G, Lerche RC, Knospe V, Zeumer H. Treatment of retinal arterial occlusion with local fibrinolysis using recombinant tissue plasminogen activator. Ophthalmology. 1999;106:768–773. doi: 10.1016/S0161-6420(99)90165-3. [DOI] [PubMed] [Google Scholar]
- Rishi P, Rishi E, Sharma T, Mahajan S. Hemi-central retinal artery occlusion in young adults. Indian J Ophthalmol. 2010;58:425–432. doi: 10.4103/0301-4738.67069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudkin AK, Lee AW, Chen CS. Vascular risk factors for central retinal artery occlusion. Eye (Lond) 2010;24:678–681. doi: 10.1038/eye.2009.142. [DOI] [PubMed] [Google Scholar]
- Salomon O, Huna-Baron R, Moisseiev J, Rosenberg N, Rubovitz A, Steinberg DM, Davidson J, Sela BA, Seligsohn U. Thrombophilia as a cause for central and branch retinal artery occlusion in patients without an apparent embolic source. Eye (Lond) 2001;15:511–514. doi: 10.1038/eye.2001.164. [DOI] [PubMed] [Google Scholar]
- Schatz H, Fong AC, McDonald HR, Johnson RN, Joffe L, Wilkinson CP, de Laey JJ, Yannuzzi LA, Wendel RT, Joondeph BC, et al. Cilioretinal artery occlusion in young adults with central retinal vein occlusion. Ophthalmology. 1991;98:594–601. doi: 10.1016/s0161-6420(91)32245-0. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Hetzel A, Geibel-Zehender A. Retinal arterial occlusion due to embolism of suspected cardiac tumors -- report on two patients and review of the topic. Eur J Med Res. 2005;10:296–304. [PubMed] [Google Scholar]
- Schmidt D, Hetzel A, Geibel-Zehender A, Schulte-Monting J. Systemic diseases in non-inflammatory branch and central retinal artery occlusion--an overview of 416 patients. Eur J Med Res. 2007;12:595–603. [PubMed] [Google Scholar]
- Schmidt DP, Schulte-Monting J, Schumacher M. Prognosis of central retinal artery occlusion: local intraarterial fibrinolysis versus conservative treatment. AJNR Am J Neuroradiol. 2002;23:1301–1307. [PMC free article] [PubMed] [Google Scholar]
- Schumacher M, Schmidt D, Jurklies B, Gall C, Wanke I, Schmoor C, Maier-Lenz H, Solymosi L, Brueckmann H, Neubauer AS, Wolf A, Feltgen N. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology. 2010;117:1367–1375. e1361. doi: 10.1016/j.ophtha.2010.03.061. [DOI] [PubMed] [Google Scholar]
- Sharma S, Pater JL, Lam M, Cruess AF. Can different types of retinal emboli be reliably differentiated from one another? An inter- and intraobserver agreement study. Can J Ophthalmol. 1998;33:144–148. [PubMed] [Google Scholar]
- Singh (Hayreh) S, Dass R. The central artery of the retina. I. Origin and course. Br J Ophthalmol. 1960a;44:193–212. doi: 10.1136/bjo.44.4.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh (Hayreh) S, Dass R. The central artery of the retina. II. A study of its distribution and anastomoses. Br J Ophthalmol. 1960b;44:280–299. doi: 10.1136/bjo.44.5.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Singh P, Singh R, Vig VK. Macular infarction following viperine snake bite. Arch Ophthalmol. 2007;125:1430–1431. doi: 10.1001/archopht.125.10.1430. [DOI] [PubMed] [Google Scholar]
- Von Graefe A. Über Embolie der Arteria centralis retinae als Ursache plötzlicher Erblindung(Albrecht von Graefe's) Archiv für Ophthalmologie. 1859;5:136–185. [Google Scholar]
- Wagener HP, Clay GE, Gipner JF. Classification of Retinal Lesions in the Presence of Vascular Hypertension: Report submitted to the American Ophthalmological Society by the committee on Classification of Hypertensive Disease of the Retina. Trans Am Ophthalmol Soc. 1947;45:57–73. [PMC free article] [PubMed] [Google Scholar]
- Wakefield MC, O'Donnell SD, Goff JM., Jr Re-evaluation of carotid duplex for visual complaints: who really needs to be studied? Ann Vasc Surg. 2003;17:635–640. doi: 10.1007/s10016-003-0073-3. [DOI] [PubMed] [Google Scholar]
- Watson PG. The treatment of acute retinal arterial occlusion. In: Cant JS, editor. Proceedings, William MacKenzie Centenary Symposium on the Ocular Circulation in Health and Disease (1968, Glasgow); Kimpton, London. 1969. pp. 234–245. [Google Scholar]
- Weber J, Remonda L, Mattle HP, Koerner U, Baumgartner RW, Sturzenegger M, Ozdoba C, Koerner F, Schroth G. Selective intra-arterial fibrinolysis of acute central retinal artery occlusion. Stroke. 1998;29:2076–2079. doi: 10.1161/01.str.29.10.2076. [DOI] [PubMed] [Google Scholar]
- Weger M, Stanger O, Deutschmann H, Leitner FJ, Renner W, Schmut O, Semmelrock J, Haas A. The role of hyperhomocysteinemia and methylenetetrahydrofolate reductase (MTHFR) C677T mutation in patients with retinal artery occlusion. Am J Ophthalmol. 2002;134:57–61. doi: 10.1016/s0002-9394(02)01471-x. [DOI] [PubMed] [Google Scholar]
- Weill A, Cognard C, Piotin M, Laloum L, Castaings L, Moret J. Intérêt persistant de la fibrinolyse intra-artérielle après 8 h ou plus d'occlusion de l'artère centrale de la rétine ou de ses branches. [Persistent value of intra-arterial fibrinolysis 8 hours or more following central retinal artery occlusion or of its branches] J Fr Ophtalmol. 1998;21:466–470. [PubMed] [Google Scholar]
- Yuzurihara D, Iijima H. Visual outcome in central retinal and branch retinal artery occlusion. Jpn J Ophthalmol. 2004;48:490–492. doi: 10.1007/s10384-004-0102-y. [DOI] [PubMed] [Google Scholar]