

Abstract
Yeast NAD+-specific isocitrate dehydrogenase (IDH) is an octameric enzyme composed of four heterodimers of regulatory IDH1 and catalytic IDH2 subunits. The crystal structure suggested that the interactions between tetramers in the octamer are restricted to defined regions in IDH1 subunits from each tetramer. Using truncation and mutagenesis, we constructed three tetrameric forms of IDH. Truncation of five residues from the amino-terminus of IDH1 did not alter the octameric form of the enzyme, but this truncation plus IDH1 G15D or IDH1 D168K residue substitutions produced tetrameric enzymes as assessed by sedimentation velocity ultracentrifugation. The IDH1 G15D substitution in the absence of any truncation of IDH1 was subsequently found to be sufficient for production of a tetrameric enzyme. The tetrameric forms of IDH exhibited ~50% reductions in Vmax and in cooperativity with respect to isocitrate relative to the wild-type enzyme, but they retained the property of allosteric activation by AMP. The truncated -5IDH1/IDH2 and tetrameric enzymes were much more sensitive than the wild-type enzyme to inhibition by the oxidant diamide and concomitant formation of a disulfide bond between IDH2 Cys-150 residues. Binding of ligands reduced the sensitivity of the wild-type enzyme to diamide but had no effect on inhibition of the truncated or tetrameric enzymes. These results suggest that the octameric structure of IDH has in part evolved for regulation of disulfide-bond formation and activity by ensuring the proximity of the amino terminus of an IDH1 subunit from one tetramer to the IDH2 Cys-150 residues in the other tetramer.
Mitochondrial NAD+-specific isocitrate dehydrogenase catalyzes a rate-limiting step in the tricarboxylic acid cycle. The affinity of yeast isocitrate dehydrogenase (IDH)1 for isocitrate is enhanced by AMP and reduced by NADH (1, 2), suggesting that rates of oxidative energy production would be increased at the level of IDH when cellular ratios of [ATP]/[AMP] and [NADH]/[NAD+] are low and attenuated when these ratios are high. The mammalian enzyme is similarly regulated by ADP and NADH, and also negatively regulated by ATP (3, 4).
Yeast IDH is a hetero-octamer composed of four IDH1 and four IDH2 subunits (5). IDH1 and IDH2 have similar molecular masses of 38,001 and 37,755, respectively, and their primary sequences are 42% identical (6, 7). Based on targeted mutagenesis studies (8–12) and two-hybrid analyses (13), it was proposed that the basic functional/structural unit of the enzyme is a heterodimer, with IDH2 contributing most of the residues in the catalytic isocitrate/Mg2+ and NAD+ binding sites and with IDH1 contributing most of the residues in homologous cooperative isocitrate and allosteric AMP binding sites. Each type of subunit, however, contributes a few residues to the active sites of the other type of subunit in each heterodimer. This basic heterodimeric structure was confirmed, and the organization of four heterodimers to form the octameric holoenzyme was defined by crystallographic analyses (14) (Figure 1A). Solution of the structures2 of ligand-free IDH and of IDH binding either the substrate analogue citrate or citrate + AMP also revealed structural transitions associated with binding of regulatory ligands including changes associated with allosteric communication.
FIGURE 1.
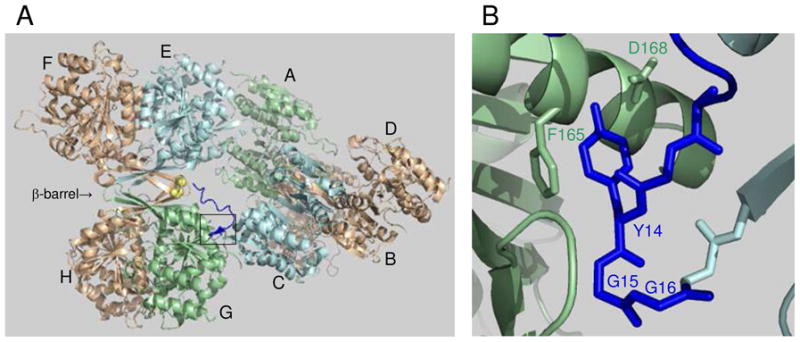
Structure of octameric yeast IDH and interactions at the tetramer interface. (A) Organization of IDH1/IDH2 heterodimers in the IDH octamer (PDB code 3BLV). Catalytic IDH2 subunits shown in light gold are located on the exterior of the octamer, with the regulatory IDH1 subunit in each heterodimer located on the interior of the octamer. Spatially equivalent IDH1 subunits are shown in light blue (C and E) or in light green (A and G). The β-barrel structure at the interface of heterodimers in the EF/GH tetramer is indicated. The amino terminus of IDH1 subunit C (shown in dark blue) extends to interact with the IDH2 Cys-150 residues (shown in yellow) located in the β-barrel structure of the other tetramer. The boxed area indicates the major site of interactions between IDH1 subunits C and G in the tetrameric interface. (B) Detail of the boxed area showing key residues in the tetrameric interface.
The interactions between IDH1 and IDH2 subunits in each heterodimer (e.g., between E and F or G and H, respectively, in Figure 1A) are extensive and similar to those described for homodimeric bacterial enzymes (15–17). The interactions between two heterodimers in IDH to form a heterotetramer (e.g., E/F and G/H in Figure 1A) are also extensive, and include β-hairpin loops from each of the four subunits that form an eight-stranded β-barrel. This β-barrel region appears to be relatively rigid and, in the ligand-free structure, could apparently be stabilized by a disulfide bond formed between Cys-150 residues (shown in yellow in Figure 1A) from two adjacent IDH2 subunits in the heterotetramer. Formation of this disulfide bond was predicted to potentially block ligand binding and to eliminate catalytic activity because AMP and (iso)citrate binding sites in the ligand-free structure are occluded or altered relative to these sites in the ligand-bound structure (14). Thus, formation of this bond could eliminate catalytic activity. We have recently shown that this disulfide bond does form in the purified IDH enzyme in response to the presence of the oxidizing agent diamide and, furthermore, the disulfide bond also forms in vivo as yeast cells enter stationary phase (18). In both cases, formation of the disulfide bond is associated with dramatic reductions in catalytic activity.
In contrast to extensive molecular interactions to form heterodimer and heterotetramer structures, the only substantial interaction between IDH heterotetramers to form the octamer involves a protrusion of ~16 residues of the amino terminus of an IDH1 subunit from one tetramer (illustrated in dark blue for C in Figure 1A) into the other tetramer. Because of a deviation from pseudo-222 symmetry in the relationship of the two heterotetramers and a twist of one heterotetramer relative to the other, the amino termini of two IDH1 subunits in the octamer (C and E in Figure 1A) are spatially equivalent. Whereas the amino termini of the other two spatially equivalent IDH1 subunits (G and A in Figure 1A) are located on the exterior of the enzyme, residues 12–16 of IDH1 subunits C and E form tight turns that insert into respective pockets in IDH1 subunits G and A in the other heterotetramers (as shown for C and G in Figure 1B). In the ligand-bound structure, the remainder of the amino termini of IDH1 subunits C and E bridge the space between the tetramers and are positioned near the IDH2 Cys-150 residues in the other tetramer. The proximity of the IDH1 amino terminus and nearby basic side chains in the ligand-bound structure was proposed to help stabilize the reduced forms of the IDH2 Cys-150 residues to facilitate catalytic activity (14).
To examine the importance of the amino termini of IDH1 subunits in stabilizing an octameric form of IDH, we used truncation and mutagenesis of these amino termini to construct stable tetrameric forms of IDH. We compared octameric and tetrameric forms of the enzyme with respect to catalytic and regulatory properties, as well as the facility for formation of the IDH2 Cys-150 disulfide bond.
EXPERIMENTAL PROCEDURES
Mutagenesis, Expression, and Purification
The template for mutagenesis was a previously described plasmid (pET-15bIDH1His/IDH2) (14) carrying yeast IDH1 and IDH2 genes and codons for a histidine tag on the 3′ end of the coding sequence of IDH1. Oligonucleotides were used to construct IDH1 genes lacking 5, 10, 15, 16, or 17 codons at the 5′ end of the coding sequence for the mature subunit. The truncated genes were amplified by polymerase chain reaction (PCR) and used to replace the IDH1 gene in pET-15b IDH1His/IDH2. Mutations were constructed using the Quickchange site-directed mutagenesis method (Stratagene) to replace codons for IDH1 Gly-15 with Ala or Asp codons or for IDH1 Asp-168 with Ala or Lys codons. All truncations and mutations were confirmed by DNA sequence analysis at the Advanced DNA Technologies facility at the University of Texas Health Science Center.
For expression and purification, plasmids were transformed into Escherichia coli strain BL21-Gold (DE3) competent cells (Stratagene). Transformants were cultivated to an optical density at A600 nm = 0.8, and expression was induced with 1 mM isopropyl 1-thio-β-D-galactopyranoside for 5 h at 37 °C. Ni2+-nitrilotriacetic acid (NTA) chromatography was used to purify enzymes from harvested cells as previously described (9). Concentrations of purified enzymes were determined by measuring A280 nm and using a molar extinction coefficient of 168,810 M−1cm−1 (19). Purified enzymes were stored at −20 °C in the elution buffer (50 mM sodium phosphate, 250 mM imidazole, and 300 mM sodium chloride, pH 8.0) containing 20% glycerol.
Analytical Ultracentrifugation
Sedimentation velocity analyses were conducted with a Beckman Optima XL-I analytical ultracentrifuge in the Center for Analytical Ultracentrifugation of Macromolecular Assemblies at the University of Texas Health Science Center. Data were analyzed with UltraScan (20) version 9.9 (21). Sedimentation velocity experiments were performed with ~2 μM samples suspended in a buffer containing 20 mM Tris-HCl, pH 7.4, and 150 mM NaCl. Measurements were taken at 20 °C using an AN60ti rotor and 2-channel titanium centerpieces (Nanolytics, Potsdam, Germany) at 25,000 rpm and at 280 nm in intensity mode or at 60,000 rpm using Rayleigh interference optics. Density and viscosity corrections were made based on methods outlined in Laue et al. (22). Partial specific volumes were estimated based on peptide sequence with UltraScan and found to be 0.7404 ccm/g for the IDH1/IDH2 enzyme, 0.7408 ccm/g for IDH1D168K/IDH2 and -5IDH1D168K/IDH2 enzymes, 0.7030 ccm/g for the IDH1G15D/IDH2 enzyme, 0.7400 for the -5IDH1/IDH2 enzyme, 0.7405 for the -5IDH1G15D/IDH2 enzyme, and 0.7404 for the -5IDH1G15A/IDH2 enzyme. Data were first analyzed by two-dimensional spectrum analysis (23) with simultaneous removal of time- and radially-invariant noise, and then refined by genetic algorithm (24) and Monte Carlo analysis (25) (see Supplementary Figure 3 for a representative data set). These analysis methods provide a whole boundary least squares fit of the experimental data and resolve individual species in a mixture to provide partial concentration, frictional ratios, sedimentation coefficients, molecular weights and diffusion coefficients for each species in the mixture. As we have demonstrated previously, unlike the C(s) method (26) implemented in the Sedfit software (www.analyticalultracentrifugation.com), these methods provide enhanced resolution and are capable of resolving heterogeneity in shape as well as molecular weight, providing more reliable molecular weight distributions than those obtained with C(s) (24, 27). Results are represented as a 2-dimensional pseudo-3D plot of the frictional coefficient versus the molecular weight or sedimentation coefficient, with a color gradient indicating the partial concentration of each species (see Supplementary Figure 1). Diffusion-corrected, model-independent sedimentation coefficient distributions were generated with the enhanced van Holde - Weischet analysis (28, 29), which provides integral G(s) distributions of the boundary fractions. When overlaid, they provide a convenient and sensitive method for comparison between different experiments (see Figure 5).
FIGURE 5.

Sedimentation velocity analysis of wild-type and mutant forms of IDH. Van Holde and Weischet integral distribution plots of sedimentation coefficients (28, 29) for affinity-purified forms of IDH are shown. The top panels (open symbols) show plots for the truncated -5IDH1/IDH2 enzyme and mutant enzymes also containing a residue substitution as indicated. The bottom panels (closed symbols) show plots for the wild-type IDH1/IDH2 and mutant enzymes containing a residue substitution as indicated. Sedimentation velocity analyses were conducted at 25,000 rpm or at 60,000 rpm as indicated.
Gel filtration was conducted using HPLC (Superdex 200 10/300 GL High Performance Column, GE Healthcare) and affinity purified enzymes (0.5 mg in 0.5 ml of a buffer of 20 mM Tris-HCl, pH 7.4, and 150 mM NaCl). Molecular size standards included carbonic anhydrase (29 kDa), albumin (66 kDa), alcohol dehydrogenase (150 kDa), amylase (200 kDa), apoferritin (443 kDa), and thyroglobulin (669 kDa).
Kinetic Analyses and Electrophoretic Methods
IDH specific activity (units/mg, with 1 unit = 1 μmol NADH/min at 24 °C) was routinely measured in 1 ml assays containing 40 mM Tris-HCl (pH 7.4), 1 mM D-isocitrate, 4 mM MgCl2, and 0.5 mM NAD+. Apparent S0.5 values and Hill coefficients with respect to isocitrate were determined in the absence or presence of 100 μM AMP using concentrations of D-isocitrate ranging from 0 to 5 mM. Apparent S0.5 values with respect to NAD+ were determined using NAD+ concentrations ranging from 0 to 1.4 mM. Apparent Vmax values are the maximum velocity obtained per mg enzyme.
Reducing gel electrophoresis was conducted using 10% polyacrylamide gels made according to Laemmli (30) and using a sample loading buffer containing 100 mM Tris-HCl (pH 6.8), 3% sodium dodecyl sulfate, 2 mM EDTA, 0.35 M β-mercaptoethanol, and 10% glycerol. Nonreducing gel electrophoresis was similarly conducted using a loading buffer lacking β-mercaptoethanol. Gels were stained with Coomassie blue.
Yeast strain and expression
To generate an idh1Δidh2Δ mutant, strain BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) (31) was used for disruption of chromosomal IDH1 (with a HIS3 gene) and of IDH2 (with a LEU2 gene). Gene replacement cassettes were constructed using PCR and selection markers from a set of loxP plasmids (32). A previously described pRS316-IDH1/IDH2 plasmid (9) designed for single-copy expression in yeast (33) was used for mutagenesis as described above. In the pRS316-IDH1/IDH2 plasmid, each IDH gene contains its authentic promoter and mitochondrial targeting sequence. DNA sequence analysis was used to confirm mutagenesis and to insure no other changes had been made in IDH1 or IDH2 genes. The plasmids were transformed into the yeast idh1Δidh2Δ strain using a lithium acetate protocol (34). Transformants were streaked onto plates containing rich YP medium (1% yeast extract and 2% Bacto-peptone) and 2% glucose. Complementation tests were conducted by streaking the transformants onto YP plates containing 2% sodium acetate as the carbon source.
Diamide treatment of purified enzymes
Diamide was freshly prepared as a 100 mM stock in water. Enzyme samples (1 μM in 50 mM Tris-HCl, pH 7.4) were incubated with various concentrations of diamide for 2 h on ice prior to determination of specific activity and electrophoresis on nondenaturing gels. These conditions were based on previous demonstration that the effects of diamide on inhibition of IDH activity and on formation of the IDH2 Cys-150 disulfide bond are both concentration and time dependent (18). For ligand protection assays, enzyme samples were pre-incubated for 15 min on ice with 4 mM MgCl2, 0.1 mM AMP, and citrate concentrations ranging from 0 to 100 mM prior to treatment for 2 h with diamide concentrations determined to produce ~50% inhibition of activity for each enzyme.
RESULTS
Construction of an IDH tetramer
The crystal structure suggested that construction of a stable tetrameric form of IDH would likely require truncation of the amino terminus of IDH1 and/or introduction of residue replacements in the region of interaction of the amino terminus of IDH1 and an IDH1 subunit in the other tetramer. Therefore, we initially introduced truncations to remove 5 or 10 amino acids from the amino terminus of IDH1 (Figure 2). Enzymes containing the IDH1 truncations were expressed in E. coli using a plasmid carrying both IDH1 and IDH2 genes and affinity purified based on the presence of a histidine tag on the carboxyl terminus of the IDH1 subunit (9). Yields of the -5IDH1/IDH2 and -10IDH1/IDH2 enzymes were comparable to that for the wild-type IDH1/IDH2 enzyme. The specific activity of the -5IDH1/IDH2 enzyme was also comparable to that of the wild-type enzyme while that of the -10IDH1/IDH2 enzyme was ~20% lower. We subsequently introduced truncations to remove 15, 16, or 17 amino acids from the amino terminus of IDH1 (Figure 2) since Gly-15 and Gly-16 contribute to a turn in the amino terminus that extends into the other tetramer (Figure 1B). However, yields of the -15IDH1/IDH2, -16IDH1/IDH2, and -17IDH1/IDH2 enzymes were ~10-fold lower than wild-type, and activity was rapidly lost after purification. Therefore, we concentrated on characterization of the -5IDH1/IDH2 enzyme.
FIGURE 2.

Amino-terminal sequence of the mature IDH1 subunit. Residue sites targeted for truncation or amino acid substitutions in the IDH1 subunit are indicated.
Sedimentation velocity experiments conducted at 25,000 rpm (Figure 3) indicated homogeneous species for the wild-type IDH1/IDH2 and truncated -5IDH1/IDH2 enzymes with similar sedimentation coefficients (10.7 and 10.9, respectively) and sizes (299 kDa and 311 kDa, respectively). Gel filtration produced comparable apparent sizes (326 kDa and 298 kDa, respectively; see below), suggesting that both enzymes are octamers. Catalytic properties were also similar, as described below.
FIGURE 3.

Analytical ultracentrifugation of IDH1/IDH2 and -5IDH1/IDH2 enzymes. Van Holde and Weischet integral distribution plots of sedimentation coefficients (28) for affinity-purified wild-type IDH1/IDH2 and truncated -5IDH1/IDH2 enzymes are shown. Sedimentation velocity runs were conducted at 25,000 rpm. Values for the wild-type enzyme were similar to those previously reported (49).
Since the -5IDH1/IDH2 enzyme apparently retains the octameric structure of the wild-type enzyme, we used the truncated enzyme to introduce residue replacements predicted from the crystal structure to alter the interaction between IDH1 subunits at the tetramer interface (e.g., between subunits C and G in Figure 1B). Based on substantial contributions to hydrogen bonding at this interface, IDH1 Gly-15 and Asp-168 residues were chosen as primary targets. Substitutions for these residues included a relatively conservative G15A replacement and G15D, D168A, and D168K replacements designed to change the charges of side chains in target residues (Figure 2). The IDH1/IDH2, -5IDH1/IDH2, -5IDH1G15A/IDH2, -5IDH1G15D/IDH2, -5IDH1D168A/IDH2, and -5IDH1D168K/IDH2 enzymes were affinity purified. As shown in Figure 4A, the truncation of IDH1 in the -5IDH1/IDH2 enzyme resulted, as expected, in slightly faster migration of this subunit than observed for IDH1 in the wild-type enzyme during denaturing gel electrophoresis. Residue substitutions for Gly-15 (in -5IDH1G15A/IDH2 and -5IDH1G15D/IDH2 enzymes) did not alter electrophoretic properties of the -5IDH1 subunit (Figure 4B), whereas substitutions for Asp-168 (in -5IDH1D168A/IDH2, and -5IDH1D168K/IDH2 enzymes) slightly increased the migration of the -5IDH1 subunit.
FIGURE 4.

Affinity-purified wild-type and mutant forms of IDH. Samples (3 μg) of affinity- purified forms of IDH as indicated were electrophoresed using denaturing polyacrylamide sodium dodecyl sulfate gels and stained with Coomassie blue.
The affinity purified enzymes were analyzed by sedimentation velocity methods, initially using a speed of 25,000 rpm. As shown in Figure 5 (top left panel), and as summarized in Table 1A, the -5IDH1/IDH2 enzyme sedimented primarily as a single species with an average sedimentation coefficient of 10.9. The major boundary species (63% and 73%, respectively) for the -5IDH1G15D/IDH2 and -5IDH1D168K/IDH2 enzymes both exhibited average sedimentation coefficients of 6.8, consistent with tetrameric forms of IDH. Genetic algorithm - Monte Carlo analysis resulted in molecular weights (155 kDa for the -5IDH1G15D/IDH2 enzyme and 141 kDa for the -5IDH1D168K/IDH2 enzyme) in excellent agreement with tetrameric forms. Most of the remaining signals consistently indicate a range of smaller molecular weight species, which likely correspond to further dissociation products of the tetramer into dimeric or monomeric species. Frictional ratios were around 1.5 for the tetrameric enzymes and 1.4 for the octameric enzymes (Supplementary Figure 1), suggesting a slightly more globular shape for the octamer (35). In contrast, the -5IDH1D168K/IDH2 enzyme also sedimented primarily as a single species with an average sedimentation coefficient of 10.6 suggesting retention of an octameric structure, whereas the broad sedimentation pattern for the -5IDH1G15A/IDH2 enzyme suggests a heterogeneity of species.
Table 1.
Sedimentation coefficients for major boundary species.
| Enzyme | 25,000 rpm |
60,000 rpm |
|||
|---|---|---|---|---|---|
| Major species (% boundary) | Sedimentation coefficient (× 1013) | Major species (% boundary) | Sedimentation coefficient (× 1013) | ||
| A | -5IDH1/IDH2 | 90 | 10.9 | 82 | 10.6 |
| -5IDH1G15D/IDH2 | 63 | 6.8 | 63 | 6.8 | |
| -5IDH1D168K/IDH2 | 73 | 6.8 | 56 | 7.0 | |
| -5IDH1G15A/IDH2 | —b | — | 50 | 9.9 | |
| -5IDH1D168A/IDH2 | 90 | 10.6 | 62 | 10.7 | |
| B | IDH1/IDH2 | 92 | 10.7 | 75 | 10.9 |
| IDH1G15D/IDH2 | 77 | 6.8 | 76 | 6.9 | |
| IDH1D168K/IDH2 | — | — | 55 | 7.0 | |
Sedimentation coefficients were determined from van Holde and Weischet integral distribution plots.
Blanks indicate broad sedimentation patterns and heterogeneity of species, precluding assignment of sedimentation coefficients.
Velocity sedimentation was subsequently conducted at a higher speed of 60,000 rpm to limit diffusion and thus sharpen the boundary fraction. This produced very little change in sedimentation patterns (Figure 5, upper right panel) or in sedimentation coefficients (Table 1) except for the -5IDH1G15A/IDH2 enzyme with 50% of the boundary species sedimenting with an average sedimentation coefficient of 9.9. This suggests this enzyme is larger than expected for a tetramer.
Given the success with construction of stable tetrameric -5IDH1G15D/IDH2 and -5IDH1D168K/IDH2 enzymes, we wished to determine if the IDH1 residue substitutions in the truncated enzyme would have similar effects if introduced into the wild-type enzyme. The IDH1 G15D and D168K residue substitutions were therefore constructed in the IDH1/IDH2 enzyme, and the enzymes were affinity purified (Figure 4C). In sedimentation velocity analyses (Figure 5, lower panels; Table 1B), the major boundary species of the IDH1G15D/IDH2 enzyme sedimented with an average sedimentation coefficient of 6.8 at 25,000 rpm and of 6.9 at 60,000 rpm (molecular weight of 165 kDa by genetic algorithm - Monte Carlo analysis), suggesting that this enzyme is also a stable tetramer. However, a broad sedimentation distribution observed for the IDH1D168K/IDH2 enzyme at both speeds suggests heterogeneity of species. Thus, it appears that the G15D substitution is sufficient for construction of a tetrameric form of IDH, whereas the D168K substitution also requires the -5 truncation of IDH1 to produce a convincing tetramer.
The sizes of apparent octameric and tetrameric enzymes were also examined using gel filtration chromatography. Molar masses measured with both methods are compared in Table 2, and detailed sedimentation velocity analyses are shown in Supplementary Table 1. The numbers determined using both methods were consistent with octameric forms for IDH1/IDH2 and -5IDH1/IDH2 enzymes and with tetrameric forms for -5IDH1G15D/IDH2, -5IDH1D168K/IDH2, and IDH1G15D/IDH2 enzymes.
Table 2.
Molecular sizes by two methods.
Finally, we examined the dependence of the tetrameric form of IDH on concentration or presence of ligands. As shown in Supplementary Figure 2 and Supplementary Table 2, similar sedimentation properties were observed for the tetrameric IDH1G15D/IDH2 enzyme when sedimentation velocity experiments were conducted with three different concentrations of the enzyme or in the presence of saturating concentrations of various ligands of IDH. Thus, there appears to be no tendency for a concentration- or ligand-induced association of the IDH1G15D/IDH2 tetramer into an octamer or other higher order species.
Kinetic parameters of IDH octamers and tetramers
Kinetic parameters of octameric enzymes (IDH1/IDH2 and -5IDH1/IDH2) and tetrameric enzymes (IDH1G15D/IDH2, -5IDH1G15D/IDH2 and -5IDH1D168K/IDH2) were compared using isocitrate and NAD+ saturation velocity curves. As previously reported (10) and as shown in Table 3, IDH (IDH1/IDH2) is highly cooperative with respect to isocitrate (Hill coefficients of 3–4) and there is a marked increase (~5-fold) in the apparent affinity for isocitrate in the presence of 100 μM AMP. As shown in Table 3, properties with respect to isocitrate were almost identical for IDH1/IDH2 and -5IDH1/IDH2 enzymes. Thus, the -5 amino terminal truncation of IDH1 does not have a major impact on apparent Vmax or other kinetic properties. For the tetrameric enzymes (Table 3), a common feature was a reduction in apparent Vmax values (~38% for IDH1G15D/IDH2 relative to IDH1/IDH2, and ~50% for -5IDH1G15D/IDH2 and -5IDH1D168K/IDH2 relative to -5IDH1/IDH2). Another common property of the tetrameric enzymes was a substantial reduction in cooperativity (Hill coefficients of 2–2.3). However, effects of the positive allosteric activator AMP on affinity for isocitrate were essentially the same for octameric and tetrameric enzymes. All of the enzymes had similar S0.5 values (0.13 to 0.19 mM) with respect to NAD+ (data not shown). These results suggest the positive allosteric AMP effect is completely operative at the level of the tetramer (or perhaps the heterodimer), but that cooperative effects with respect to isocitrate extend from one tetramer to the other in the octameric enzyme, and that maximum velocity requires communication between tetramers in the octamer.
Table 3.
Kinetic parameters of octameric and tetrameric forms of IDH.
| Enzyme | Vmax (units/mg) | S0.5 (mM) | Hill coef. |
|---|---|---|---|
| −/+ AMPa | −/+ AMP | −/+ AMP | |
| IDH1/IDH2 | 32.3/32.3 (±0.7/0.4)b | 0.53/0.10 (±0.10/0) | 3.1/3.5 (±0.1/0.3) |
| IDH1G15D/IDH2 | 20.8/21.9 (±0.5/1.7) | 0.60/0.13 (±0.13/0.01) | 2.3/2.0 (±0.2/0.1) |
| -5IDH1/IDH2 | 30.0/30.3 (±0.9/0.9) | 0.52/0.11 (±0.04/0.01) | 3.1/3.5 (±0.2/0.1) |
| -5IDH1G15D/IDH2 | 14.3/15.4 (±1.7/0.7) | 0.64/0.14 (±0.03/0.04) | 2.2/2.0 (±0.4/0.5) |
| -5IDH1D168K/IDH2 | 14.8/15.3 (±1.2/1.9) | 0.61/0.13 (±0.13/0.04) | 2.2/2.0 (±0/0.6) |
Velocity saturation curves with respect to isocitrate were conducted in the absence or presence of 100 μM AMP as described in Materials and Methods.
Differences from the mean are shown for values determined in two independent experiments.
Loss or dysfunction of IDH in yeast results in an inability to grow with acetate as the carbon source (8, 9), a phenotype shared with some other yeast tricarboxylic acid cycle mutants (36). Therefore, to test activities of mutant enzymes in vivo, we constructed truncated and mutated IDH1 genes in a yeast plasmid also carrying the wild-type IDH2 gene and transformed the plasmids into an idh1Δidh2Δ yeast disruption mutant. The transformants were tested for growth on plates with rich YP medium and acetate as the carbon source. As shown in Figure 6, the idh1Δidh2Δ strain failed to grow under these conditions, whereas transformants carrying plasmids expressing either octameric (IDH1/IDH2 and -5IDH1/IDH2) or tetrameric forms of IDH (IDH1G15D/IDH2, -5IDH1G15D/IDH2 and -5IDH1D168K/IDH2) were able to grow as well on this carbon as the parental strain. These results indicate that the tetrameric forms of IDH are functional in vivo.
FIGURE 6.
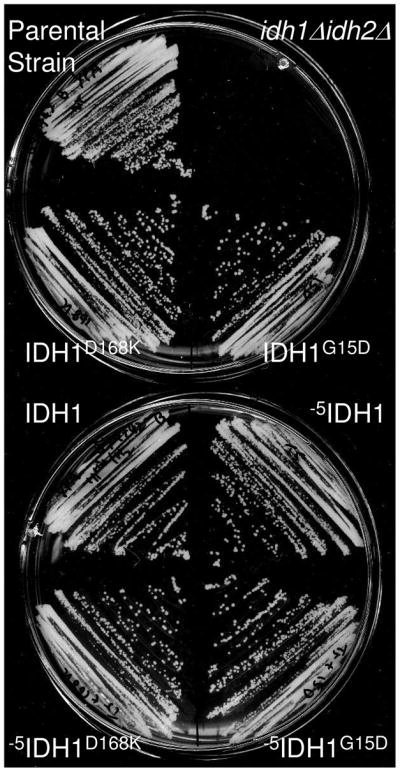
Growth of yeast transformants on plates with acetate as the carbon source. The parental strain, an idh1Δidh2Δ mutant, and transformants of the idh1Δidh2Δ strain expressing IDH enzymes composed of IDH2 and the indicated form of IDH1 were streaked onto YP agar plates containing acetate as the carbon source. Plates were incubated at 30°C for 4 days.
Diamide inhibition of activity and formation of the Cys-150 disulfide bond
As we previously reported (18), a disulfide bond can be formed between Cys-150 residues in adjacent IDH2 subunits (as illustrated in Figure 1A) both in vitro in response to incubation of purified IDH with diamide and in vivo as yeast cells enter stationary phase. Since the structures determined for the enzyme suggested that the amino terminus from an IDH1 subunit extends from one tetramer and helps to stabilize the reduced form of the IDH2 Cys-150 residues, we predicted that truncation of the IDH1 subunit would facilitate formation of the Cys-150 disulfide bond and, furthermore, that a tetrameric form of IDH would be particularly susceptible to formation of this bond. Therefore, we incubated the purified octameric and tetrameric forms of IDH with increasing amounts of diamide and examined effects on activity and on formation of the disulfide bond using nondenaturing gel electrophoresis. An example of results from the latter (Figure 7) indicates no formation of this bond for the IDH1/IDH2 enzyme following incubation with no or a low concentration of diamide, whereas traces of the disulfide bond are present in the IDH1G15D/IDH2 even in the absence of the oxidizing agent and most of the IDH2 subunit is present as the disulfide bond form following incubation with 10 μM diamide.
FIGURE 7.

Disulfide-bond formation in IDH1/IDH2 and IDH1G15D/IDH2 enzymes. Affinity purified enzymes were treated for 2 h with 0 or 10 μM diamide prior to electrophoresis of 3.5 μg samples on nondenaturing PAGE gels and staining with Coomassie blue. The identity of the IDH2 band containing the Cys-150 disulfide bond was previously verified (18).
The effects of increasing concentrations of diamide on activity of the IDH1/IDH2 and -5IDH1/IDH2 enzymes are illustrated in Figure 8A, and concentrations of diamide producing 50% reductions in catalytic activity are summarized in Table 4. The activity of the -5IDH1/IDH2 enzyme was ~3-fold more sensitive than the IDH1/IDH2 enzyme to incubation with diamide, indicating that the close proximity of the amino-terminus of an IDH1 subunit from the other tetramer in the wild-type enzyme does in fact help to stabilize the reduced form of the IDH1/IDH2 enzyme. In similar experiments (Figure 8B and Table 4), the tetrameric forms of IDH (IDH1G15D/IDH2, -5IDH1G15D/IDH2 and -5IDH1D168K/IDH2) exhibited dramatic increases in sensitivity to diamide (38- to 77-fold relative to the IDH1/IDH2 enzyme). These results indicate that tetrameric forms of IDH are particularly sensitive to inactivation in the presence of an oxidizing agent. This inactivation corresponds to formation of the IDH2 Cys-150 bond (as illustrated in Supplementary Figure 4).
FIGURE 8.

Effects of diamide on catalytic activity. (A) Affinity purified octameric enzymes as indicated were incubated with increasing concentrations of diamide prior to activity assays as described in Experimental Procedures. (B) Affinity purified tetrameric enzymes as indicated were incubated with lower concentrations of diamide prior to activity assays. Activities for the octameric enzymes are also shown to emphasize small responses to lower concentrations of diamide. Maximum activity measured for each untreated enzyme was set as 100%.
Table 4.
Concentrations of diamide required for 50% inhibition of catalytic activity.
| Enzyme | μM Diamidea | Enzyme | μM Diamide |
|---|---|---|---|
| IDH1/IDH2 | 500 | -5IDH1/IDH2 | 160 |
| IDH1G15D/IDH2 | 6.5 | -5IDH1G15D/IDH2 | 7.5 |
| -5IDH1D168K/IDH2 | 13.0 | ||
Values were determined from graphs presented in Figure 8.
As described above, in the ligand-free structure of IDH, the IDH2 Cys-150 side chains are in sufficient proximity for formation of a disulfide bond and the ligand-binding sites for isocitrate and AMP appear to be occluded or altered relative to corresponding ligand-bound sites (14). In contrast, in the citrate- or citrate/AMP- bound structure, the distance between Cys-150 side chains is greater. This suggests that, in the holo-octameric enzyme, the presence of bound ligands might preclude or disfavor formation of the disulfide bond. We tested this possibility by pre-incubation of the wild-type IDH1/IDH2 enzyme with ligands (AMP, Mg2+, and increasing amounts of citrate) prior to treatment with a sufficient amount of diamide to produce a substantial inhibition of catalytic activity (see Table 4). As illustrated in Figure 9A, in the absence of citrate in this experiment, 0.5 mM diamide produced a 63% reduction in activity of the IDH1/IDH2 enzyme. Preincubation with increasing concentrations of citrate (and fixed levels of other ligands) prior to exposure to diamide progressively reduced the level of inhibition by the oxidant until no inhibition by diamide was observed following preincubation with 100 mM citrate. Nonreducing gels demonstrated a corresponding progressive reduction in formation of the IDH2 Cys-150 disulfide bond species following preincubation with substrates as well as the absence of this species following preincubation with 100 mM citrate (Supplementary Figure 5). Thus, the presence of a substrate analog reduces inhibition of wild-type IDH by diamide.
FIGURE 9.

Ligand protection from diamide-induced inactivation. (A) The wild-type IDH1/IDH2 enzyme was pre-incubated with 4 mM MgCl2, 100 μM AMP, and the indicated concentrations of citrate for 15 min prior to treatment with 0.5 mM diamide and activity assays. (B) The indicated enzymes were similarly pre-incubated with ligands prior to treatment with concentrations of diamide shown in Table 4 estimated to produce a 50% reduction in activity for each enzyme.
Since the truncated -5IDH1/IDH2 enzyme is more susceptible to diamide inhibition than the wild-type enzyme, we tested if preincubation of this enzyme with increasing amounts of citrate would affect this inhibition. As shown in Figure 9B, treatment of the -5IDH1/IDH2 enzyme with 0.16 mM diamide produced a 43% reduction in activity, and preincubation with increasing amounts of citrate had essentially no effect on the extent of diamide inhibition of activity (or on the level of the IDH2 Cys-150 disulfide bond, Supplementary Figure 5). Similar results were obtained for the tetrameric forms of IDH (IDH1G15D/IDH2, -5IDH1G15D/IDH2 and -5IDH1D168K/IDH2) using diamide concentrations designed to produce 40–50% reductions in activity of each enzyme (Figure 9B and Table 4). Thus, the presence of a substrate analogue had no effect on diamide-induced inhibition nor did it eliminate formation of the IDH2 Cys-150 disulfide bond in truncated or tetrameric forms of IDH (Supplementary Figure 4). These results support the structural prediction that, in the presence of ligands, extension of the amino termini of IDH1 subunits into adjacent tetramers in the IDH1/IDH2 octamer is important for maintaining a reducing environment near the IDH2 Cys-150 side chains and thus for maintaining a catalytically active form of the enzyme.
DISCUSSION
Yeast IDH is very stable in that the affinity-purified enzyme retains full activity as a holo-octamer upon long-term storage. However, we have constructed tetrameric forms of the enzyme by introducing a single residue substitution (IDH1 G15D) or by removing 5 residues from the amino terminus of IDH1 in conjunction with introducing another residue substitution (IDH1 D168K). These changes were directly based on predictions from crystallographic structures determined for IDH (14). Clearly, other residues (e.g. IDH1 Tyr-14, Gly-16, or Phe-165 in Figure 1B) in the tetramer interface could also be targets for substitution that might produce similar results.
The three tetrameric forms of IDH (IDH1G15D/IDH2, -5IDH1G15D/IDH2, and -5IDH1D168K/IDH2) generated in this study share similar kinetic properties distinct from those of the octameric forms of IDH (IDH1/IDH2 and -5IDH1/IDH2), suggesting that these properties are generic for the tetramer and not reflective of specific alterations. A comparison of properties of the tetramers and octamers suggest that the octameric form of IDH has evolved to maximize catalytic activity and cooperativity with respect to isocitrate. In addition, the tetrameric enzymes are extremely sensitive to oxidation and formation of the IDH2 Cys-150 disulfide bond, and the truncated -5IDH1/IDH2 enzyme is also 3-fold more sensitive to inhibition by diamide than the IDH1/IDH2 enzyme. This suggests that the octameric holo-enzyme has evolved to support regulation of disulfide bond formation in each component tetramer by amino termini of IDH1 subunits from the other tetramer. This notion is supported by the ability of ligands to attenuate diamide inhibition of the IDH1/IDH2 enzyme but not of truncated or tetrameric forms of the enzyme.
As mentioned above, the deviation from pseudo-222 symmetry exhibited by the yeast IDH octamer (14) results in spatial asymmetry for regulatory IDH1 subunits, so that amino termini of two IDH1 subunits (e.g., C and E in Figure 1) extend into adjacent tetramers whereas the amino termini of the other two IDH1 subunits (A and G in Figure 1) are located on the exterior of the octamer. The similarity in kinetic properties of the IDH1/IDH2 and -5IDH1/IDH2 enzymes suggests that the truncation of all IDH1 subunits in the octamer has no major effect on catalytic or regulatory properties. Thus, effects on disulfide-bond formation are likely due to truncation of the interacting subunits (e.g., C and E) and not to truncation of the exterior subunits (A and G).
With respect to the asymmetry of regulatory IDH1 subunits in yeast IDH, it should be noted that mammalian isocitrate dehydrogenase is reported to be an octameric enzyme composed of four catalytic α subunits and two each of regulatory β and subunits (37, 38). Sizes of the mammalian enzyme subunits are quite similar to those of yeast IDH, and respective catalytic and regulatory subunits share 40–55% sequence identity (39, 40). Comparison of sequences for the regulatory subunits shows that the mammalian subunit (but not the β subunit) contains a sequence (-Ala-Lys-Tyr-Gly-Gly-Arg-) that aligns with a sequence (-Lys-Lys-Tyr-Gly-Gly-Arg) in IDH1 containing the Gly-15 residue (underlined), whereas the mammalian β subunit (but not the subunit) contains a sequence (-Phe-Ala-Phe-Asp-Tyr-Ala-) that aligns with a sequence (-Phe-Ala-Phe-Asp-Phe-Ala-) in yeast IDH1 containing the Asp-168 residue (underlined). Thus, we would predict that heterodimers in the mammalian octameric enzyme may be organized with subunits corresponding to IDH1 subunits C and E and with β subunits corresponding with IDH1 subunits A and G in Figure 1. Structural modeling of the mammalian enzyme shows no likelihood for formation of a disulfide bond corresponding with that formed by IDH2 Cys-150 residues, so regulation of properties of the octameric mammalian enzyme may involve other interactions between component tetramers.
Yeast idh1Δidh2Δ transformants expressing truncated and tetrameric forms of IDH at levels comparable to that of the wild-type enzyme in the parental strain are able to grow with acetate as the carbon source (Figure 6). Thus, these forms of the enzyme are active in vivo. This was not an unexpected result, since previous studies (8, 10, 12) have indicated that only mutant forms of IDH with substantial reductions in specific activity prevent steady-state growth with acetate as the carbon source. In contrast, expression of mutant forms of IDH with primary regulatory defects results in slow transitions from growth with glucose to growth with a nonfermentable carbon source (41). We are testing effects of expression of mutant enzymes from the current study in similar transitions. We note that estimated concentrations of IDH in the mitochondrial matrix of yeast cells range from 1.7 to 4.3 mg/ml.3 While it is not possible to assess oligomeric state in vivo, these values are in the range of concentrations (1 to 3 mg/ml) used in in vitro centrifugation and gel filtration analyses. Also, the in vitro experiments were conducted with highly purified enzymes in contrast with conditions in vivo that would presumably be unfavorable for oligomerization of tetrameric forms of IDH.
Since we have previously shown that formation of the IDH2 Cys-150 disulfide bond occurs in yeast cells during entry into stationary phase, we are testing effects on viability of expression of mutant enzymes from this study that are particularly susceptible to formation the disulfide bond in vitro. The transition from logarithmic growth to stationary phase in yeast cells involves metabolic changes necessary for utilization of carbon sources like ethanol and acetate for anabolic processes. Formation of a disulfide bond and reduced activity of IDH under these conditions could potentially enhance export of citrate and isocitrate from the mitochondrial matrix into the cytosol for utilization in gluconeogenesis and in the glyoxylate pathway. The glyoxylate pathway, present in nonmitochondrial compartments of yeast and plant cells and in some bacteria, permits net assimilation of two-carbon precursors into four-carbon metabolites.
The idea that IDH could be a central point for control of metabolic changes is not a novel idea since there is a solid precedent for reversible down-regulation, albeit via a different mechanism, of isocitrate dehydrogenase in Eshcerichia coli. When E. coli cells are shifted to medium with acetate as the carbon source, ~80% of the isocitrate dehydrogenase molecules are rapidly inactivated by phosphorylation of a serine residue in the catalytic site (43, 44). This modification results in redirection of much of the total carbon flux from the tricarboxylic acid cycle into biosynthetic pathways (45–47). A specific kinase/phosphatase controls the extent of reversible modification of bacterial isocitrate dehydrogenase (48). We have found no evidence for phosphorylation of yeast IDH and propose that disulfide bond formation may have a similar, reversible catalytic effect. The mammalian enzyme may be regulated by yet another mechanism to effect similar metabolic changes. We are currently investigating these possibilities for yeast and mammalian mitochondrial enzymes.
Supplementary Material
Acknowledgments
We thank Dr. P. John Hart for structural predictions. We also acknowledge the support of the UTHSCSA Center for Macromolecular Interactions, which is supported by the Cancer Therapy and Research Center through the NIH-NCI P30 award CA054174, as well as by Texas State funds provided through the Office of the Vice President for Research of the UTHSCSA.
Footnotes
This work was supported by National Institutes of Health Grants GM051265 (L.M-H.) and RR022200 (B.D.). Supercomputer allocations were provided by National Science foundation TG-MCB070038 (B.D.).
Abbreviations: IDH1, yeast NAD+-specific isocitrate dehydrogenase; YP, rich medium containing yeast extract and Bacto-peptone; NTA, nitrilotriacetic acid; EDTA, ethylenediaminetetraacetic acid; HPLC, high performance liquid chromatography; PDB, Protein Data Bank.
Accession codes for IDH structures in the Protein Data Bank are 3BLX for the ligand-free structure, 3BLV for the citrate-bound structure, and 3BLW for the citrate plus AMP-bound structure.
Estimates of in vivo concentrations were based on quantitative immunoblot analyses showing that IDH represents 0.2–0.5% of the total cellular protein in yeast cells grown with a nonfermentable carbon source, on a value (6 pg/fL) for the total protein concentration in yeast cells (42), and on an assumption that the mitochondrial concentration of a mitochondrial protein would be ~10-fold greater than the total cellular concentration.
SUPPORTING INFORMATION AVAILABLE
Supplementary Figures 1–5 and Supplementary Tables 1 and 2. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hathaway JA, Atkinson DE. The effect of adenylic acid on yeast nicotinamide adenine dinucleotide isocitrate dehydrogenase, a possible control mechanism. J Biol Chem. 1963;238:2875–2881. [PubMed] [Google Scholar]
- 2.Barnes LD, McGuire JJ, Atkinson DE. Yeast diphosphopyridine nucleotide specific isocitrate dehydrogenase. Regulation of activity and unidirectional catalysis. Biochemistry. 1972;11:4322–4329. doi: 10.1021/bi00773a019. [DOI] [PubMed] [Google Scholar]
- 3.Chen RF, Plaut GWE. Activation and inhibition of DPN-linked isocitrate dehydrogenase of heart by certain nucleotides. Biochemistry. 1963;2:1023–1032. doi: 10.1021/bi00905a020. [DOI] [PubMed] [Google Scholar]
- 4.Gabriel JL, Zervos PR, Plaut GW. Activity of purified NAD-specific isocitrate dehydrogenase at modulator and substrate concentrations approximating conditions in mitochondria. Metabolism. 1986;35:661–667. doi: 10.1016/0026-0495(86)90175-7. [DOI] [PubMed] [Google Scholar]
- 5.Keys DA, McAlister-Henn L. Subunit structure, expression, and function of NAD(H)-specific isocitrate dehydrogenase in Saccharomyces cerevisiae. J Bacteriol. 1990;172:4280–4287. doi: 10.1128/jb.172.8.4280-4287.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cupp JR, McAlister-Henn L. NAD+-dependent isocitrate dehydrogenase. Cloning, nucleotide sequence, and disruption of the IDH2 gene from Saccharomyces cerevisiae. J Biol Chem. 1991;266:22199–22205. [PubMed] [Google Scholar]
- 7.Cupp JR, McAlister-Henn L. Cloning and characterization of the gene encoding the IDH1 subunit of NAD+-dependent isocitrate dehydrogenase from Saccharomyces cerevisiae. J Biol Chem. 1992;267:16417–16423. [PubMed] [Google Scholar]
- 8.Cupp JR, McAlister-Henn L. Kinetic analysis of NAD+-isocitrate dehydrogenase with altered isocitrate binding sites: contribution of IDH1 and IDH2 subunits to regulation and catalysis. Biochemistry. 1993;32:9323–9328. doi: 10.1021/bi00087a010. [DOI] [PubMed] [Google Scholar]
- 9.Zhao WN, McAlister-Henn L. Affinity purification and kinetic analysis of mutant forms of yeast NAD+-specific isocitrate dehydrogenase. J Biol Chem. 1997;272:21811–21817. doi: 10.1074/jbc.272.35.21811. [DOI] [PubMed] [Google Scholar]
- 10.Lin AP, McCammon MT, McAlister-Henn L. Kinetic and physiological effects of alterations in homologous isocitrate-binding sites of yeast NAD+-specific isocitrate dehydrogenase. Biochemistry. 2001;40:14291–14301. doi: 10.1021/bi0111707. [DOI] [PubMed] [Google Scholar]
- 11.Lin AP, McAlister-Henn L. Isocitrate binding at two functionally distinct sites in yeast NAD+-specific isocitrate dehydrogenase. J Biol Chem. 2002;277:22475–22483. doi: 10.1074/jbc.M202534200. [DOI] [PubMed] [Google Scholar]
- 12.Lin AP, McAlister-Henn L. Homologous binding sites in yeast isocitrate dehydrogenase for cofactor (NAD+) and allosteric activator (AMP) J Biol Chem. 2003;278:12864–12872. doi: 10.1074/jbc.M300154200. [DOI] [PubMed] [Google Scholar]
- 13.Panisko EA, McAlister-Henn L. Subunit interactions of yeast NAD+-specific isocitrate dehydrogenase. J Biol Chem. 2001;276:1204–1210. doi: 10.1074/jbc.M005056200. [DOI] [PubMed] [Google Scholar]
- 14.Taylor AB, Hu G, Hart PJ, McAlister-Henn L. Allosteric motions in structures of yeast NAD+-specific isocitrate dehydrogenase. J Biol Chem. 2008;283:10872–10880. doi: 10.1074/jbc.M708719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hurley JH, Dean AM, Sohl JL, Koshland DE, Jr, Stroud RM. Regulation of an enzyme by phosphorylation at the active site. Science. 1990;249:1012–1016. doi: 10.1126/science.2204109. [DOI] [PubMed] [Google Scholar]
- 16.Hurley JH, Dean AM, Koshland DE, Jr, Stroud RM. Catalytic mechanism of NADP+-dependent isocitrate dehydrogenase: implications from the structures of magnesium-isocitrate and NADP+ complexes. Biochemistry. 1991;30:8671–8678. doi: 10.1021/bi00099a026. [DOI] [PubMed] [Google Scholar]
- 17.Imada K, Sato M, Tanaka N, Katsube Y, Matsuura Y, Oshima T. Three-dimensional structure of a highly thermostable enzyme, 3-isopropylmalate dehydrogenase of Thermus thermophilus at 2.2 A resolution. J Mol Biol. 1991;222:725–738. doi: 10.1016/0022-2836(91)90508-4. [DOI] [PubMed] [Google Scholar]
- 18.Garcia JA, Minard KI, Lin AP, McAlister-Henn L. Disulfide bond formation in yeast NAD+-specific isocitrate dehydrogenase. Biochemistry. 2009;48:8869–8878. doi: 10.1021/bi900968a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demeler B. UltraScan: A Comprehensive Data Analysis Software Package for Analytical Ultracentrifugation Experiments. In: Scott D, Harding S, Rowe A, editors. Modern Analytical Ultracentrifugation: Techniques and Methods. Royal Society of Chemistry; Cambridge, U.K: 2005. pp. 210–229. [Google Scholar]
- 21.Demeler B. UltraScan: A comprehensive data analysis software package for analytical ultracentrifugation experiments. 2009 http://www.utrascan.uthscsa.edu/
- 22.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, editors. Analytical Ultracentrifugation in Biochemistry and Polymer Science. Royal Society of Chemistry; Cambridge, U.K: 1992. pp. 90–125. [Google Scholar]
- 23.Brookes E, Cao W, Demeler B. A two-dimensional spectrum analysis for sedimentation velocity experiments of mixtures with heterogeneity in molecular weight and shape. Eur Biophys J. 2009;39:405–414. doi: 10.1007/s00249-009-0413-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brookes E, Demeler B. Parsimonius Regularization using Genetic Algorithms Applied to the Analysis of Analytical Ultracentrifugation Experiments. GECCO Proceedings ACM 978-1-59593-697-4/07/0007.2007. [Google Scholar]
- 25.Demeler B, Brookes E. Monte Carlo analysis of sedimentation experiments. Colloid Polym Sci. 2008;286:129–137. [Google Scholar]
- 26.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brookes E, Demeler B. Parallel computational techniques for the analysis of sedimentation velocity experiments in UltraScan. Colloid Polym Sci. 2008;286:138–148. [Google Scholar]
- 28.Demeler B, van Holde KE. Sedimentation velocity analysis of highly heterogeneous systems. Anal Biochem. 2004;335:279–288. doi: 10.1016/j.ab.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 29.Demeler B, Saber H, Hansen JC. Identification and interpretation of complexity in sedimentation velocity boundaries. Biophysical Journal. 1997;72:397–407. doi: 10.1016/S0006-3495(97)78680-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 32.Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002;30:e23. doi: 10.1093/nar/30.6.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gietz D, St Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demeler B. Methods for the design and analysis of sedimentation velocity and sedimentation equilibrium experiments with proteins. Curr Protoc Protein Sci. 2010;Chapter 7(Unit 7.13) doi: 10.1002/0471140864.ps0713s60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCammon MT. Mutants of Saccharomyces cerevisiae with defects in acetate metabolism: isolation and characterization of Acn− mutants. Genetics. 1996;144:57–69. doi: 10.1093/genetics/144.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramachandran N, Colman RF. Chemical characterization of distinct subunits of pig heart DPN-specific isocitrate dehydrogenase. J Biol Chem. 1980;255:8859–8864. [PubMed] [Google Scholar]
- 38.Soundar S, Park JH, Huh TL, Colman RF. Evaluation by mutagenesis of the importance of 3 arginines in alpha, beta, and gamma subunits of human NAD-dependent isocitrate dehydrogenase. J Biol Chem. 2003;278:52146–52153. doi: 10.1074/jbc.M306178200. [DOI] [PubMed] [Google Scholar]
- 39.Nichols BJ, Hall L, Perry AC, Denton RM. Molecular cloning and deduced amino acid sequences of the gamma-subunits of rat and monkey NAD+-isocitrate dehydrogenases. Biochem J. 1993;295(Pt 2):347–350. doi: 10.1042/bj2950347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nichols BJ, Perry AC, Hall L, Denton RM. Molecular cloning and deduced amino acid sequences of the alpha- and beta- subunits of mammalian NAD+-isocitrate dehydrogenase. Biochem J. 1995;310(Pt 3):917–922. doi: 10.1042/bj3100917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu G, Lin AP, McAlister-Henn L. Physiological consequences of loss of allosteric activation of yeast NAD+-specific isocitrate dehydrogenase. J Biol Chem. 2006;281:16935–16942. doi: 10.1074/jbc.M512281200. [DOI] [PubMed] [Google Scholar]
- 42.Sherman F. Getting started with yeast. Meth Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- 43.Thorsness PE, Koshland DE., Jr Inactivation of isocitrate dehydrogenase by phosphorylation is mediated by the negative charge of the phosphate. J Biol Chem. 1987;262:10422–10425. [PubMed] [Google Scholar]
- 44.Dean AM, Lee MH, Koshland DE., Jr Phosphorylation inactivates Escherichia coli isocitrate dehydrogenase by preventing isocitrate binding. J Biol Chem. 1989;264:20482–20486. [PubMed] [Google Scholar]
- 45.LaPorte DC, Walsh K, Koshland DE., Jr The branch point effect. Ultrasensitivity and subsensitivity to metabolic control. J Biol Chem. 1984;259:14068–14075. [PubMed] [Google Scholar]
- 46.Walsh K, Koshland DE., Jr Determination of flux through the branch point of two metabolic cycles. The tricarboxylic acid cycle and the glyoxylate shunt. J Biol Chem. 1984;259:9646–9654. [PubMed] [Google Scholar]
- 47.Walsh K, Koshland DE., Jr Branch point control by the phosphorylation state of isocitrate dehydrogenase. A quantitative examination of fluxes during a regulatory transition. J Biol Chem. 1985;260:8430–8437. [PubMed] [Google Scholar]
- 48.LaPorte DC, Chung T. A single gene codes for the kinase and phosphatase which regulate isocitrate dehydrogenase. J Biol Chem. 1985;260:15291–15297. [PubMed] [Google Scholar]
- 49.Anderson SL, Schirf V, McAlister-Henn L. Effect of AMP on mRNA binding by yeast NAD+-specific isocitrate dehydrogenase. Biochemistry. 2002;41:7065–7073. doi: 10.1021/bi0200662. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.