

Abstract
The p38 MAP kinases are stress-activated MAP kinases whose induction is often associated with the onset of heart failure. This study investigated the role of p38 MAP kinase isoforms in the regulation of myocardial contractility and ischemia/reperfusion injury using mice with cardiac-specific expression of kinase dead (dominant negative) mutants of p38α (p38αdn) or p38β (p38βdn). Hearts were subjected to 20 min ischemia and 40 min reperfusion. Immunofluorescence staining for p38αdn and p38βdn protein was performed on neonatal cardiomyocytes infected with adenovirus expressing flag-tagged p38αdn and p38βdn protein. Basal contractile function was increased in both p38αdn and p38βdn hearts compared to WT. Ischemic injury was increased in p38βdn vs. WT hearts, as indicated by lower post-ischemic recoveries of contractile function and ATP. However, despite a similar increase in contractility, ischemic injury was not increased in p38αdn vs. WT hearts. Immunohistological analysis of cardiomyocytes with comparable levels of protein overexpression show that p38αdn and p38βdn proteins were co-localized with sarcomeric α-actinin, however, p38αdn was detected in the nucleus while p38βdn was exclusively detected in the cytosol. In summary, attenuated p38 activity led to increased myocardial contractility; specific isoforms of p38 and their sub-cellular localization may have different roles in modulating ischemic injury.
Keywords: p38 MAP kinase, contractile function, ischemia, NMR spectroscopy, energetics
The p38 mitogen-activated protein (MAP) kinases are stress-activated kinases involved in inflammation, cell growth, cell injury and apoptosis (10, 21, 22). There are four isoforms of p38 MAP kinase: α, β, γ and δ. p38α and p38β MAP kinases predominate in heart (27).
In previous studies, p38 MAP kinases are reported to be activated in heart failure patients (6) as well as other hypertrophy and heart failure models (11, 15, 28). Activation of p38 in myocytes has been related to hypertrophy and apoptosis in cultured myocytes, and development of restrictive cardiomyopathy in vivo. Our recent study also demonstrated that inhibiting p38 activity in isolated adult myocytes augmented contractility without affecting membrane calcium currents or intracellular calcium transients (16). Yet, the effect of inhibiting specific p38 isoforms on whole heart contractility has not been tested.
Since hypertrophy and heart failure often arise from initial ischemic events, the activation of p38 MAP kinases during ischemia and reperfusion and the role of p38 MAP kinases in preconditioning has been studied intensely in recent years (1, 2, 5, 9, 17, 18, 25). However, the results of these studies are contradictory and the role of p38 MAP kinases in ischemia remains highly controversial. A number of studies have demonstrated that the pharmacological agents SB 202190 or SB 203580, which inhibit both p38α and p38β MAP kinases, protect against ischemic injury, implying that p38 MAP kinase activity is detrimental (2, 17, 18, 25). However, although conflicting data exist (4), some studies have shown that p38 MAP kinases are activated during the cardioprotective phenomenon of preconditioning (1, 9, 20, 29) and that the cardioprotective effects of preconditioning can be blocked by SB 202190 or SB 203580 (1, 20), implying that p38 MAP kinase activity is protective. Others have found that preconditioning causes an initial activation of p38 but with repetitive cycles of ischemic preconditioning, p38 activity is reduced (23) and remains low during the subsequent sustained period of ischemia (25, 26), suggesting that reducing p38 activity is protective. Some of the controversy may be due to non-specific effects of SB 202190 or SB 203580 (13). Alternatively, the controversy may be due to differential effects of the two cardiac isoforms, p38α and p38β, in different species and models, given that both the pharmacological inhibitors and the common techniques of p38 activity measurement are not isoform-specific.
To determine the effect of specific p38 isoform inhibition on cardiac contractility and the specific role of p38α vs. p38β in the ischemic heart, two transgenic mouse models were created. One model, p38αdn, was established by cardiac-specific expression of a kinase dead mutant (dominant negative) of p38α while the other model, p38βdn, was generated in the same manner using a kinase dead mutant (dominant negative) of p38β. In previous studies, it has been shown that over-expression of these mutant proteins blocked pressure-overload induced p38 activation (30, 31). While these studies supported a role for p38 isoforms in different aspects of cardiac hypertrophy, gene expression, and apoptotic cell death, the question remains unanswered about their ability to regulate cardiac contractility in the intact heart. By monitoring basal contractile function, we aimed to determine whether attenuation of p38 activity can modulate cardiac contractility in the whole heart under physiological perfusion conditions. By subjecting hearts from these transgenic mice to ischemia and reperfusion while monitoring contractile function and energetics, we aimed to determine the roles of p38α vs. p38β in contractile dysfunction following ischemic injury.
Materials and Methods
Animals
Transgenic mice were created by microinjection of a transgene construct, containing mutant cDNA under the control of the α-myosin heavy chain promoter, into the pronuclei of fertilized mouse oocytes. The mutant cDNA was created by substitution of the amino acids AGA for TGY in the catalytic domain of either p38α or p38β MAP kinase of human origin. This mutation destroys the catalytic activity of the kinase. Expression of these mutant proteins reduces endogenous p38α and p38β kinase activities by competing for kinase substrates under upstream stimulation (30, 31). After embryo implantation, the presence of the transgene in the resultant founder mice was confirmed by Southern blot analysis while expression of mutant mRNA was determined by Northern analysis and protein expression was determined by Western blotting (30, 31).
The present study used 6 transgenic p38α dominant negative mice (p38α(-) Tr) and 5 of their wild-type littermates (p38α(-) WT). In addition, 6 transgenic p38β dominant negative mice (p38β(-) Tr) and 5 of their wild-type littermates (p38β(-) WT) were studied. All animals were treated in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
RNA and protein analysis
RNA was prepared from snap-frozen mouse ventricles using the Trizol method as described (15). Ten μg of total RNA from each sample was analyzed by Northern blot using 32P-labeled human p38α and p38β cDNA probes, respectively. Immunoblots were performed with protein samples extracted from snap-frozen ventricles as described (15). Fifty μg of protein from each sample was separated on 4–12% NuPAGE Bis-Tris gel by electrophoresis using MOPS buffer (Invitrogen). p38 isoforms were detected by polyclonal anti-p38α (C-20) and anti-p38β (C-16) antibodies, respectively (Santa Cruz Biotechnology, CA).
Ischemia/reperfusion protocol
Hearts were isolated and perfused in the Langendorff mode, at constant perfusion pressure, as described previously (7). Hearts were then subjected to 20 min of global, no-flow ischemia followed by 40 min reperfusion. Left-ventricular developed pressure (LVDP), ±dP/dt and heart rate were monitored via a water-filled latex balloon in the left ventricle. Rate-pressure product (RPP: LVDP·heart rate) was then calculated. Recovery of contractile function was assessed by measurement of LVDP at the end of reperfusion and expressed as a percentage of pre-ischemic LVDP. LVEDP was set at 20 cm of water at the start of the perfusion, and there were no significant differences in LVEDP among the groups during the pre-ischemic perfusion period, immediately prior to the start of ischemia.
NMR spectroscopy
Relative changes in concentrations of phosphorus metabolites were observed during the ischemia/reperfusion protocols by acquiring consecutive 31P-NMR spectra as described previously (8). The areas of the spectral peaks were expressed as a percentage of the peak areas of an initial, pre-ischemic control spectrum from each heart. Intracellular pH was estimated from the chemical shift of the Pi peak relative to PCr using previously obtained titration curves.
Neonatal cardiomyocyte culture and adenoviral infection
Neonatal cardiomyocytes were prepared using an established method as described (28). Myocytes from 1–2-day-old Sprague-Dawley rats were plated on laminin coated chamber slides in serum containing media (4:1 Dulbecco's modified Eagle's medium: medium 199, 10% horse serum, 5% fetal bovine serum, 100 units/ml penicillin, 100 ∝g/ml streptomycin and 10 ∝M glutamine) overnight. Subsequently, the cells were changed into low serum media containing 1% horse serum and 0.5% fetal bovine serum, and infected with Adv-p38αdn or Adv-p38βdn at a multiplicity of infection (MOI) of 100 particles/cell for 12 hours. The cells were then cultured in serum free media for an additional 36 to 70 hours before analysis.
Immunofluorescent imaging
Fresh heart tissue was frozen in a slush of liquid N2, mounted on a chuck and sectioned at 10 ∝m thickness on a Riechert-Jung cryostat and placed on glass slides. Additionally, cultured myocytes on chamber slides were fixed briefly in ethanol at −20 °C followed by 2% paraformaldehyde at 4 °C. Sections or cells were pre-treated with PBS containing 5% normal goat serum and 3% BSA. After overnight incubation with primary antibodies at 4 °C and subsequent washes, fluorescent dye conjugated secondary antibody was added and further incubated for 2 hours at room temperature, followed by final wash steps to remove unbound antibodies. Images from tissue slides were recorded on a Zeiss 410 confocal laser scanning microscope and images from cultured cells were recorded from a Zeiss inverted fluorescent microscope.
Statistics
Results are expressed as means ± SEM. Significance (p≤0.05) was determined by analysis of variance (ANOVA) followed by a Fisher's post-hoc test.
Results
Characterization of p38α and p38β dominant negative transgenic mice
Out of four independent transgenic founder lines established for each construct, significant expression of the transgene mRNA was detected in the ventricular tissue from two p38αdn lines and one p38βdn line based on Northern blot analysis (Fig. 1A). Overexpression of the dominant negative mutant p38α and β protein was detected in mouse hearts from these lines based on Western blot analysis. Endogenous expression of p38α was detected in these hearts (Fig. 1B), however, endogenous expression of p38β was below the limit of detection by the isoform-specific anti-p38β MAPK antibody, as reported earlier (15). No significant changes in heart weight or gross morphology were observed in the transgenic animals compared to their non-transgenic littermates at 10 weeks of age (data not shown). The founder lines (the line 3 for p38αdn and the line 4 for p38βdn) with the highest level of transgene expression were used for further study; the p38αdn and p38βdn lines had comparable levels of transgene overexpression based on Northern blot analysis (Fig. 1A). Each of the two transgenic lines was further bred into Blackswiss background. As demonstrated in previous studies, both p38α and p38β activity was increased by pressure-overload in wild-type mouse hearts, and the induction of p38 activity was significantly blunted in respective transgenic animals, suggesting an effective inhibition of each p38 isoform by its respective mutant protein in the intact heart (30, 31).
Fig. 1.
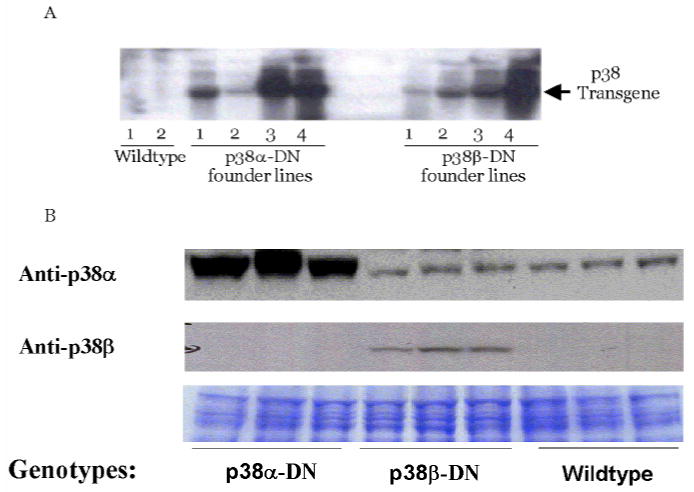
Expression of p38α-DN and p38β-DN mutants in transgenic hearts. A. Northern blot showing human p38α-DN and p38β-DN mRNA levels in hearts from four independent founder lines for each transgene and wildtype control as indicated. Ten ∝g of total RNA was loaded for each sample, equal loading and RNA integrity was confirmed by 28/18 rRNA bands stained by ethidium bromide dye B. Immunoblot showing expression of the p38α-DN and p38β-DN proteins in the mouse ventricles of the high expressor line for each transgene. Fifty ∝g of protein from each sample, including transgenic mice and their littermate wildtype controls was used and probed with anti-p38α (C-20) and anti-p38β (C-16) antibodies (Santa Cruz, CA) as indicated
Inhibition of p38 MAP kinase activity enhances basal contractility in intact heart
During the pre-ischemic period, LVDP was higher in transgenic p38α(-) hearts, at 168 cm H2O, than in wild-type littermate hearts, at 123 cm H2O (p<0.001; Table I). The rate of contraction (+dP/dt), at 4.9 cm H2O/ms in transgenic vs. 3.6 cm H2O/ms in wild-type, and the rate of relaxation (-dP/dt), at −5.0 cm H2O/ms in transgenic vs. −3.2 cm H2O/ms in wild-type, were also higher in transgenic p38α(-) hearts than in wild-type hearts (p<0.05; Table I). Heart rate did not differ between transgenic p38α(-) hearts and wild-type hearts, both at ∼400 bpm. However, RPP was higher in transgenic p38α(-) hearts, at 71·103 cm H2O·bpm, than in wild-type hearts, at 48·103 cm H2O·bpm (p<0.01). Thus basal myocardial contractility was increased by expression of the dominant negative mutant p38α, implying that p38α attenuates contractility in the intact heart.
Table I. Myocardial contractile function during normoxic perfusion.
| Group | Heart rate (bpm) | Left-ventricular developed pressure (cm H2O) | Rate-pressure product (cm H2O·bpm·10−3) | +dP/dt (cm H2O·ms−1) | -dP/dt (cm H2O·ms−1) | n |
|---|---|---|---|---|---|---|
| p38α(-) | ||||||
| Wild-type | 386±43 | 123±6 | 48±6 | 3.6±0.3 | −3.2±0.2 | 5 |
| transgenic | 427±32 | 168±4* | 71±5* | 4.9±0.4* | −5.0±0.3* | 6 |
| P38β(-) | ||||||
| Wild-type | 407±18 | 113±4 | 46±3 | 3.9±0.2 | −3.3±0.3 | 5 |
| transgenic | 386±10 | 160±13‡ | 62±7‡ | 4.7±0.6 | −4.5±0.6‡ | 6 |
Data are means ± SEM. p38α(-) transgenic: dominant negative (loss of function) mutant for the α isoform of p38 MAP kinase. p38α(-) wild-type: wild-type littermates of the p38α(-) transgenic mice. p38β(-) transgenic: dominant negative (loss of function) mutant for the β isoform of p38 MAP kinase. p38β(-) wild-type: wild-type littermates of the p38β(-) transgenic mice. For p38α(-) transgenic,
significantly different from p38α(-) wild-type (p<0.05). For p38β(-) transgenic,
significantly different from p38β wild-type (p<0.05)
Similarly, LVDP was higher in transgenic p38β(-) hearts, at 160 cm H2O, than in wild-type hearts, at 113 cm H2O, during the pre-ischemic period (p<0.001; Table I). The rate of relaxation (-dP/dt), at −4.5 cm H2O/ms in transgenic vs. −3.3 cm H2O/ms in wild-type, was also significantly higher in transgenic p38β(-) hearts than in wild-type hearts (p<0.05; Table I). Heart rate did not differ between transgenic p38β(-) hearts and wild-type hearts, both at ∼400 bpm. However, RPP was higher in transgenic p38β(-) hearts, at 62·103 cm H2O·bpm, than in wild-type hearts, at 46·103 cm H2O·bpm (p<0.05). Thus basal myocardial contractility was increased by expression of the dominant negative mutant p38β, implying that p38β, as well as p38α, attenuates contractility in the intact heart.
Post-ischemic functional recovery
Heart rate was not different among the groups during reperfusion. End-diastolic pressure was similar in the groups during reperfusion, ranging from 22±3 to 27±3 cm of water after 20 min ischemia and 40 min reperfusion.
Recovery of contractile function after 20 min ischemia and 40 min reperfusion was the same in transgenic p38α(-) hearts as in wild-type hearts, both at ∼35% initial LVDP (Fig. 2).
Fig. 2.
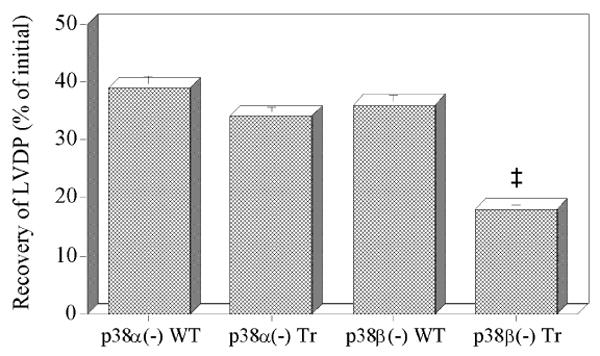
Recovery of left ventricular developed pressure (LVDP) as a percentage of initial LVDP after 20 min ischemia and 40 min reperfusion in hearts from p38α and p38β MAP kinase dominant negative mice (p38α(-) Tr; p38β(-) Tr) and their respective wild-type littermates (p38α(-) WT; p38β(-) WT). Data are means ± SEM. For p38β(-) Tr, ‡: significantly different from p38β(-) WT (p<0.01)
However, recovery of contractile function after 20 min ischemia and 40 min reperfusion was lower in transgenic p38β(-) hearts, at 18% initial LVDP, than in wild-type hearts, at 36% initial LVDP (p<0.01; Fig. 2). These findings imply that p38α and p38β MAP kinases have different roles in ischemia/reperfusion injury.
Phosphate metabolites and intracellular pH
Phosphate metabolites and intracellular pH were measured in the transgenic and wild-type hearts to determine whether expression of the dominant negative mutant p38α and p38β had different effects on myocardial energetics and pH regulation. Baseline PCr/ATP ratios, prior to ischemia, were not significantly different among the groups, at ∼1.7, indicating that myocardial energetics under aerobic conditions were not affected by the mutant proteins. The PCr/ATP ratio is the same in the transgenic and wild-type hearts, indicating there must be a matched increase in energy production to compensate for the increased high energy phosphate utilization in the transgenic hearts to support the hypercontractile state.
ATP levels
During ischemia, ATP depletion was similar in transgenic p38α(-) and wild-type hearts, reaching ∼20% initial ATP by the end of ischemia in both groups (Fig. 3a). During reperfusion, ATP levels were also similar in transgenic p38α(-) and wild-type hearts, reaching ∼40% initial ATP by the end of reperfusion in both groups (Fig. 3a).
Fig. 3.
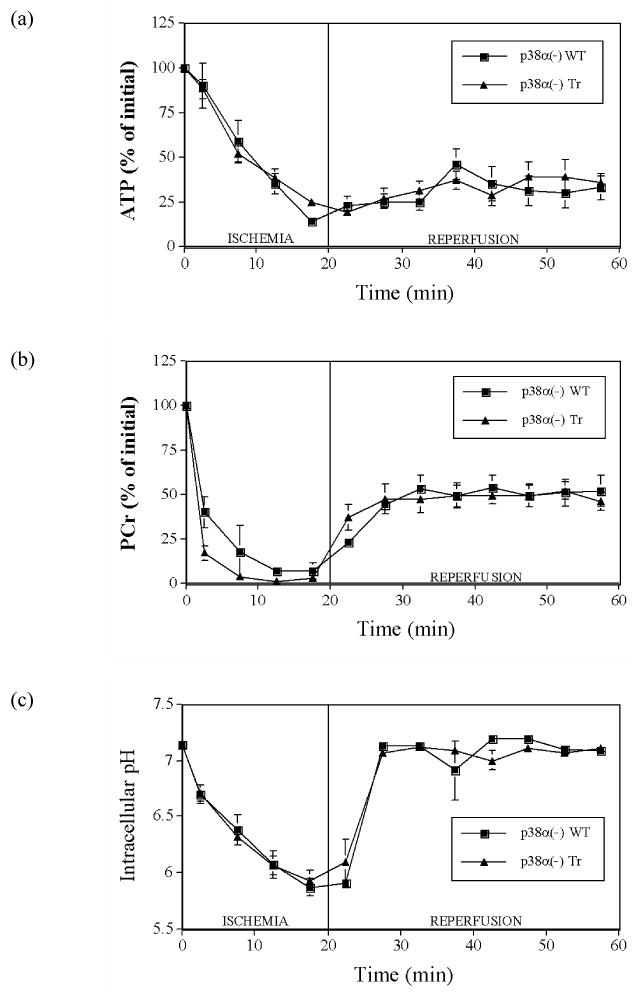
Myocardial intracellular levels of (a) ATP, (b) PCr, and (c) intracellular pH during 20 min ischemia and 40 min reperfusion in hearts from p38α MAP kinase dominant negative mice (p38α(-) Tr) and their wild-type littermates (p38α(-) WT). Points are means ± SEM
In contrast, ATP depletion was greater in transgenic p38β(-) hearts, reaching 15% initial ATP by the end of ischemia, compared to wild-type hearts, which reached 24% by the end of ischemia (p<0.05; Fig. 4a). During reperfusion, ATP recovered less in transgenic p38β(-) hearts, reaching 24% initial ATP by the end of 40 min reperfusion, than in wild-type hearts, which reached ∼40% initial ATP (p<0.05; Fig. 4a).
Fig. 4.
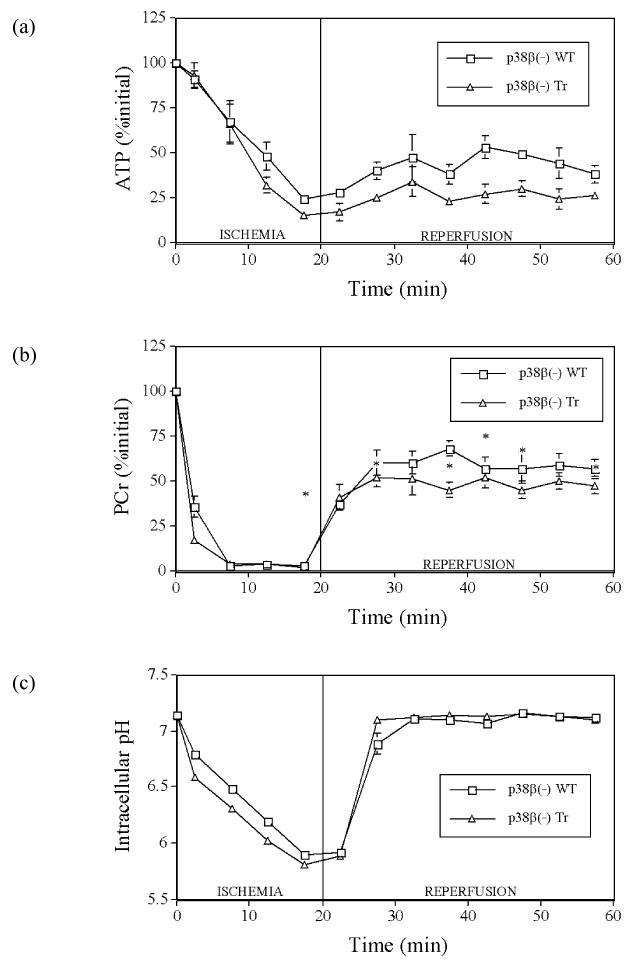
Myocardial intracellular levels of (a) ATP, (b) PCr, and (c) intracellular pH during 20 min ischemia and 40 min reperfusion in hearts from p38β MAP kinase dominant negative mice (p38β(-) Tr) and their wild-type littermates (p38β(-) WT). Points are means ± SEM. *: significantly different from p38β(-) Tr (p<0.05)
PCr and inorganic phosphate levels
During ischemia, PCr levels fell in all hearts. At the end of ischemia, there were no differences in PCr levels between transgenic p38α(-) hearts vs. hearts from their wild-type littermates, both at ∼5% initial PCr (Fig. 3b), nor between transgenic p38β(-) hearts vs. hearts from their respective wild-type littermates, both also at ∼5% of initial PCr (Fig. 4b). During reperfusion, PCr levels increased in all hearts. By the end of reperfusion, there were also no differences in PCr levels between transgenic p38α(-) hearts and hearts from their wild-type littermates, both at ∼50% initial PCr (Fig. 3b), nor between transgenic p38β(-) hearts vs. hearts from their respective wild-type littermates, both also at ∼50% of initial PCr (Fig. 4b).
During ischemia, intracellular inorganic phosphate levels rose in proportion to the decrease in ATP and PCr, and recovered partially during reperfusion, but remained elevated in all groups. No significant differences were observed among the groups. At the end of reperfusion, intracellular inorganic phosphate ranged from 38±11% to 73±15% (no significant differences by ANOVA) of initial ATP, with the transgenic hearts having virtually the same value (68±26% of initial ATP for transgenic p38α(-) hearts versus 73±15% for transgenic p38β(-) hearts) but very different degrees of functional recovery.
Intracellular pH
During ischemia, pH decreased in all hearts. At the end of ischemia, there were no differences in pH levels between transgenic p38α(-) hearts vs. hearts from their wild-type littermates, both at ∼pH 5.90 (Fig. 3c), nor between transgenic p38β(-) hearts vs. hearts from their respective wild-type littermates, both at ∼pH 5.85 (Fig. 4c). During reperfusion, pH increased in all hearts back to pre-ischemic values. At the end of reperfusion, there were also no differences in pH levels, at ∼pH 7.10, between transgenic p38α(-) hearts vs. hearts from their wild-type littermates (Fig. 3c) or between transgenic p38β(-) hearts vs. hearts from their respective wild-type littermates, both also at ∼pH 7.10 (Fig. 4c).
Differential localization of p38α and p38β isoforms in cardiomyocytes
By immunohistochemistry, using isoform specific antibodies, both p38αdn and p38βdn mutant proteins were detected on the Z-lines of the sarcomere in adult myocytes (Fig. 5) co-localizing with α-actinin (data not shown). A similar localization pattern was observed for endogenous p38α protein as demonstrated in wildtype samples. p38βdn mutant protein was also co-localized with endogenous wildtype p38α protein, while the expression of endogenous p38β protein was too low for co-immunostaining analysis (Fig. 5). In the nucleus, however, p38α and β mutant proteins showed significantly different patterns of localization. As best illustrated in neonatal myocytes infected with adenovirus expressing flag-tagged p38αdn and p38βdn, anti-flag antibodies showed clear nuclear localization for p38αdn while p38βdn signal was excluded from the nucleus (Fig. 6). Both vectors were used at the same titer and western blots showed equal levels of overexpression of the target genes (data not shown). Within one experiment, different levels of expression were detected among the infected neonatal cardiomyocytes in the culture, but the p38αdn mutant protein was never observed to be excluded from the nuclei among all of the cardiomyocytes analyzed, whereas the p38βdn mutant protein was absent from the nuclei in all cardiomyocytes examined (Fig. 6).
Fig. 5.
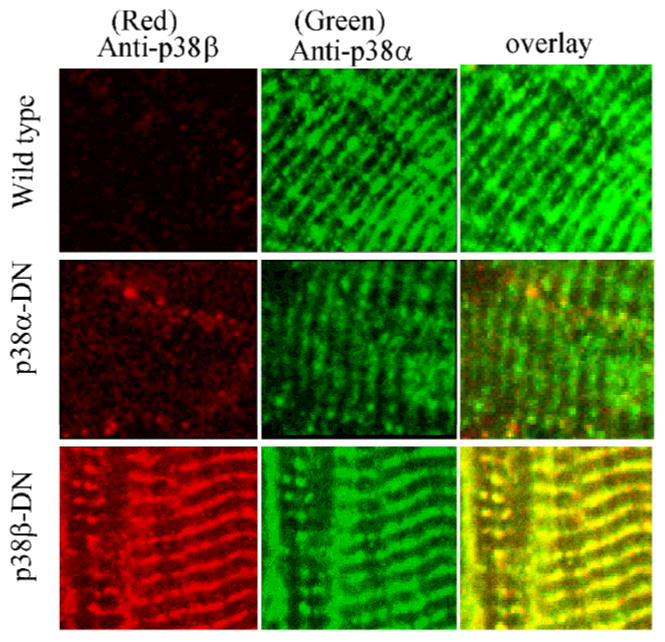
Intracellular localization of p38α-DN and p38β-DN proteins in myocytes. Representative immunofluorescent images from frozen sections of wild-type and transgenic ventricles probed simultaneously with rabbit polyclonal anti-p38α (C-20) and goat polyclonal anti-p38β (C-16) antibodies and detected with Alexa 488 conjugated anti-rabbit and Alexa 568 conjugated anti-goat secondary antibodies (Molecular Probes). Images from 488 and 568 channels were digitally recorded on a confocal laser scanning microscope from the same slides and merged to produce the overlay images
Fig. 6.
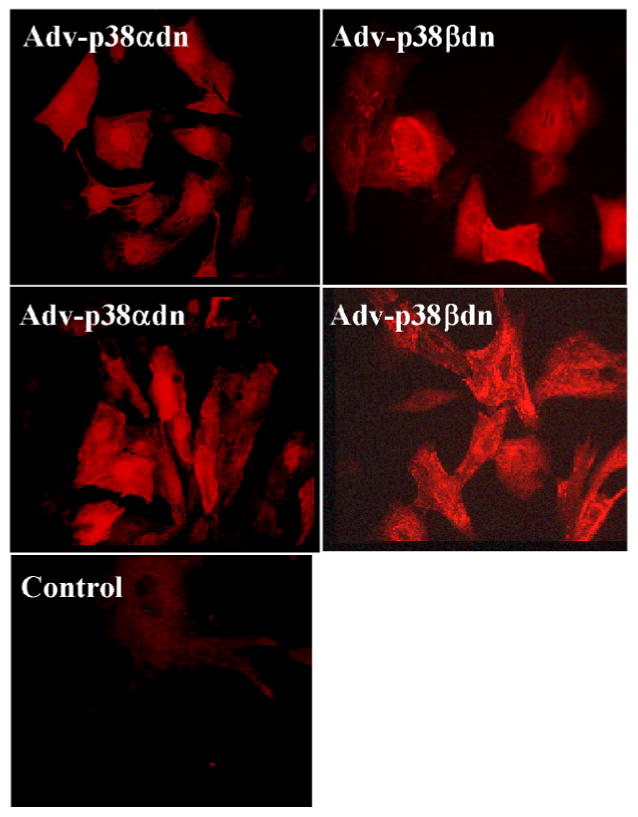
Differential localization of p38αdn and p38βdn mutant proteins in cultured myocytes. Neonatal myocytes were transfected with adenoviruses expressing Flag-tagged p38αdn and p38βdn, respectively and immunostained after 48 hours of incubation using an anti-Flag antibody (M2, Sigma) and rhodamine conjugated secondary antibody. Images were recorded digitally from a fluorescent microscope with identical exposing parameters
The co-localization of the two p38 isoforms on the Z-line of the sarcomere vs. their different nuclear localization pattern in the myocyte suggests potential overlapping function in contractile regulation in the cytosol but a differential function in other stress responses involving gene expression as reported by Zhang et al. (30).
Discussion
Role of p38α vs. p38β in modulation of cardiac contractility
In the present study, peak contraction (LVDP), the rate of contraction (+dP/dt) and the rate of relaxation (−dP/dt) were higher in perfused hearts from transgenic p38α(-) mice than in hearts from their wild-type littermates (Table I). Similarly, LVDP and -dP/dt were also higher in perfused hearts from transgenic p38β(-) mice than in hearts from their respective wild-type littermates (Table I). These findings imply that both p38α and p38β MAP kinases attenuate contractility in the intact heart, and inhibiting either results in increased contractile function.
These results are consistent with previous findings that the p38α/β inhibitor SB 202190 increased contractility in perfused rat hearts (25) and isolated myocytes (16) and that activation of p38 α/β in adult myocytes resulted in decreased myocyte contractility (16). The fact that p38 inhibition enhances myocyte contractility without altering intracellular calcium transients or membrane calcium currents suggests the possibility that p38 MAP kinases play a role in regulating calcium sensitivity of myofilaments in cardiomyocytes. Consistent with these findings, immunohistochemistry from these transgenic hearts identified a previously unrecognized sub-cellular localization of both p38 isoform mutant proteins at the Z-line of the sarcomere. Although this study indicates a common role of p38α and p38β isoforms in regulating myocyte contractility, the major isoform of p38 detected in heart appears to be the α isoform. Our study could not exclude the possibility that the observed effect of p38βdn protein on contractility was due to cross-inhibition of the endogenous p38α protein. Further studies will be needed to measure directly the effect of p38 activity on sarcomere calcium sensitivity and the underlying molecular mechanism.
The p38αdn and p38βdn hearts both have increased contractility but no change in the creatine phosphate to ATP ratio at baseline. This indicates that the increased metabolic demand created by the hypercontractile phenotype must be balanced by an increase in energy production under aerobic conditions, and suggests that p38 manipulation does not impair the response to increased metabolic demand.
Role of p38α vs. p38β in post-ischemic contractile dysfunction and energetics
To determine the roles of p38α vs. p38β in ischemia, the transgenic p38αdn, transgenic p38βdn and their wild-type littermate hearts were subjected to 20 min ischemia and 40 min reperfusion. During ischemia, the decrease in ATP, PCr and intracellular pH was the same in transgenic p38α(-) hearts as in hearts from their wild-type littermates (Fig. 2). During reperfusion, recovery of the energy metabolites, ATP and PCr, was the same in the transgenic p38α-DN hearts as in the wild-type littermate hearts (Fig. 2). In addition, post-ischemic contractile dysfunction was also the same in the transgenic p38αdn hearts as in the wild-type hearts (Fig. 1). It appears, therefore, that p38α MAP kinase activity plays no significant role in ischemic injury or energetics in the present model although it cannot be excluded that the expression level of the dominant negative mutant is not sufficient to attenuate the deleterious effects of the endogenous p38; however, in the contractility measurements, the dominant negative p38α was present in sufficient amount to alter contractile function.
In contrast, ischemic ATP depletion was increased in the transgenic p38β-DN hearts compared to their respective wild-type littermates (Fig. 3). This does not appear to be due to a decrease in glycolytic ATP production since the fall in intracellular pH is the same in the transgenic p38β-DN hearts and the hearts from their wild-type littermates, and lactic acid production is the main determinant of the magnitude of the fall in intracellular pH during total global ischemia. This suggests that p38β signaling plays a role in limiting ATP consumption during ischemia. During reperfusion, recovery of ATP and contractile function was also less in the transgenic p38β-DN hearts compared to the wild-type littermate hearts (Figs 1 and 3). These findings imply that, in vivo, p38β MAP kinase activity may attenuate ischemic ATP depletion and protect the myocardium from post-ischemic contractile and energetic dysfunction. Consistent with our finding in this study that p38β may exert a protective function in heart and modulate myocyte energetics, the p38β isoform has been shown to promote myocyte survival and to antagonize the effects of p38α in neonatal myocytes (28). Similarly, p38 activity has also been shown to influence the control of metabolic pathways (3).
Previous studies using genetic manipulation of specific p38 isoform activity during myocardial ischemia in vivo or simulated ischemia in vitro suggest that inhibition of p38α protects against cell death (12, 24). However, overexpression of MKK6, which should result in joint activation of p38α and p38β, is also protective in an in vivo model of ischemia-reperfusion (19), whereas acute treatment of cardiomyocytes with an activated MKK6 by adenovirus transfection increases cell death (12) and induces inflammatory genes (14). These studies generally support the concept that the dominant p38 isoform in the heart, p38α, is detrimental when activated during ischemia-reperfusion, but that concomitant activation of p38β may mitigate or even reverse this detrimental effect. However, the amount of p38β in the heart is very low, and no statistically significant increase in p38β activity was found in the MKK6 overexpressor mouse hearts (19). The differential effects of the different p38 isoforms may explain some of the conflicts in the literature between studies finding that p38 activation is beneficial versus those that report detrimental effects of p38 activation. Furthermore, acute or continuous effects of activation of different p38 isoforms may have very different effects from transient activation.
Our results differ from, but are not completely inconsistent with, those of Saurin et al. (24). Saurin et al. (24) transfected myocytes with an adenovirus containing the same mutant p38α dominant negative construct used to generate the transgenic mice in our present study. Myocytes were then subjected to simulated ischemia. Transfection with p38α-DN increased cell survival, indicating that p38α activation was detrimental. Comparing our present study to that of Saurin et al., it appears that p38α activation may play a greater role in cell death induced by ischemia than in ischemia-induced contractile dysfunction. Saurin et al. also demonstrated that activation of p38α was increased, while activation of p38β was decreased, during ischemia. Furthermore, Saurin et al. also demonstrated that preconditioning decreased p38α activation and had no effect on p38β activation. The results of Kaiser et al. (12) likewise suggest that inhibition of p38α limits infarct size in hearts subjected to ischemia-reperfusion in vivo. Taken together, our current results and those of Wang et al. (28), Saurin et al. (24), and Kaiser et al. (12) indicate that p38α activation is either detrimental or has no effect, depending upon the ischemic model and the endpoint, while p38β activation is protective. The opposing effects of p38α and p38β and the different contributions of p38α in different models may explain the previous contradictory findings regarding the role of p38 in ischemic injury.
Conclusions
In summary, by measuring cardiac contractility in isolated hearts from p38α MAP kinase and p38β MAP kinase dominant negative transgenic mice, we found evidence that both p38α MAP kinase and p38β MAP kinase can modulate contractility. This is the first study to assess the isoform-specific effects of p38α vs. p38β on cardiac contractility in the intact heart. By subjecting the transgenic hearts to ischemia and reperfusion, we also demonstrated that p38β MAP kinase can modulate ischemic energetics and post-ischemic contractile and energetic dysfunction and may be protective. In contrast, in our perfused heart ischemia model, p38αdn appears to have no effect on ischemia/reperfusion injury. Taken together, these results imply that p38α MAP kinase and p38β MAP kinase have common roles in contractile regulation but different roles in ischemia. The finding that p38α and p38β have significant impact on cardiac contractility and behave differently during ischemia suggest that isoform specific inhibition of p38 may represent a valid strategy for drug intervention to improve contractile function and protect against ischemia/reperfusion injury in heart.
Acknowledgments
The authors thank the National Institutes of Health and American Heart Association for grant support (to CS and YW). The authors also thank Dr. Robert E. London for use of the NMR facilities, and Ms. Haiying Pu for technical assistance.
References
- 1.Armstrong SC, Delacey M, Ganote CE. Phosphorylation state of hsp27 and p38 MAPK during preconditioning and protein phosphatase inhibitor protection of rabbit cardiomyocytes. J Mol Cell Cardiol. 1999;31:555–567. doi: 10.1006/jmcc.1998.0891. [DOI] [PubMed] [Google Scholar]
- 2.Barancik M, Htun P, Strohm C, Kilian S, Schaper W. Inhibition of the cardiac p38-MAPK pathway by SB203580 delays ischemic cell death. J Cardiovasc Pharmacol. 2000;35:474–483. doi: 10.1097/00005344-200003000-00019. [DOI] [PubMed] [Google Scholar]
- 3.Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem. 2001;276:44495–44501. doi: 10.1074/jbc.M105945200. [DOI] [PubMed] [Google Scholar]
- 4.Behrends M, Schulz R, Post H, Alexandrov A, Belosjorow S, Michel MC, Heusch G. Inconsistent relation of MAPK activation to infarct size reduction by ischemic preconditioning in pigs. Am J Physiol Heart Circ Physiol. 2000;279:H1111–H1119. doi: 10.1152/ajpheart.2000.279.3.H1111. [DOI] [PubMed] [Google Scholar]
- 5.Bogoyevitch MA, Gillespie BJ, Ketterman AJ, Fuller SJ, Ben LR, Ashworth A, Marshall CJ, Sugden PH. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart. p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circ Res. 1996;79:162–173. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- 6.Cook SA, Sugden PH, Clerk A. Activation of c-Jun N-terminal kinases and p38-mitogen-activated protein kinases in human heart failure secondary to ischaemic heart disease. J Mol Cell Cardiol. 1999;31:1429–1434. doi: 10.1006/jmcc.1999.0979. [DOI] [PubMed] [Google Scholar]
- 7.Cross HR, Lu L, Steenbergen C, Philipson KD, Murphy E. Overexpression of the cardiac Na+/Ca2+ exchanger increases susceptibility to ischemia/reperfusion injury in male, but not female, transgenic mice. Circ Res. 1998;83:1215–1223. doi: 10.1161/01.res.83.12.1215. [DOI] [PubMed] [Google Scholar]
- 8.Cross HR, Steenbergen C, Lefkowitz RJ, Koch WJ, Murphy E. Overexpression of the β2-adrenergic receptor and a βARK1 inhibitor both increase contractility but have differential effects on susceptibility to ischemic injury. Circ Res. 1999;85:1077–1084. doi: 10.1161/01.res.85.11.1077. [DOI] [PubMed] [Google Scholar]
- 9.Dana A, Skarli M, Papakrivopoulou J, Yellon DM. Adenosine A(1) receptor induced delayed preconditioning in rabbits: induction of p38 mitogen-activated protein kinase activation and Hsp27 phosphorylation via a tyrosine kinase- and protein kinase C-dependent mechanism [see comments] Circ Res. 2000;86:989–997. doi: 10.1161/01.res.86.9.989. [DOI] [PubMed] [Google Scholar]
- 10.English J, Pearson G, Wilsbacher J, Swantek J, Karandikar M, Xu S, Cobb MH. New insights into the control of MAP kinase pathways. Exp Cell Res. 1999;253:255–270. doi: 10.1006/excr.1999.4687. [DOI] [PubMed] [Google Scholar]
- 11.Izumi Y, Kim S, Murakami T, Yamanaka S, Iwao H. Cardiac mitogen-activated protein kinase activities are chronically increased in stroke-prone hypertensive rats. Hypertension. 1998;31:50–56. doi: 10.1161/01.hyp.31.1.50. [DOI] [PubMed] [Google Scholar]
- 12.Kaiser RA, Bueno OF, Lips DJ, Doevendans PA, Jones F, Kimball TF, Molkentin JD. Targeted inhibition of p38 mitogen-activated protein kinase antagonizes cardiac injury and cell death following ischemia-reperfusion in vivo. J Biol Chem. 2004;279:15524–15530. doi: 10.1074/jbc.M313717200. [DOI] [PubMed] [Google Scholar]
- 13.Lali FV, Hunt AE, Turner SJ, Foxwell BM. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J Biol Chem. 2000;275:7395–7402. doi: 10.1074/jbc.275.10.7395. [DOI] [PubMed] [Google Scholar]
- 14.Li M, Georgakopoulos D, Lu G, Hester L, Kass DA, Hasday J, Wang Y. p38 MAP kinase mediates inflammatory cytokine induction in cardiomyocytes and extracellular matrix remodeling in heart. Circulation. 2005;111:2494–2502. doi: 10.1161/01.CIR.0000165117.71483.0C. [DOI] [PubMed] [Google Scholar]
- 15.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao RP, Kass DA, Wang Y. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci USA. 2001;98:12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao P, Wang SQ, Wang S, Zheng M, Zheng M, Zhang SJ, Cheng H, Wang Y, Xiao RP. p38 Mitogen-activated protein kinase mediates a negative inotropic effect in cardiac myocytes. Circ Res. 2002;90:190–196. doi: 10.1161/hh0202.104220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma XL, Kumar S, Gao F, Louden CS, Lopez BL, Christopher TA, Wang C, Lee JC, Feuerstein GZ, Yue TL. Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation. 1999;99:1685–1691. doi: 10.1161/01.cir.99.13.1685. [DOI] [PubMed] [Google Scholar]
- 18.Mackay K, Mochly-Rosen D. An inhibitor of p38 mitogen-activated protein kinase protects neonatal cardiac myocytes from ischemia. J Biol Chem. 1999;274:6272–6279. doi: 10.1074/jbc.274.10.6272. [DOI] [PubMed] [Google Scholar]
- 19.Martindale JJ, Wall JA, Martinez-Longoria DM, Aryal P, Rockman HA, Guo Y, Bolli R, Glembotski CC. Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J Biol Chem. 2005;280:669–676. doi: 10.1074/jbc.M406690200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakano A, Baines CP, Kim SO, Pelech SL, Downey JM, Cohen MV, Critz SD. Ischemic preconditioning activates MAPKAPK2 in the isolated rabbit heart: evidence for involvement of p38 MAPK. Circ Res. 2000;86:144–151. doi: 10.1161/01.res.86.2.144. [DOI] [PubMed] [Google Scholar]
- 21.Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- 22.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 23.Ping P, Zhang J, Huang S, Cao X, Tang XL, Li RC, Zheng YT, Qiu Y, Clerk A, Sugden P, Han J, Bolli R. PKC-dependent activation of p46/p54 JNKs during ischemic preconditioning in conscious rabbits. Am J Physiol. 1999;277:H1771–H1785. doi: 10.1152/ajpheart.1999.277.5.H1771. [DOI] [PubMed] [Google Scholar]
- 24.Saurin AT, Martin JL, Heads RJ, Foley C, Mockridge JW, Wright MJ, Wang Y, Marber MS. The role of differential activation of p38-mitogen activated protein kinase in preconditioned ventricular myocytes. FASEB J. 2000;14:2237–2246. doi: 10.1096/fj.99-0671com. [DOI] [PubMed] [Google Scholar]
- 25.Schneider S, Chen W, Hou J, Steenbergen C, Murphy E. Inhibition of p38 MAPK alpha/beta reduces ischemic injury and does not block protective effects of preconditioning. Am J Physiol Heart Circ Physiol. 2001;280:H499–H508. doi: 10.1152/ajpheart.2001.280.2.H499. [DOI] [PubMed] [Google Scholar]
- 26.Steenbergen C. The role of p38 mitogen-activated protein kinase in myocardial ischemia/reperfusion injury; relationship to ischemic preconditioning. Basic Res Cardiol. 2002;97:276–285. doi: 10.1007/s00395-002-0364-9. [DOI] [PubMed] [Google Scholar]
- 27.Sugden PH, Clerk A. “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ Res. 1998;83:345–352. doi: 10.1161/01.res.83.4.345. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Huang S, Sah VP, Ross J, Jr, Brown JH, Han J, Chien KR. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem. 1998;273:2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- 29.Weinbrenner C, Liu GS, Cohen MV, Downey JM. Phosphorylation of tyrosin 182 of p38 mitogen-activated protein kinase correlates with the protection of preconditioning in the rabbit heart. J Mol Cell Cardiol. 1997;29:2383–2391. doi: 10.1006/jmcc.1997.0473. [DOI] [PubMed] [Google Scholar]
- 30.Zhang S, Ren J, Zhang CE, Treskov I, Wang Y, Muslin AJ. Role of 14-3-3-mediated p38 mitogen-activated protein kinase inhibition in cardiac myocyte survival. Circ Res. 2003;93:1026–1028. doi: 10.1161/01.RES.0000104084.88317.91. [DOI] [PubMed] [Google Scholar]
- 31.Zhang S, Weinheimer C, Courtois M, Kovacs A, Zhang CE, Cheng AM, Wang Y, Muslin AJ. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J Clin Invest. 2003;111:833–841. doi: 10.1172/JCI16290. [DOI] [PMC free article] [PubMed] [Google Scholar]