

Abstract
β-Site amyloid precursor protein-cleaving enzyme 1 (BACE1)—the neuronal β-secretase responsible for producing β-amyloid (Aβ) peptides—emerged as one of the key therapeutic targets of Alzheimer's disease (AD). Although complete ablation of the BACE1 gene prevents Aβ formation, we reported that BACE1 knock-out mice display severe presynaptic deficits at mossy fiber (MF)-to-CA3 synapses in the hippocampus, a major locus of BACE1 expression. We also found that the deficits are likely due to abnormal presynaptic Ca2+ regulation. Cholinergic system has been implicated in AD, in some cases involving Ca2+-permeable α7-nicotinic acetylcholine receptors (nAChRs). Here we report that brief application of nicotine, via α7-nAChRs, can restore MF long-term potentiation in BACE1 knock-outs. Our data suggest that activating α7-nAChRs can recover the presynaptic deficits in BACE1 knock-outs.
Introduction
Alzheimer's disease (AD) is the most prevalent form of senile dementia with limited treatment options (Vassar et al., 2009). A current hypothesis of AD states that overexpression of amyloid-β (Aβ) peptide initiates a cascade of events leading to its pathology (Walsh and Selkoe, 2007). β-Site amyloid precursor protein-cleaving enzyme 1 (BACE1), the neuronal β-secretase, is the first enzyme involved in the sequential cleavage of amyloid precursor proteins (APPs) to produce Aβ (Vassar et al., 2009). High levels of BACE1 are correlated with an increase in Aβ in sporadic AD (Hébert et al., 2008; O'Connor et al., 2008). Knocking out BACE1 abolishes Aβ peptide production (Cai et al., 2001), prevents amyloid plaque deposition, and rescues memory deficits in APP transgenic lines (Luo et al., 2003; Ohno et al., 2004). These observations encourage the development of BACE1 inhibition strategies for AD treatment. However, studies revealed that BACE1 knock-outs (KOs) display behavior deficits (Harrison et al., 2003; Laird et al., 2005; Savonenko et al., 2008) and specific synaptic dysfunctions in the CA1 of hippocampus (Laird et al., 2005). Moreover, at the MF-to-CA3 synapses, where high levels of BACE1 are expressed (Laird et al., 2005), BACE1 KOs display severe presynaptic dysfunctions (Wang et al., 2008). The deficits include a reduction in presynaptic release and an absence of mossy fiber long-term potentiation (mfLTP), which are due to abnormal presynaptic Ca2+ signaling (Wang et al., 2008). These studies caution against the use of BACE1 inhibitors as a practical treatment for AD.
Cholinergic system modulates neurotransmitter release from glutamatergic and GABAergic terminals via the action of nicotinic acetylcholine receptors (nAChRs) (Gray et al., 1996; Radcliffe et al., 1999; Giocomo and Hasselmo, 2005; Jiang and Role, 2008; Bancila et al., 2009). Among them, α7-nAChR is a Ca2+-permeable homopentameric ion channel highly expressed in the hippocampus and cerebral cortex (Séguéla et al., 1993). Several studies have linked α7-nAChR with neurodegenerative disorders, including AD (Perry et al., 2000). We present data that activating α7-nAChRs, by nicotine (Nic) or a specific agonist, PNU282987, can restore presynaptic function and mfLTP in BACE1 KOs via recruiting calcium-induced calcium release (CICR).
Materials and Methods
Animals.
All mice used (BACE1 +/+ and −/−) were derived from heterozygous breeders (+/−) as described previously (Laird et al., 2005). The Institutional Animal Care and Use Committees of both University of Maryland and Johns Hopkins University approved all procedures involving animals.
Electrophysiological recordings.
Hippocampal slices (400 μm thick) were prepared from adult (3–6 months old) male BACE1 KOs and wild types (WTs) as previously described (Wang et al., 2008). Briefly, hippocampi were sliced in ice-cold dissection buffer (in mm: 212.7 sucrose, 2.6 KCl, 1.23 NaH2PO4, 26 NaHCO3, 10 dextrose, 3 MgCl2, and 1 CaCl2; 5% CO2/95% O2). Recordings were done in a submersion-type chamber perfused with artificial CSF (ACSF, in mm: 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 dextrose, 1.5 MgCl2, and 2.5 CaCl2; 5% CO2/95% O2, 29.5–30.5°C, 2 ml/min). Synaptic responses were evoked through glass bipolar stimulating electrodes placed in the dentate granule cell layer to activate MFs with pulse duration of 0.2 ms (at 0.067 Hz), and recorded extracellularly in the stratum lucidum of CA3. Paired-pulse facilitation (PPF) was measured at 25, 50, 100, 200, 400, 1000, and 2000 ms interstimulus intervals (ISIs). To induce mfLTP, three trains of 100 Hz (1 s) stimuli were given at 20 s intervals. We used α7-nAChR agonists (−)-nicotine (Sigma-Aldrich) and PNU282987 (Tocris Bioscience), and an antagonist α-bungarotoxin (Tocris Bioscience). To block intracellular Ca2+ release, ruthenium red (Tocris Bioscience) or ryanodine (Tocris Bioscience) was applied. All experiments were done in the presence of 100 μm d,l-2-amino-5-phosphonovaleric acid (d,l-APV) (Sigma-Aldrich) to isolate the presynaptic NMDAR-independent mfLTP (Nicoll and Schmitz, 2005). At the end of each experiment, 1 μm (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV) (Tocris Bioscience) was added, and responses blocked by ≥80% were taken to be MF inputs. Field potential slopes were measured, and data are expressed as mean ± SEM.
Results
Nicotine restores presynaptic function at MF synapses in BACE1 KOs
We first examined the effect of nicotine on the presynaptic function of MFs in BACE1 KOs by measuring PPF. The results showed that nicotine decreased PPF ratio in a dose-dependent manner at 25 and 50 ms ISIs in KOs (n = 7 slices/3 mice; ANOVA: p < 0.05) (Fig. 1A), and 10 μm was the lowest concentration that significantly decreased PPF ratio in both genotypes (KO: control = 4.81 ± 0.16, nicotine = 4.05 ± 0.22, n = 15 slices/10 mice, paired t test: p < 0.001; WT: control = 3.77 ± 0.43, nicotine = 3.50 ± 0.40, n = 10 slices/9 mice, paired t test: p < 0.001) (Fig. 1B). We previously showed that BACE1 KOs display a significant increase in PPF ratio at MF synapses indicating a reduction in presynaptic release (Wang et al., 2008). Nicotine at 10 μm concentration decreased the PPF ratio of KOs to a similar level of WTs (t test: p = 0.57) without affecting synaptic transmission in either genotype (KO: 100 ± 1% of baseline at 20 min after nicotine, n = 15 slices/10 mice; paired t test: p = 0.97; WT: 99 ± 1%, n = 10 slices/9 mice; paired t test: p = 0.54) (Fig. 1B). These results suggest that 10 μm nicotine reverses PPF deficits in BACE1 KOs without affecting synaptic strength. Therefore, 10 μm nicotine was used in subsequent experiments.
Figure 1.

Nicotine recovers deficits in PPF at MF synapses in BACE1 KOs. A, Nicotine reduced PPF ratio in a dose-dependent manner, which was significant at 25 and 50 ms ISIs. *ANOVA, p < 0.05; Fisher's PLSD, p < 0.05 between control and 10, 50, 100 μm nicotine groups. B, Nicotine (10 μm) significantly decreased PPF ratio in both genotypes, but did not influence basal synaptic transmission. Top, Representative FP traces of paired-pulse stimulation (50 ms ISI) before (thin traces) and after (thick traces) nicotine. Calibration: KO, 1 mV; WT, 0.5 mV, 10 ms. Bottom left, No change in basal synaptic strength with nicotine (KO, black circles; WT, open squares). Bottom right, Comparison of PPF ratio (50 ms ISI) before (C) and after (N) nicotine application. *Paired t test, p < 0.001.
Nicotine rescues mfLTP in BACE1 KOs without affecting mfLTP in WTs
Consistent with our previous results, KOs lacked mfLTP under control conditions, but 10 μm nicotine applied during the whole duration of the experiment restored mfLTP [control: 95 ± 4% at 1 h after high-frequency stimulation (HFS), n = 6 slices/4 mice; nicotine: 133 ± 7%, n = 8 slices/7 mice; t test: p < 0.001] (Fig. 2A). Nicotine-induced rescue of mfLTP was accompanied by a significant decrease in PPF ratio (50 ms ISI; baseline: 4.36 ± 0.26, 1 h after HFS: 3.01 ± 0.27, paired t test: p < 0.001) (Fig. 2A, inset), suggesting presynaptic expression. Interestingly, 10 μm nicotine did not alter the magnitude of mfLTP in WTs (control: 148 ± 3% at 1 h after HFS, n = 5 slices/3 mice; nicotine: 144 ± 6%, n = 7 slices/6 mice; t test: p = 0.52) (Fig. 2B).
Figure 2.

Nicotine rescues mfLTP in BACE1 KOs without effects in WTs. A, KO slices treated with 10 μm nicotine (black circles) showed significant mfLTP compared to control slices without nicotine (open circles). B, The magnitude of mfLTP in WT slices treated with 10 μm nicotine (black squares) was similar to that of control WT slices (open squares). C, Transient application of nicotine (10 μm, 10 min; gray bar) before and during HFS rescued mfLTP in KOs (black circles). D, The same transient nicotine (10 μm, 10 min; gray bar) application did not influence mfLTP in WT (black squares). Insets: A, B, Changes in PPF ratio with HFS [ΔPPF ratio = (PPF ratio at time b) − (PPF ratio at time a)] for control (Ctl) and Nic; C, D, ΔPPF ratio with nicotine application [(PPF ratio at time b) − (PPF ratio at time a)] and with HFS [(PPF ratio at time c) − (PPF ratio at time a)]. Bars, Average ± SEM. Open circles, Individual data points. *Paired t test, p < 0.001. Arrow, HFS (100 Hz, 1 s × 3). Right panels, Superimposed FP traces taken at times indicated in the left panels. Calibration: 0.5 mV, 5 ms.
To investigate whether nicotine affects the induction mechanisms of mfLTP, we transiently applied nicotine for 10 min before and during the HFS. KOs displayed significant mfLTP, which was similar in magnitude with that evoked in WTs (KO = 147 ± 2% at 1 h after HFS, n = 8 slices/5 mice, paired t test: p < 0.001; WT: 157 ± 8%, n = 8 slices/5 mice, paired t test: p < 0.001) (Fig. 2C,D). Furthermore, mfLTP was accompanied by a significant decrease in PPF ratio (50 ms ISI) in both genotypes (WT: baseline = 3.66 ± 0.16, 1 h after HFS = 2.55 ± 0.21, paired t test: p < 0.001; KO: baseline = 4.59 ± 0.35, 1 h after HFS = 2.95 ± 0.33, paired t test: p < 0.001) (Fig. 2C,D, insets), consistent with an increase in presynaptic release. These results demonstrate that nicotine specifically rescues the induction mechanisms of mfLTP in BACE1 KOs.
Nicotine-induced rescue of mfLTP in BACE1 KOs is mediated by α7-nAChRs
We showed that presynaptic dysfunction of MF synapses in BACE1 KOs is at the level of Ca2+ regulation (Wang et al., 2008). To determine whether nicotine acts via the Ca2+-permeable α7-nAChRs, we used a specific agonist, PNU282987 (Bodnar et al., 2005). A brief application of PNU282987 (500 nm, 10 min) before and during HFS recovered mfLTP in KOs (1 h after HFS: 167 ± 19%, n = 8 slices/5 mice; paired t test: p < 0.05) (Fig. 3A) for up to 2 h (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Furthermore, PPF ratio decreased significantly after PNU282987 application and further by LTP induction (baseline: 6.29 ± 0.77, +PNU282987: 5.81 ± 0.76, 1 h after HFS: 4.80 ± 0.69) (Fig. 3A, inset). PNU282987 alone did not produce changes in synaptic strength (1 h after PNU282987: 105 ± 4%, n = 4 slices/2 mice; paired t test: p = 0.30) (Fig. 3A).
Figure 3.
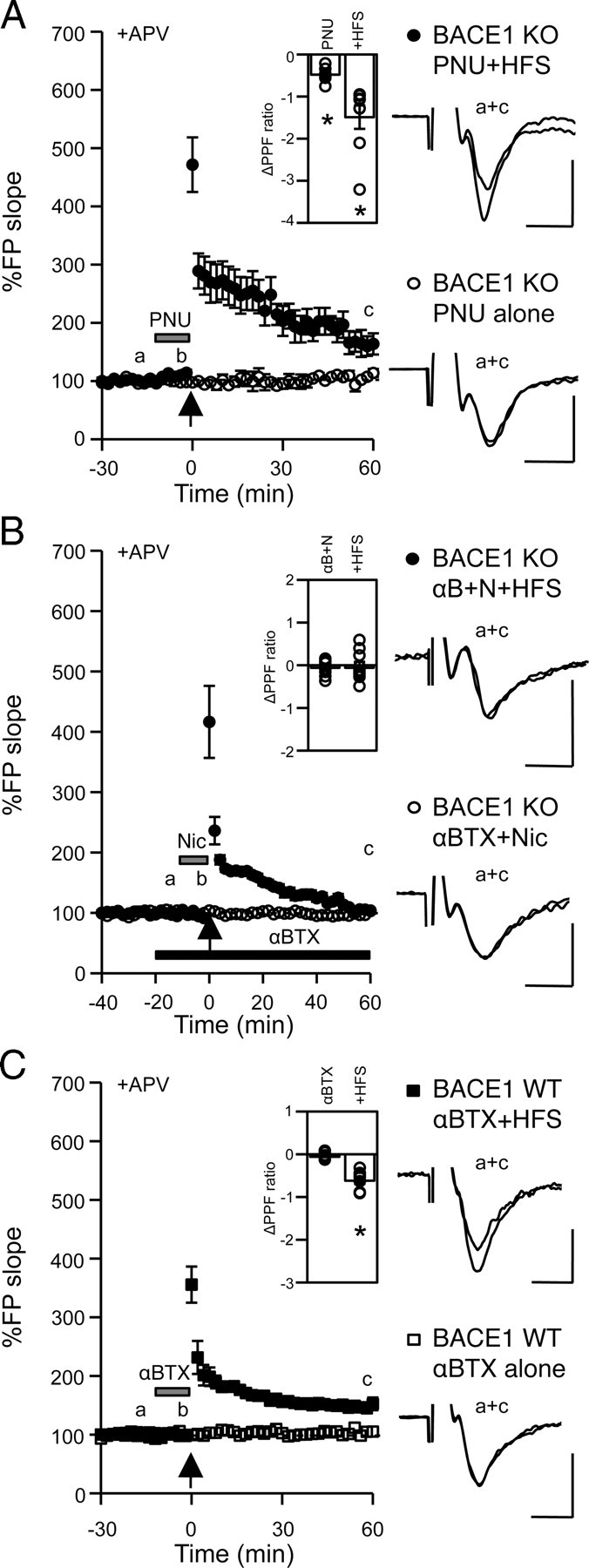
Nicotine-induced rescue of mfLTP in BACE1 KO is mediated by α7-nAChRs. A, Transient bath application of PNU282987 (PNU: 500 nm, 10 min; gray bar) rescued mfLTP in KOs (black circles). PNU282987 alone did not alter synaptic transmission (open circles). Inset, ΔPPF ratio in KO PNU+HFS experiments. ΔPPF ratio with PNU282987 application [(PPF at b) − (PPF at a)]; ΔPPF ratio with HFS [(PPF at c) − (PPF at a)], *paired t test: p < 0.01. B, Application of αBTX (100 nm, black bar) blocked nicotine-induced rescue of mfLTP in KOs (black circles). Application of αBTX and nicotine without HFS did not influence basal synaptic transmission (open circles). Inset, ΔPPF ratio in KO αBTX+Nic+HFS experiments. ΔPPF ratio with nicotine application in the presence of αBTX [(PPF at b) − (PPF at a)]; ΔPPF ratio with HFS [(PPF at c) − (PPF at a)]. C, mfLTP in wild type is not blocked by αBTX. αBTX alone (100 nm, 10 min; gray bar) did not affect synaptic transmission (open squares). Black squares, αBTX+HFS. Inset (for αBTX+HFS experiments), ΔPPF ratio with αBTX [(PPF at b) − (PPF at a)]; ΔPPF ratio with HFS [(PPF at c) − (PPF at a)]; *Paired t test, p < 0.001. Right, FP traces. Calibration: 0.5 mV, 5 ms.
To further test whether nicotine-induced rescue of mfLTP was mediated by α7-nAChRs, we applied 100 nm α-bungarotoxin (αBTX), a selective antagonist. αBTX abolished the effect of nicotine on mfLTP (1 h after HFS: 105 ± 4%, n = 10 slices/6 mice; paired t test: p > 0.05) (Fig. 3B) and PPF ratio (αBTX: 5.22 ± 0.65, αBTX+nicotine: 5.17 ± 0.65, 1 h after HFS: 5.19 ± 0.73) (Fig. 3B, inset) in KOs. Application of αBTX and nicotine in the absence of HFS did not alter synaptic transmission (1 h after αBTX+Nic: 100 ± 1%, n = 4 slices/2 mice; paired t test: p = 0.86) (Fig. 3B). These results suggest that nicotine-induced rescue of presynaptic deficits in BACE1 KOs is mediated by α7-nAChRs.
Finally, we tested whether α7-nAChRs are required for mfLTP in WTs. A brief application of αBTX (10 min) before and during HFS failed to block mfLTP in WTs (1 h after HFS: 148 ± 6%, n = 9 slices/7 mice; paired t test: p < 0.001) (Fig. 3C). This indicates that activation of α7-nAChRs is not necessary for mfLTP induction in WTs, hence the rescue of mfLTP in KOs by α7-nAChR activation is probably via recruitment of an alternative pathway not normally used in WTs.
CICR is involved in nicotine-induced rescue of mfLTP in BACE1 KOs
Activation of α7-nAChRs enhances CICR from ryanodine-sensitive Ca2+ stores (Sharma and Vijayaraghavan, 2003; Sharma et al., 2008). To investigate whether CICR is also involved in nicotine-induced rescue of mfLTP in KOs, we used 20 μm ruthenium red (RR) or 100 μm ryanodine (Ryan), which are blockers of ryanodine-sensitive stores. Both drugs completely abolished nicotine-induced recovery of PPF ratio [RR: 5.00 ± 0.69, RR+Nic: 4.97 ± 0.70, 1 h after HFS: 4.62 ± 0.74 (Fig. 4A, inset); Ryan: 5.01 ± 0.20, Ryan+Nic: 5.03 ± 0.23, 1 h after HFS: 4.93 ± 0.25] and mfLTP (1 h after RR+Nic: 91 ± 5%, n = 9 slices/5 mice; paired t test: p = 0.18; 1 h after Ryan+Nic: 100 ± 2%, n = 6 slices/3 mice; paired t test: p = 0.66) (Fig. 4A) in KOs without influencing basal synaptic transmission. mfLTP was present in WTs treated with RR (1 h after HFS: 124 ± 5%, n = 9 slices/5 mice; paired t test: p < 0.01) (Fig. 4B), but was significantly less than that in control WTs (t test: p < 0.01), suggesting that CICRs are only partially involved.
Figure 4.
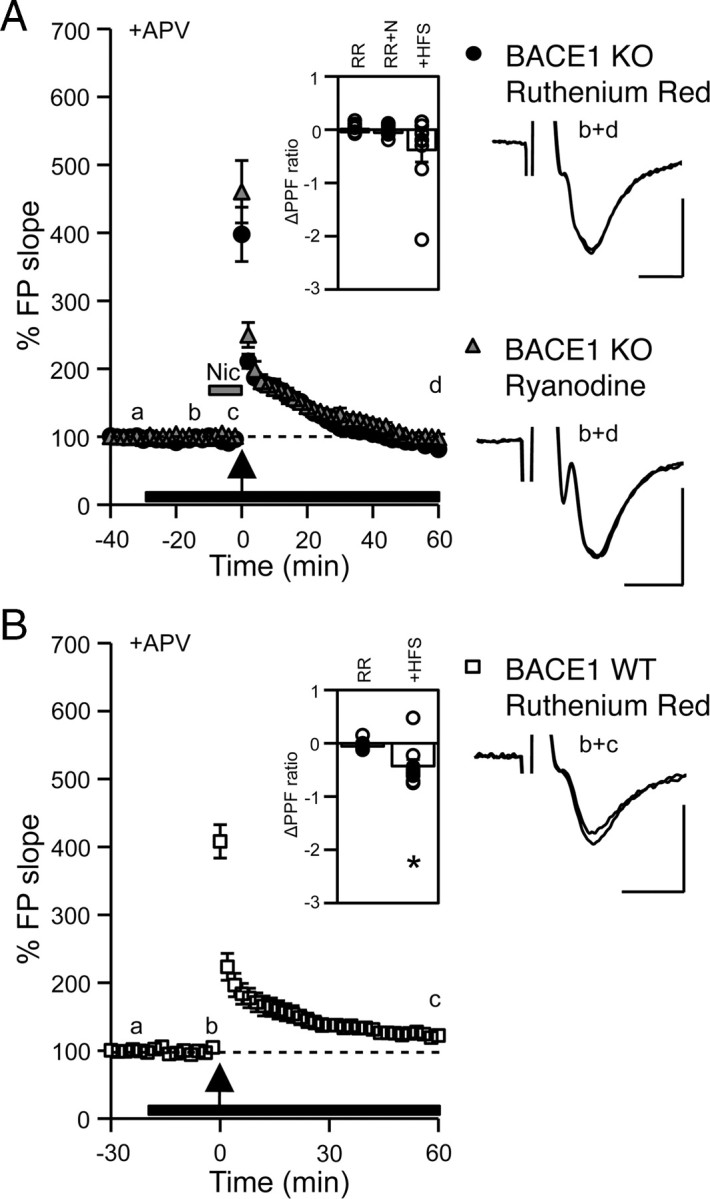
Nicotine-induced rescue of mfLTP in BACE1 KOs requires CICR. A, Application (black bar) of RR (20 μm) or Ryan (100 μm) abolished nicotine-induced rescue of mfLTP in KOs (RR: black circles, Ryan: gray triangles). Inset, ΔPPF ratio of RR application [(PPF at b) − (PPF at a)], +nicotine [(PPF at c) − (PPF at b)], and +HFS [(PPF at d) − (PPF at b)]. B, RR (20 μm; black bar) reduced mfLTP in WTs (open squares). Inset, ΔPPF ratio of RR application [(PPF at b) − (PPF at a)] and +HFS [(PPF at c) − (PPF at b)]. *Paired t test, p < 0.01. Right, FP traces. Calibration: 0.5 mV, 5 ms.
Discussion
We found that nicotine restores PPF and LTP at MF-to-CA3 synapses in BACE1 KOs. The nicotine effect was mimicked by α7-nAChR-specific agonist PNU282987, and blocked by α7-nAChR antagonist αBTX. We have evidence that nicotine acts via recruiting CICR. These results suggest nicotine and α7-nAChR agonists as potential pharmacological means to circumvent the presynaptic deficits caused by BACE1 inhibition.
mfLTP is presynaptically expressed, requiring an increase in presynaptic Ca2+ and a subsequent activation of cAMP–PKA signaling pathway (Nicoll and Schmitz, 2005). We previously demonstrated that presynaptic dysfunction seen in BACE1 KOs is at the level of Ca2+ regulation, but the downstream PKA signaling is intact (Wang et al., 2008). These results predict that restoring presynaptic Ca2+ signaling should recover mfLTP in BACE1 KOs. Presynaptic α7-nAChR elevates the intracellular concentration of free Ca2+ (Vijayaraghavan et al., 1992) and enhances glutamate release at MF terminals (Sharma and Vijayaraghavan, 2003; Sharma et al., 2008; Bancila et al., 2009). The nicotine-induced rescue of PPF and mfLTP without much effect on basal synaptic transmission is likely via the recruitment of CICR, which is known to preferentially amplify use-dependent release (Shimizu et al., 2008). Short-term presynaptic plasticity, including PPF, does not depend on CICR at MF terminals (Carter et al., 2002). Consistent with this, inhibiting CICRs in WTs did not alter PPF ratio, but reduced mfLTP magnitude, which suggests that HFS recruits CICR. In the case of KOs, it is clear that the CICR triggered by α7-nAChR activation is needed to rescue mfLTP. Although we cannot rule out the possible involvement of α7-nAChRs on interneurons, the detection of α7-nAChR immunoreactivity in the MF input region (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) provides a substrate for α7-nAChR agonists to act on MF terminals. This is further corroborated by a recent electron microscopy study, which localized α7-nAChRs on MF terminals (Bancila et al., 2009). Interestingly, the α7-nAChRs were present away from the active zone, suggesting an indirect regulation of presynaptic release.
It is known that α7-nAChRs can rapidly desensitize upon agonist binding in a dose-dependent manner (Peng et al., 1994). Because nicotine-induced rescue of mfLTP was blocked by αBTX, we suspect residual α7-nAChR activity even with the prolonged application of nicotine used in our study. Interestingly, the increase in glutamate release at MF terminals with α7-nAChR activation is rather slow and involves presynaptic Ca2+ increase via CICR from internal stores (Sharma and Vijayaraghavan, 2003; Sharma et al., 2008). In synaptosomes isolated from the prefrontal cortex, α7-nAChR agonist-induced glutamate release is dependent on CICR and a downstream activation of extracellular signal-regulated kinase (ERK) signaling (Dickinson et al., 2008). These results suggest that presynaptic signaling of α7-nAChRs leading to glutamate release may outlast the initial activation of the receptor.
The regulation of α7-nAChRs has been implicated in the pathology of AD. There are studies reporting high-affinity binding between Aβ42 and α7-nAChRs (Wang et al., 2000a,b), which either inhibit (Guan et al., 2001; Liu et al., 2001; Pettit et al., 2001) or activate (Dineley et al., 2001) α7-nAChR signaling. It is possible that Aβ42 may facilitate α7-nAChRs at low concentration, but may inhibit nAChRs when the burden of Aβ peptides increases (Dineley et al., 2001; Dougherty et al., 2003). The concentration-dependent dual role of Aβ42 is evident in a study showing that picomolar range of Aβ42 facilitates, but nanomolar range abolishes, LTP in CA1 and learning via its action on α7-nAChRs (Puzzo et al., 2008). It is unlikely that endogenous Aβ42 acts in this manner to influence mfLTP, because blocking α7-nAChRs with α-BTX did not affect mfLTP in WTs. This result indirectly argues that the lack of mfLTP in BACE1 KOs may not be a strict consequence of lacking Aβ. Interestingly, BACE1 has been found to regulate neuregulin-1 (NRG1) cleavage (Hu et al., 2006; Willem et al., 2006), and indeed this process is affected in BACE1 KOs (Savonenko et al., 2008). NRG1 is critically involved in maintaining surface expression of presynaptic α7-nAChRs (Hancock et al., 2008; Zhong et al., 2008). However, in isolated CA3 slices, we did not see a change in the total or cell surface levels of α7-nAChRs and NRG1 in the KOs (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Furthermore, our ability to rescue mfLTP in KOs with α7-nAChR agonists suggests sufficient presence of functional α7-nAChRs.
Several potential methods are being developed to overcome dysfunctions caused by complete BACE1 inhibition, such as partial BACE1 inhibition (Vassar et al., 2009). While our results might reflect a developmental loss of BACE1, they suggest that combining α7-nAChR agonists with BACE1 inhibitors may be another alternative.
Footnotes
This work was supported by a grant from the National Institutes of Health (P01-NS047308) to P.C.W. and H.-K.L., the Graduate Student Summer Research Fellowship [University of Maryland (UMD)] and the Robert and Florence Deutsch Graduate Award (UMD) to H.W., and the Howard Hughes Medical Institute Undergraduate Research Fellowship to A.L.
References
- Bancila V, Cordeiro JM, Bloc A, Dunant Y. Nicotine-induced and depolarisation-induced glutamate release from hippocampus mossy fibre synaptosomes: two distinct mechanisms. J Neurochem. 2009;110:570–580. doi: 10.1111/j.1471-4159.2009.06169.x. [DOI] [PubMed] [Google Scholar]
- Bodnar AL, Cortes-Burgos LA, Cook KK, Dinh DM, Groppi VE, Hajos M, Higdon NR, Hoffmann WE, Hurst RS, Myers JK, Rogers BN, Wall TM, Wolfe ML, Wong E. Discovery and structure-activity relationship of quinuclidine benzamides as agonists of alpha7 nicotinic acetylcholine receptors. J Med Chem. 2005;48:905–908. doi: 10.1021/jm049363q. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Carter AG, Vogt KE, Foster KA, Regehr WG. Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci. 2002;22:21–28. doi: 10.1523/JNEUROSCI.22-01-00021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson JA, Kew JN, Wonnacott S. Presynaptic alpha 7- and beta 2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol. 2008;74:348–359. doi: 10.1124/mol.108.046623. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JJ, Wu J, Nichols RA. β-Amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci. 2003;23:6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giocomo LM, Hasselmo ME. Nicotinic modulation of glutamatergic synaptic transmission in region CA3 of the hippocampus. Eur J Neurosci. 2005;22:1349–1356. doi: 10.1111/j.1460-9568.2005.04316.x. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Guan ZZ, Miao H, Tian JY, Unger C, Nordberg A, Zhang X. Suppressed expression of nicotinic acetylcholine receptors by nanomolar beta-amyloid peptides in PC12 cells. J Neural Transm. 2001;108:1417–1433. doi: 10.1007/s007020100017. [DOI] [PubMed] [Google Scholar]
- Hancock ML, Canetta SE, Role LW, Talmage DA. Presynaptic type III neuregulin1-ErbB signaling targets alpha7 nicotinic acetylcholine receptors to axons. J Gen Physiol. 2008;131:i4. doi: 10.1085/JGP1316OIA4. [DOI] [PubMed] [Google Scholar]
- Harrison SM, Harper AJ, Hawkins J, Duddy G, Grau E, Pugh PL, Winter PH, Shilliam CS, Hughes ZA, Dawson LA, Gonzalez MI, Upton N, Pangalos MN, Dingwall C. BACE1 (beta-secretase) transgenic and knockout mice: identification of neurochemical deficits and behavioral changes. Mol Cell Neurosci. 2003;24:646–655. doi: 10.1016/s1044-7431(03)00227-6. [DOI] [PubMed] [Google Scholar]
- Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Jiang L, Role LW. Facilitation of cortico-amygdala synapses by nicotine: activity-dependent modulation of glutamatergic transmission. J Neurophysiol. 2008;99:1988–1999. doi: 10.1152/jn.00933.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Kawai H, Berg DK. β-Amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci U S A. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Damore MA, Fitzpatrick D, Liu H, Zhang J, Yan Q, Vassar R, Citron M. BACE1 (beta-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol Dis. 2003;14:81–88. doi: 10.1016/s0969-9961(03)00104-9. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, De Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Peng X, Katz M, Gerzanich V, Anand R, Lindstrom J. Human alpha 7 acetylcholine receptor: cloning of the alpha 7 subunit from the SH-SY5Y cell line and determination of pharmacological properties of native receptors and functional alpha 7 homomers expressed in Xenopus oocytes. Mol Pharmacol. 1994;45:546–554. [PubMed] [Google Scholar]
- Perry E, Martin-Ruiz C, Lee M, Griffiths M, Johnson M, Piggott M, Haroutunian V, Buxbaum JD, Nãsland J, Davis K, Gotti C, Clementi F, Tzartos S, Cohen O, Soreq H, Jaros E, Perry R, Ballard C, McKeith I, Court J. Nicotinic receptor subtypes in human brain ageing, Alzheimer and Lewy body diseases. Eur J Pharmacol. 2000;393:215–222. doi: 10.1016/s0014-2999(00)00064-9. [DOI] [PubMed] [Google Scholar]
- Pettit DL, Shao Z, Yakel JL. β-Amyloid1–42 peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci. 2001;21:RC120. doi: 10.1523/JNEUROSCI.21-01-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe KA, Fisher JL, Gray R, Dani JA. Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Ann N Y Acad Sci. 1999;868:591–610. doi: 10.1111/j.1749-6632.1999.tb11332.x. [DOI] [PubMed] [Google Scholar]
- Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci U S A. 2008;105:5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron. 2003;38:929–939. doi: 10.1016/s0896-6273(03)00322-2. [DOI] [PubMed] [Google Scholar]
- Sharma G, Grybko M, Vijayaraghavan S. Action potential-independent and nicotinic receptor-mediated concerted release of multiple quanta at hippocampal CA3-mossy fiber synapses. J Neurosci. 2008;28:2563–2575. doi: 10.1523/JNEUROSCI.5407-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Fukaya M, Yamasaki M, Watanabe M, Manabe T, Kamiya H. Use-dependent amplification of presynaptic Ca2+ signaling by axonal ryanodine receptors at the hippocampal mossy fiber synapse. Proc Natl Acad Sci U S A. 2008;105:11998–12003. doi: 10.1073/pnas.0802175105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC. The β-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaraghavan S, Pugh PC, Zhang ZW, Rathouz MM, Berg DK. Nicotinic receptors that bind alpha-bungarotoxin on neurons raise intracellular free Ca2+ Neuron. 1992;8:353–362. doi: 10.1016/0896-6273(92)90301-s. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers—a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Song L, Laird F, Wong PC, Lee HK. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J Neurosci. 2008;28:8677–8681. doi: 10.1523/JNEUROSCI.2440-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lee DH, Davis CB, Shank RP. Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem. 2000a;75:1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. β-Amyloid1-42 binds to α7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000b;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Zhong C, Du C, Hancock M, Mertz M, Talmage DA, Role LW. Presynaptic type III neuregulin 1 is required for sustained enhancement of hippocampal transmission by nicotine and for axonal targeting of α7 nicotinic acetylcholine receptors. J Neurosci. 2008;28:9111–9116. doi: 10.1523/JNEUROSCI.0381-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]