

Abstract
Background
We investigated the effect of polymorphisms in the renin-angiotensin-aldosterone system (RAAS) genes on ventricular remodeling, growth, renal function and response to enalapril in infants with single ventricle.
Methods and Results
Single ventricle infants enrolled in a randomized trial of enalapril were genotyped for polymorphisms in 5 genes: angiotensinogen, angiotensin-converting enzyme, angiotensin II type 1 receptor, aldosterone synthase, and chymase. Alleles associated with RAAS upregulation were classified as risk alleles. Ventricular mass, volume, somatic growth, renal function using estimated glomerular filtration rate (eGFR), and response to enalapril were compared between patients with ≥2 homozygous risk genotypes (high-risk), and those with <2 homozygous risk genotypes (low-risk) at two time points - before the superior-cavopulmonary-connection (pre-SCPC) and at age 14 months. Of 230 trial subjects, 154 were genotyped: 38 were high-risk, 116 were low-risk. Ventricular mass and volume were elevated in both groups pre-SCPC. Ventricular mass and volume decreased and eGFR increased after SCPC in the low-risk (p<0.05) but not the high-risk group. These responses were independent of enalapril treatment. Weight and height z-scores were lower at baseline and height remained lower in the high-risk group at 14 months especially in those receiving enalapril (p<0.05).
Conclusions
RAAS-upregulation genotypes were associated with failure of reverse remodeling after SCPC surgery, less improvement in renal function, and impaired somatic growth, the latter especially in patients receiving enalapril. RAAS genotype may identify a high-risk subgroup of single ventricle patients who fail to fully benefit from volume unloading surgery. Follow-up is warranted to assess longterm impact.
Clinical Trial Registration
Clinical Trials.gov Identifier NCT00113087
Infants with single ventricle lesions account for a significant proportion of the health burden related to congenital heart defects1. In infancy, these lesions are characterized by a single functioning ventricle that supports both the systemic and pulmonary circulations, resulting in significant ventricular volume overload. Two subsequent palliative surgeries are performed, one at 4–6 months of age, the superior cavopulmonary connection (SCPC) and another between 18–36 months of age, the Fontan procedure. The SCPC diverts superior vena cava flow directly to the lungs thus partially unloading the ventricle2. It is not until after the Fontan procedure that there is a complete separation of the pulmonary and systemic circulations3. The single ventricle therefore remains exposed to chronic hypoxia and increased volume load throughout infancy. This is usually associated with an increase in ventricular mass that is disproportionate to the increase in volume. This can have a detrimental effect on cardiac function by causing an imbalance in myocardial oxygen demand and supply with resultant myocardial damage4–6. Ultimately, the burden of the abnormal physiology coupled with the maladaptive response of the ventricular muscle contributes to poor long term outcomes including growth impairment, heart failure and reduced survival7–9.
An important mediator of the ventricular response to hemodynamic load is the renin-angiotensin-aldosterone system (RAAS). Persistent RAAS upregulation can have a detrimental effect by causing peripheral vasoconstriction as well as promoting cellular apoptosis and fibrosis 10. While angiotensin converting enzyme (ACE) inhibitor therapy has shown beneficial anti-hypertrophic effects in patients with pressure overload-induced hypertrophy11–14, the recently concluded Pediatric Heart Network randomized trial failed to show a beneficial effect of ACE inhibition on the 14-month outcomes of ventricular mass or somatic growth in infants with single ventricle15. Variations in RAAS genes are an important determinant of the ventricular hypertrophic response in physiologic and pathologic states16–19. These variations can also influence the response to ACE inhibitor or angiotensin receptor blocker therapy in patients with other conditions like systemic hypertension20–22. The Pediatric Heart Network therefore undertook a pharmacogenetic sub-study to investigate if polymorphisms in RAAS genes influence the cardiac phenotype and the response to ACE inhibitor therapy in infants with single ventricle.
The primary objective of this analysis was to investigate associations between RAAS gene polymorphisms and the ventricular remodeling response to enalapril in infants with single ventricle lesions. The five RAAS genes included angiotensinogen (AGT), angiotensin-converting enzyme (ACE), angiotensin II type 1 receptor (AGTR1), aldosterone synthase (CYP11B2), and cardiac chymase A (CMA1). We also studied the potential influence of RAAS genotypes on somatic growth and measures of renal function23, 24.
METHODS
Study Design
The study was performed as part of a randomized, double-blind, placebo-controlled clinical trial by the Pediatric Heart Network comparing the effects of enalapril versus placebo on somatic growth in infants with single ventricles. (ClinicalTrials.gov Identifier NCT00113087) 15. Patients ≤ 45 days of age with single ventricle physiology, stable systemic and pulmonary blood flow, and planned SCPC were enrolled from August 2003, through May 2007 at 10 centers in the United States and Canada. Detailed descriptions of the study design, methods and results have been published15,25. Patients were randomly assigned to enalapril (target dose 0.4 mg/kg/day) or placebo and followed from enrollment until 14 months of age to assess the effects on growth for at least 6 months after the SCPC surgery. Patients were consented for the genetic study prior to the SCPC surgery. The study protocol was approved by local Institutional Review Boards and written informed consent was obtained from a parent/guardian.
RAAS genotyping
A blood sample was obtained and genomic DNA was isolated from whole blood using PureGene kits (Gentra Systems). Patients were genotyped for polymorphisms in five RAAS genes: 1) a G/A missense variant at position −235 in angiotensinogen (AGT) (rs11568053); 2) a 287 base pair intron 16 deletion variant of angiotensin converting enzyme (ACE); 3) an A/C substitution at position 1166 in the 3’ untranslated region of angiotensin II type 1 receptor (AGTR1) (rs5186); 4) a C/T polymorphism at position −344 in aldosterone synthase (CYP11B2) (rs1799998); and 5) an A/G polymorphism at position −1903 of cardiac chymase A (CMA1) (rs1800875)26. RAAS genotypes were determined by pyrosequencing assays for AGTR1, CYP11B2, AGT, and CMA1 and electrophoresis of PCR products for the ACE assay as previously described17. Polymorphisms were selected based on previous association studies, functional effects, and population allele frequencies16–19, 27. Alleles previously associated with RAAS upregulation were classified as risk alleles with high-risk defined as homozygosity for the risk alleles. The clinicians caring for the patients were unaware of patient genotypes.
Clinical Phenotype
All subjects underwent assessment of weight, height, head circumference z-scores, ventricular mass, volumes, ejection fraction (EF) on two-dimensional echocardiography, Ross heart failure (HF) class, systolic and diastolic blood pressure (BP) at echocardiography visits, and B-type natriuretic peptide (BNP) levels pre-SCPC and at 14 months. For each of the height, weight, and head circumference measurements, the z-score represents the number of standard deviations from the mean value for age compared to normative values published by the World Health Organization and the Centers for Disease Control28. Serial serum creatinine was used to estimate glomerular filtration rate (eGFR) using the Schwartz equation, the only validated method for the pediatric population29. Since the Schwartz equation can underestimate renal insufficiency, we analyzed change score in eGFR for our study30. Echocardiograms were analyzed by a single core laboratory observer as detailed previously25, 31. Ventricular mass, volume, and mass:volume ratio were expressed as z-scores relative to body surface area32. BNP concentration was measured in a core laboratory. Endpoints were measured at the study visit immediately prior to the SCPC surgery (mean age, 5.1±1.6 months) and at the final study visit (mean age, 14.1±0.9 months).
Statistical Analysis
We ascertained if the genotype frequencies were in Hardy-Weinberg equilibrium using Pearson chi-square test. We combined risk genotypes in order to analyze the compound effect of multiple risk genotypes within the same biologic pathway. Due to sample size limitations, for the primary analysis, we divided subjects into two pre-specified subgroups -the high-risk group with ≥2 homozygous risk genotypes and low-risk group with <2 risk genotypes. This was based on our previous reports that showed an association of ≥2 RAAS high-risk homozygous genotypes with ventricular hypertrophy in cardiomyopathy and transplant patients17, 25. The following outcomes were compared by risk group at the pre-SCPC and final study visit: z-scores for weight, height, head circumference, ventricular mass, ventricular end-diastolic volume (EDV), mass/volume ratio, EF, systolic and diastolic BP, Ross HF score, BNP levels and eGFR. The change in outcomes between the two time-points was also compared. Mean z-scores were compared to the normal mean (zero) using a one-sample t-test or Wilcoxon signed rank test. We also analyzed the interaction of systemic ventricular morphology with genotype on ventricular mass by ANOVA. When significant associations by risk group were found, we assessed the effect of each individual genotype using the recessive and dominant models. We examined association between treatment, outcomes and level of genetic risk by fitting the number of high-risk genes (0, 1, 2, and ≥3) as a linear term. We used linear regression with a treatment assignment and high vs. low risk interaction term to assess whether there was a differential effect of treatment (enalapril vs. placebo) on outcome by risk subgroup. Statistical analyses were performed using SAS Statistical Software v.9.2 (SAS Institute, Cary NC) and S-Plus 8.0 (Insightful Corp., Seattle, WA). We estimated effect size for the two-factor interaction (treatment X genetic risk) at a power of 0.80 for p<0.05 assuming equal variance for z-scores (variance=1.16). The minimum effect size at 80% power for the primary outcome measure of weight z-score was 0.61z between high and low-risk genotype groups, 0.97z between high-risk enalapril vs placebo and 0.63z between low-risk enalapril vs placebo33, 34.
RESULTS
Genotype frequencies
Of 230 subjects enrolled in the trial, 31 died or underwent cardiac transplantation. Of the remaining, 195 were approached, 164 (84%) consented for the genetic sub-study, 159 submitted a blood sample, and 154 had adequate DNA samples for genotyping. As samples were obtained prior to SCPC, the study cohort was biased towards patients who survived beyond stage 1 palliation as seen by the higher incidence of death/transplant in the non-genotyped vs genotyped patients (33% vs 4%, p<0.001). The allele and genotype frequencies are shown in Table 1 and were comparable to the general population17, 21, 22, 27. Genotype frequencies were in Hardy-Weinberg equilibrium with no gender/racial/ethnic-based differences. Forty-six patients (30%) had no homozygous risk genotypes, 70 (45%) had one, 27 (17%) two, 10 (7%) three and 1 (1%) had four risk genotypes. Thirty-eight patients (25%) were classified as high-risk (≥2 homozygous risk genotypes), and 116 patients as low-risk (<2 homozygous risk genotypes).
Table 1.
Frequency of high-risk alleles and risk genotypes (n=154)
| Gene ID | High risk allele frequency |
Homozygous high risk genotype frequency |
|---|---|---|
| AGT (%) | C (0.56) | CC (0.32) |
| ACE | D (0.54) | DD (0.27) |
| AGTR1 | C (0.27) | CC (0.05) |
| CYP11B2 | C (0.44) | CC (0.20) |
| CMA1 | A (0.44) | AA (0.18) |
AGT = Angiotensinogen; ACE = Angiotensin converting enzyme; AGTR1 = Angiotensin II type 1 receptor; CYP11B2 = Aldosterone synthase; CMA1 = Chymase
Patient characteristics
There were no differences in demographic characteristics at enrollment between the genotype groups, except for an older age (mean, 24 vs 20 days) and lower weight and height z-scores at enrollment in the high-risk group (Table 2). Clinical and echocardiographic characteristics at the pre-SCPC and final study visits are shown in Table 3. The average age at SCPC surgery was not different between the high and low-risk groups (5.4±1.7 vs. 5.6±1.6 mo, p=0.72). There were no differences in echocardiographic ventricular volumes, EF, incidence of moderate-severe AVVR, BNP levels, systolic and diastolic BP, Ross HF score and incidence of death/transplant between the two groups at the pre-SCPC and final study visits. Ventricular mass was higher in the high risk group at the final study visit.
Table 2.
Patient characteristics at enrollment (n=154)
| High Risk | Low risk | p | ||
|---|---|---|---|---|
| n | 38 | 116 | ||
| Age at randomization (days) | 24 ± 11 | 20 ± 9 | 0.05 | |
| Gestational age at birth (weeks) | 38.3 ± 1.5 | 38.4 ± 1.4 | 0.58 | |
| Birth weight (kg) | 3.21 ± 0.51 | 3.28 ± 0.52 | 0.67 | |
| Birth weight for gestational age percentile | 45.7 ± 29.1 | 50.2 ± 29.1 | 0.37 | |
| Male | 74% | 66% | 0.43 | |
| Race | 0.80 | |||
| Caucasian | 79% | 81% | ||
| African American | 18% | 14% | ||
| Asian | 0% | 1% | ||
| Other | 3% | 4% | ||
| Hispanic | 8% | 19% | 0.20 | |
| Hypoplastic left heart syndrome | 61% | 58% | 0.07 | |
| Weight for age z-score | −1.70 ± 1.40 | −1.14 ± 1.24 | 0.02 | |
| Height for age z-score | −1.44 ± 1.39 | −0.94 ± 1.20 | 0.03 | |
| Head circumference for age z-score | −2.11 ± 1.61 | −1.65 ± 1.38 | 0.09 | |
Table 3.
Patient characteristics at pre-SCPC and final study visits
| Pre-SCPC | High-risk | Low-risk | P | Final visit | High-risk | Low-risk | P |
|---|---|---|---|---|---|---|---|
| N †† | 38 | 116 | N | 37 | 110 | ||
| Weight z* | −1.9±1.0 | −1.5±1.2 | 0.07 | Weight z* | −0.7±1.1 | −0.4±1.1 | 0.28 |
| Height z* | −1.6±1.3 | −1.1±1.2 | 0.03 | Height z* | −1.3±1.1 | −0.8±1.1 | 0.02 |
| HC z* | −1.5±1.3 | −1.3±1.2 | 0.50 | HC z | −0.4±1.2 | −0.1±1.5 | 0.17 |
| Median BNP | Median BNP | ||||||
| 80 (44–155) | 71 (30–161) | 0.67 | 40 (18–109) | 29 (16–58) | 0.30 | ||
| (pg/ml) (IQR) | (pg/ml) (IQR) | ||||||
| EDVz* | 1.7±2.6 | 2.1±2.4 | 0.46 | EDVz* | 1.6±3.5 | 1.0±1.9 | 0.99 |
| ESVz* | 2.7±3.8 | 3.3±3.6 | 0.29 | ESVz* | 3.1±6.1 | 2.0±2.8 | 0.91 |
| EF% | 59±9 | 58±10 | 0.37 | EF% | 58±14 | 59±9 | 0.89 |
| Mass z* | 4.1±3.5 | 4.2±2.8 | 0.67 | Mass z* | 3.7±2.9 | 2.5±2.3 | 0.02 |
| Mass/vol | 1.33±0.55 | 1.22±0.48 | 0.30 | Mass/vol | 1.29±0.45 | 1.15±0.41 | 0.09 |
| Mass/vol z* | 2.7±3.6 | 2.0±3.1 | 0.30 | Mass/vol z* | 2.5±2.9 | 1.6±2.7 | 0.09 |
| Mod AVVR | 27% | 25% | 0.83 | ≥Mod AVVR | 14% | 17% | 0.80 |
| O2 sats (%) | 75±8 | 76±7 | 0.35 | O2 sats (%) | 84±4 | 83±10 | 0.55 |
| ††EDP | 8±3 | 8±4 | 0.94 | ||||
| (mmHg) | |||||||
| Stroke vol | 13±5 | 14±6 | 0.37 | Stroke vol | 17±8 | 17±5 | 0.87 |
| HR (bpm) | 118±18 | 123±19 | 0.26 | HR (bpm) | 112±14 | 114±14 | 0.61 |
| SBP (mmHg) | 88±11 | 88±14 | 0.43 | SBP (mmHg) | 89±15 | 90±17 | 0.95 |
| SBPz | 0.29±1.11 | 0.23±1.37 | 0.79 | SBPz | −0.13±1.48 | −0.08±1.64 | 0.87 |
| DBP (mmHg) | 43±13 | 46±12 | 0.08 | DBP (mmHg) | 48±15 | 49±13 | 0.70 |
| DBPz | −0.11±1.4 | 0.18±1.29 | 0.24 | DBPz | 0.24±1.62 | 0.33±1.47 | 0.77 |
| Ross HF | Ross HF | ||||||
| 0.26 | 0.39 | ||||||
| score | score | ||||||
| I | 68% | 50% | I | 76% | 82% | ||
| II | 13% | 28% | II | 11% | 9% | ||
| III | 13% | 21% | III | 13% | 9% | ||
| IV | 6% | 1% | IV | 0% | 0% |
z score differs from zero, p<0.05
At the pre-SCPC visit, sample size varies from 30–38 for the high-risk and 107–116 for the low-risk group. At the final study visit, sample size ranges from 33–37 in the high-risk and 105–110 in the low-risk group, with the exception of BNP, which had group sizes of 27 and 85, respectively.
SCPC = superior cavopulmonary connection; HC = head circumference; BNP = B-type natriuretic peptide; EDV = end-diastolic volume; ESV = end-systolic volume; Mod AVVR = moderate atrio-ventricular valve regurgitation; EF = ejection fraction; O2 sats = Oxygen saturation;
EDP = end-diastolic pressure (only available at pre-SCPC cardiac catheterization); HR = Heart rate; bpm = beats per minute; SBP = systolic blood pressure; DBP = diastolic blood pressure; HF = Heart failure; IQR=interquartile range
RAAS genotype and response to volume unloading
Ventricular mass, volume and mass/volume ratio were significantly elevated compared to normative values for age in both groups at the pre-SCPC visit (Table 3). Ventricular EDV z-score decreased by an average of 1.2 units in the low-risk group from pre-SCPC to 14 months (p<0.001) but not in the high-risk group (p=0.02 vs. low-risk group, Figure 1a). Similarly, ventricular mass z-score decreased after the SCPC surgery in the low-risk (p<0.001) but not the high-risk group and remained elevated above normal at the final study visit (p=0.049 vs low-risk group; Figure 1b).
Figure 1.
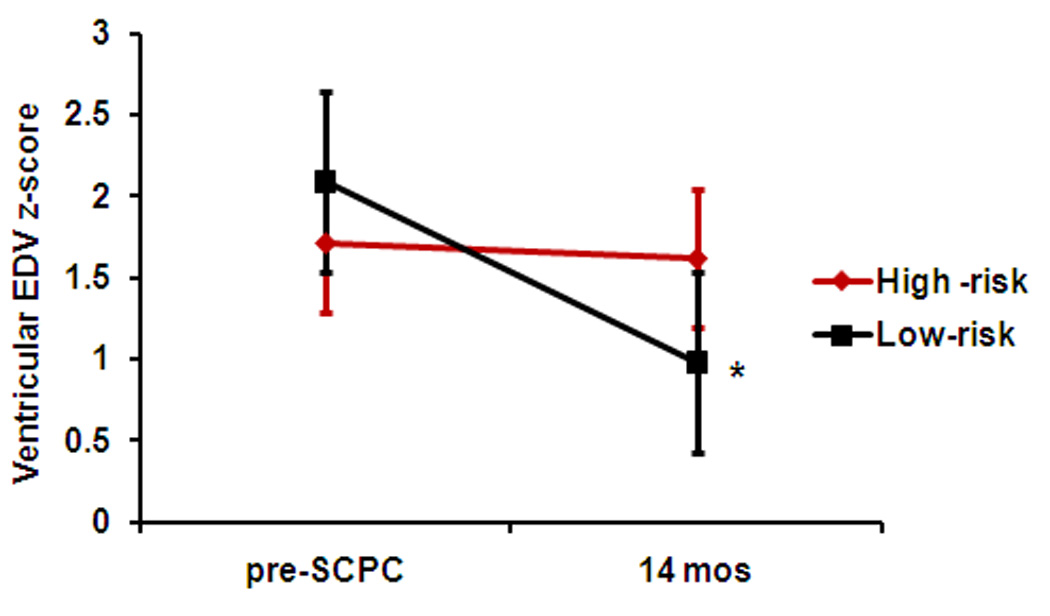
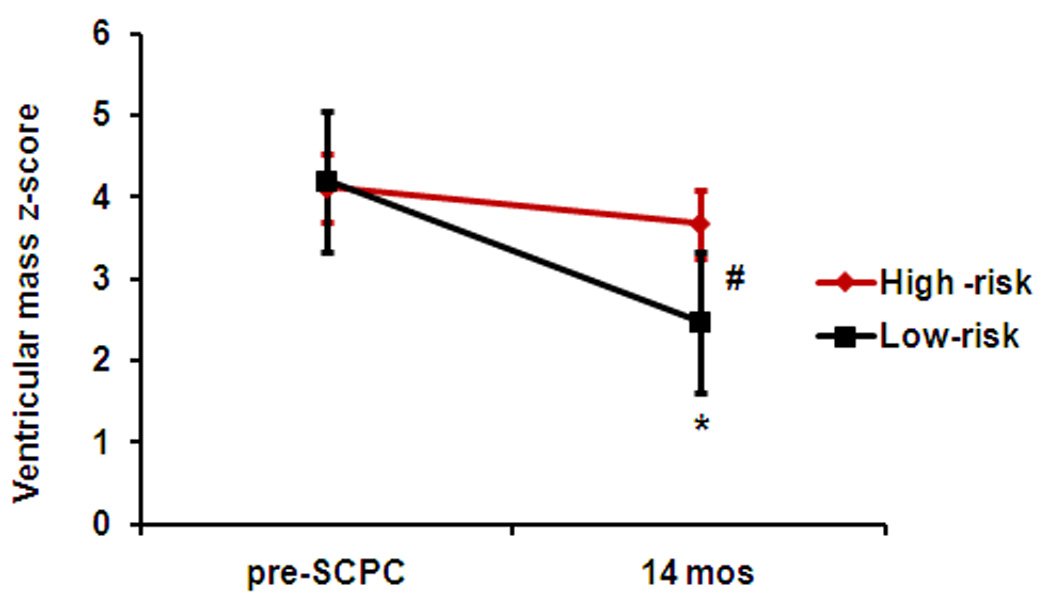
(a) Ventricular EDV z-scores, and (b) Ventricular mass z-scores decreased in the low-risk (black, n=116) but not in the high-risk (red, n=38) genotype groups from pre-SCPC to final study visit. High-risk, ≥ 2 homozygous risk genotypes; Low-risk, < 2 homozygous risk genotypes. *p<0.05 from pre-SCPC; # p<0.05 from low-risk group at 14 months. SCPC = superior cavopulmonary connection; EDV = end-diastolic volume
Ventricular mass/volume ratio remained significantly elevated compared to normative values for age in both groups pre-SCPC and at 14 months (Table 3). There was a positive association between number of RAAS-upregulation genotypes and ventricular mass z-score at 14 months (0.55±0.24 increase in z-score per risk genotype, p=0.015). When analyzed by individual genotypes, this association was significant for the AGTR1 risk genotype (Figure 2). The mass/volume ratio at 14 months also showed a positive correlation with a higher number of risk genotypes (0.50±0.27 increase in z-score per risk genotype, p=0.05). There was no significant interaction between systolic and diastolic BP (or BP z-scores) and genotype on ventricular mass z-score; systemic vascular resistance was not measured. The frequency of recurrent coarctation was also similar between the two risk groups (5.3% in high-risk and 5.2% in the low-risk group). There was no significant interaction of risk genotype group and ventricular morphology on the ventricular mass z-score at the pre-SCPC (p=0.53) or the 14-month visit (p=0.35).
Figure 2.
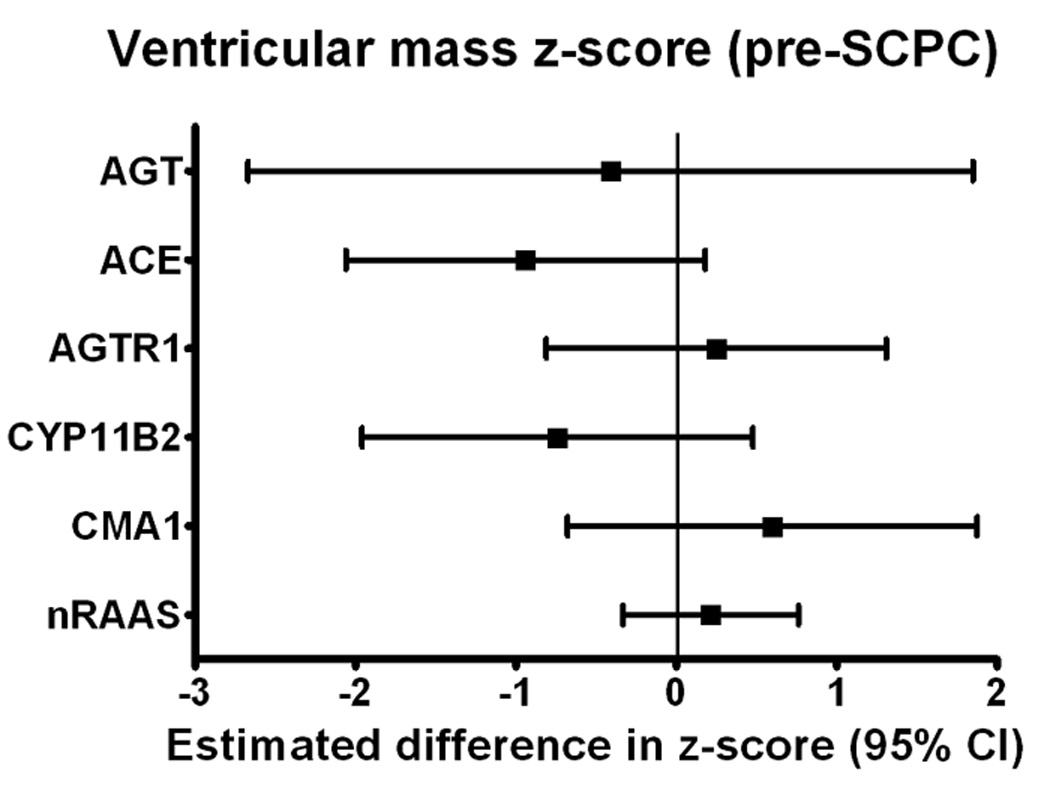
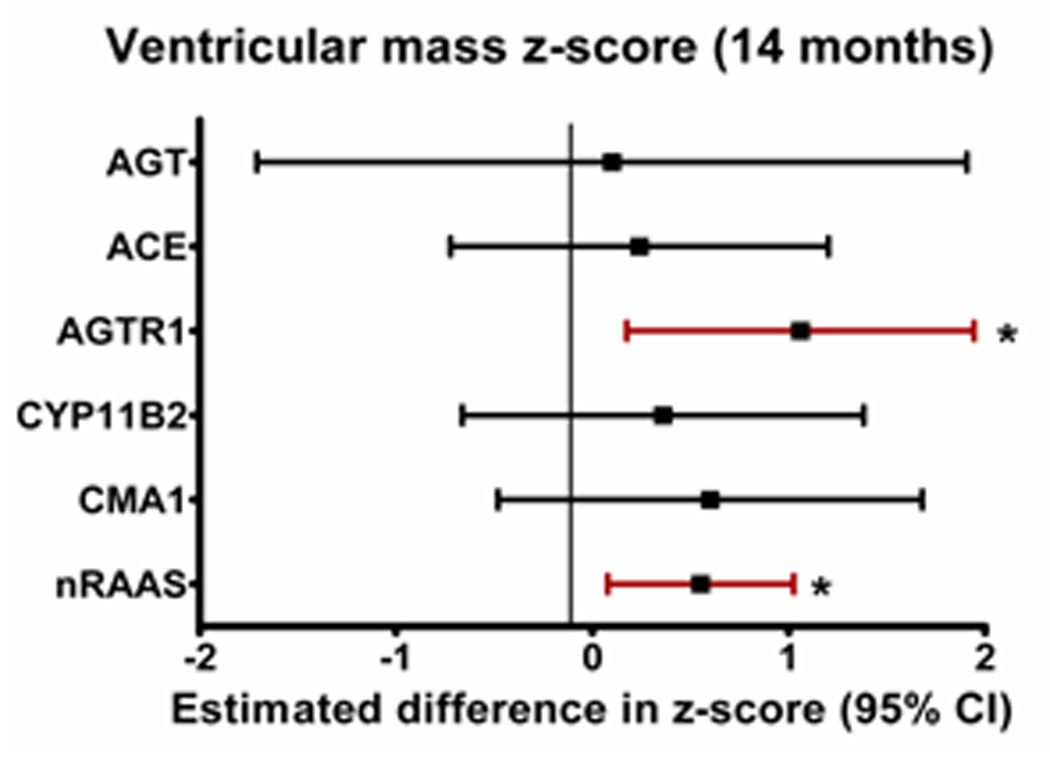
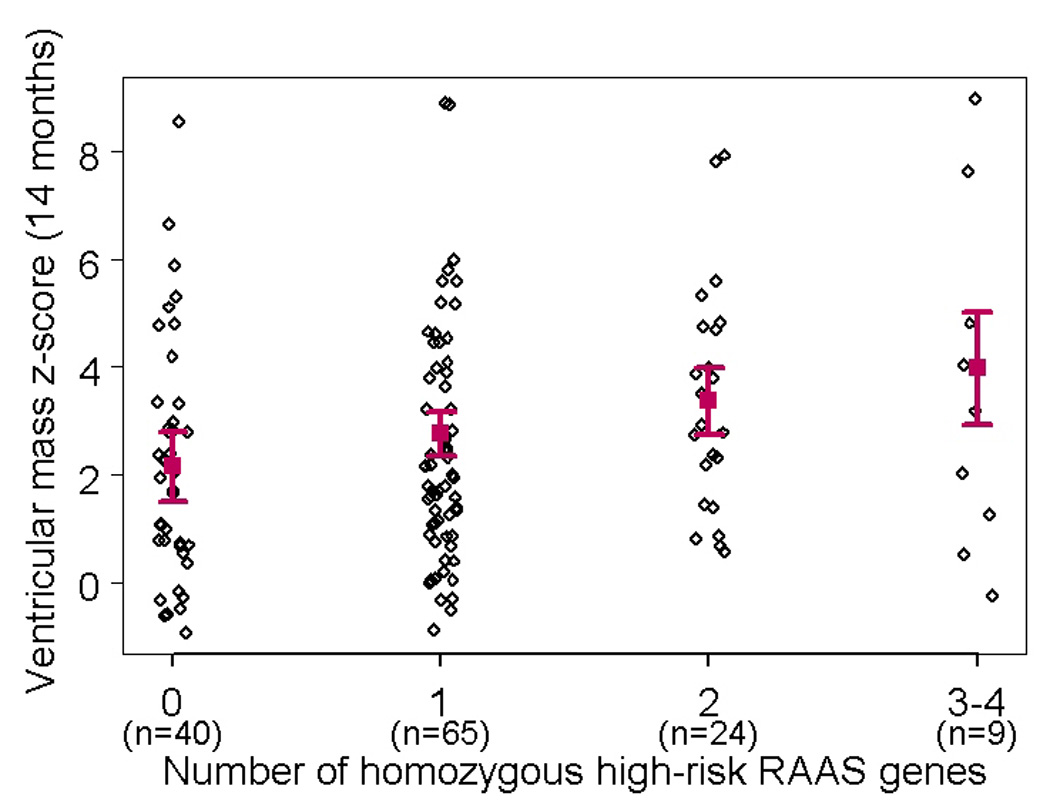
Difference (and 95% confidence intervals) in ventricular mass z-scores at (a) pre-SCPC and (b) final study visit between individual risk genotypes (high-risk minus no high-risk) using recessive model (*p<0.05 vs low risk genotype) showing a trend towards higher mass z-scores at 14 months in patients with high-risk genotypes with strongest association with AGTR1. (c) Linear regression model (mean± 95% confidence limits) showing incremental effect of increasing number of RAAS-upregulation genotypes on ventricular mass z-score at 14 months. 3 cases with mass z-scores outside the extreme physiologic range were excluded. AGT = Angiotensinogen; ACE = Angiotensin converting enzyme; AGTR1 = Angiotensin II type 1 receptor; CYP11B2 = Aldosterone synthase; CMA1 = Chymase; nRAAS = total number of high risk renin-angiotensin-aldosterone system genotypes; SCPC = superior cavopulmonary connection
RAAS genotype and somatic growth
Infants with high-risk genotypes had lower weight, and height z-scores compared to the low-risk group at enrollment (p<0.05) (Figure 3). Weight and head circumference z-scores increased in both groups during study follow-up (p<0.01 for both) so that by 14 months, there was no difference in weight and head circumference between the groups. Even though change in height z-scores was similar between the two groups from pre-SCPC to 14 months, height z-scores at 14 months remained significantly lower in high-risk compared to low-risk patients (−1.3±1.1 vs. −0.8±1.1, p=0.01).
Figure 3.
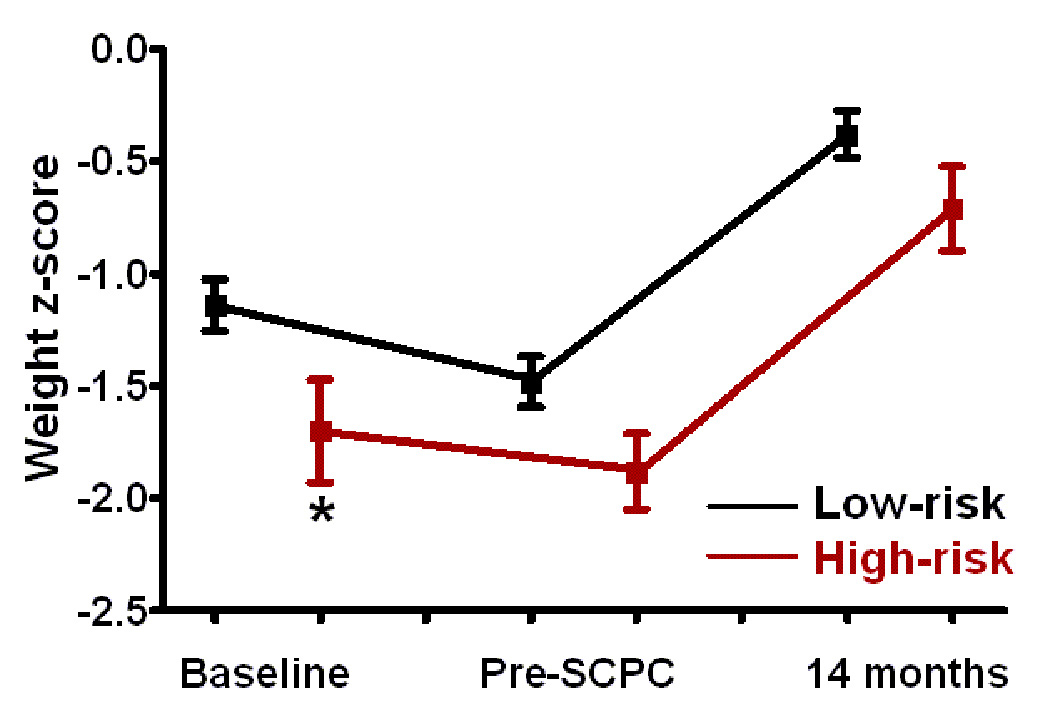
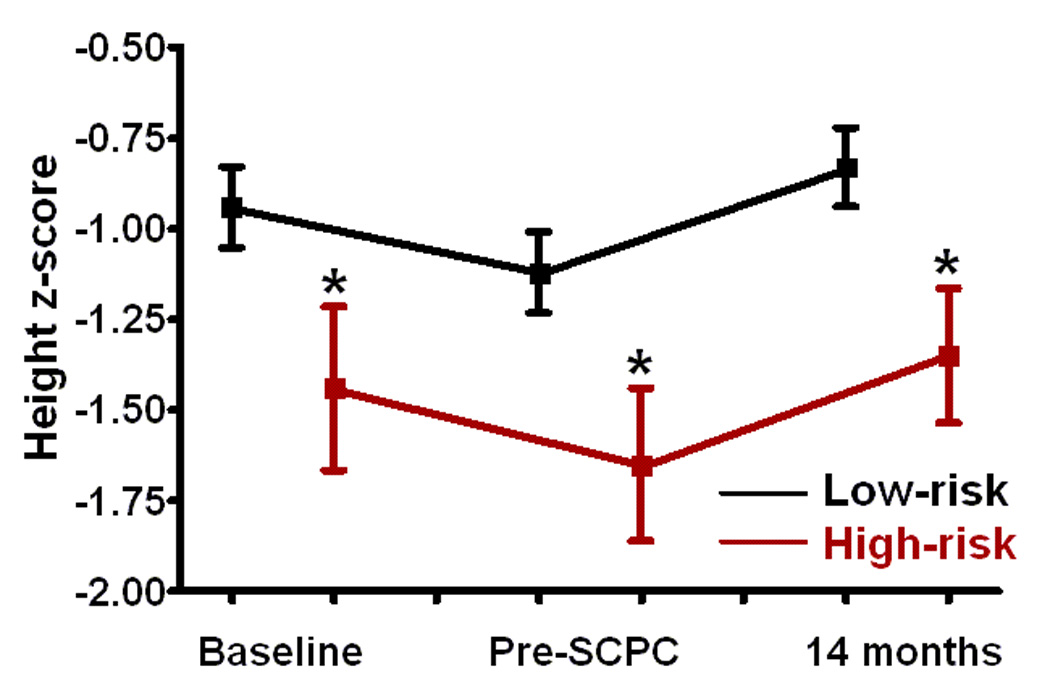
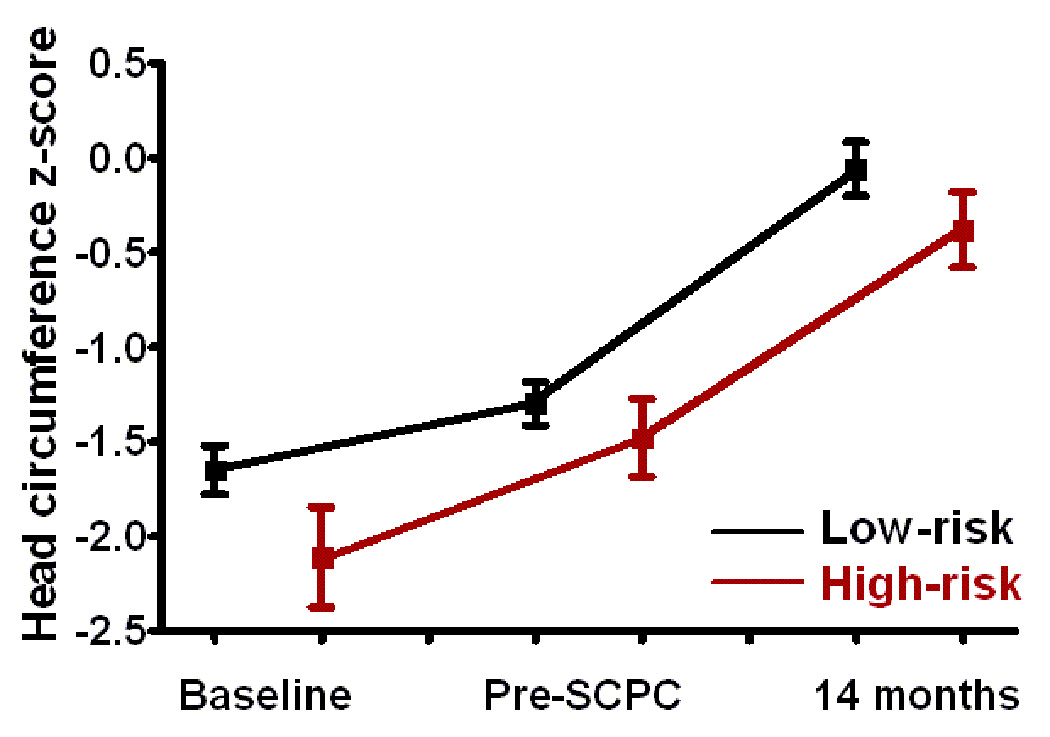
(a) Weight, (b) height, and (c) head circumference z-scores at baseline i.e. enrollment, pre-SCPC and final study visit by genotype; Low-risk (black, n=116); high-risk (red, n=38). The offset at the different time points between the genotype groups is for better visualization of standard errors. Squares = mean value; whiskers = standard error. * p<0.05 from low risk group.
SCPC = Superior cavopulmonary connection
RAAS genotype and response to enalapril
The proportion of subjects assigned to enalapril was 47% in high-risk and 54% in low-risk group (p=0.57; mean dose, 0.31±0.13 mg/kg/day). There were no differences in 14-month outcomes of ventricular mass, volume, function, BNP concentration, or Ross HF class between the enalapril and placebo groups regardless of genotype. However, high-risk patients treated with enalapril had lower weight, height and head circumference z-cores pre-SCPC compared to those assigned to placebo (interaction p<0.05; data not shown). After adjusting for baseline z-scores, this difference remained significant for height with a lower mean height z-score at both time-points in high-risk patients on enalapril (p<0.05) (Figures 4a, 4b; interaction p<0.05).
Figure 4.
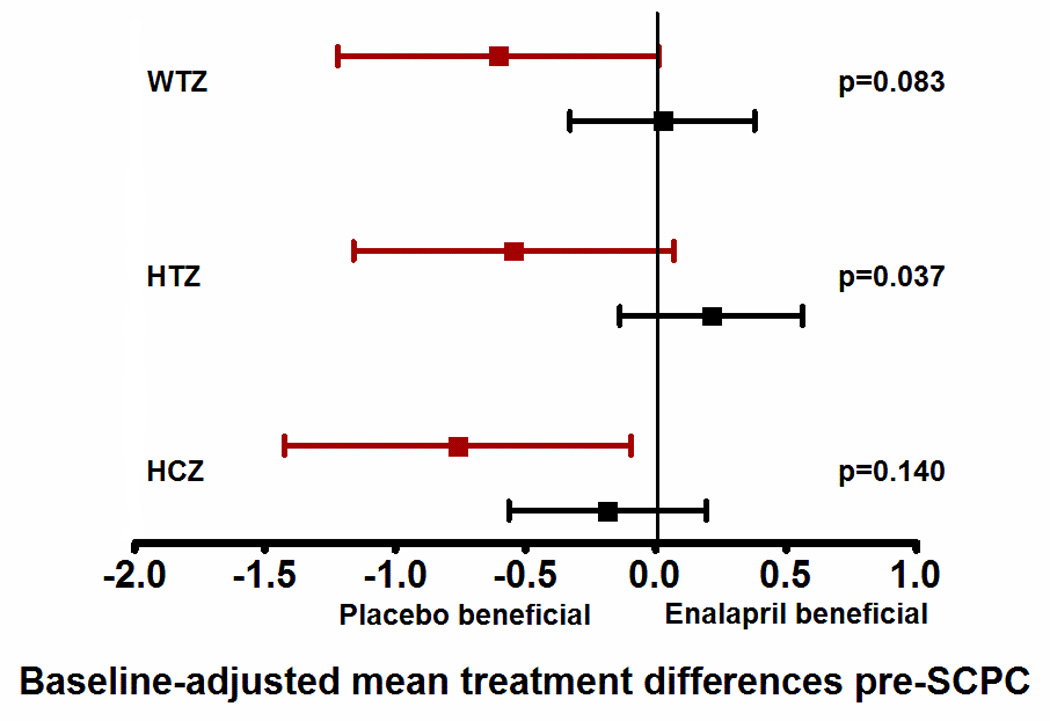
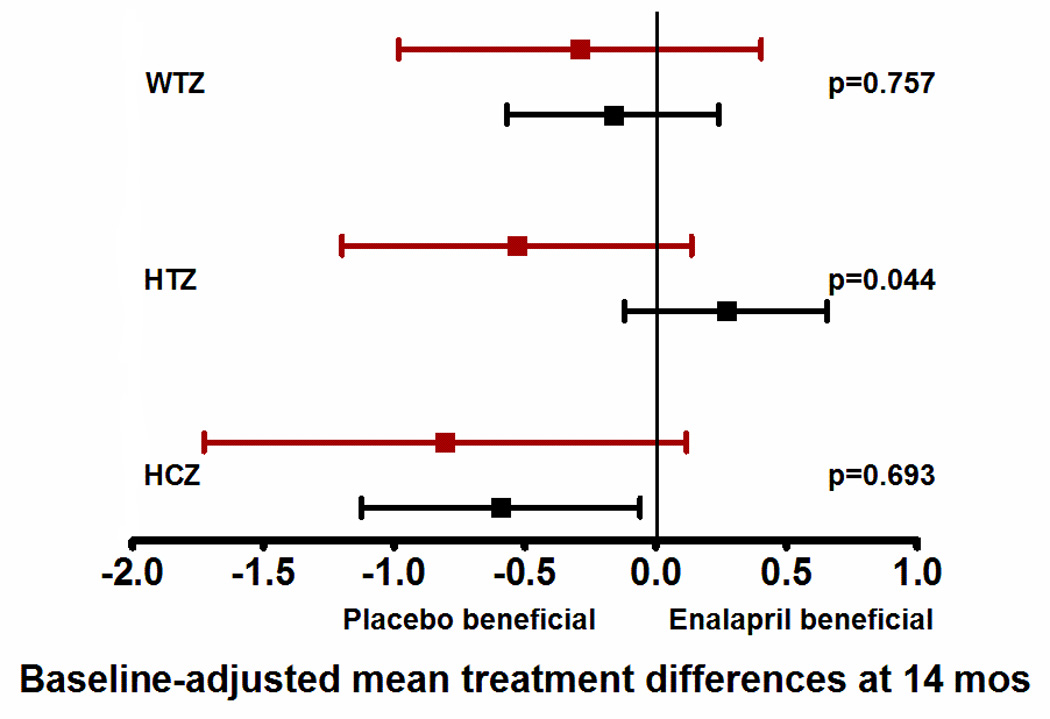
This figure shows the differences in growth z-scores between the enalapril and placebo treated patients in the two risk groups (high-risk shown in red, and low-risk shown in black) at two time points - at pre-SCPC and final study visits. Data are shown as mean and 95% confidence intervals, adjusted for baseline z-scores. Mean values to the left of zero indicate lower z-scores in enalapril-treated patients i.e. placebo-beneficial; mean values to the right of zero indicate higher z-scores in the enalapril-treated patients i.e enalapril-beneficial. The interaction p values represent the differences in treatment effect between the high and low risk groups. There was no treatment effect on weight, height or head circumference in the low-risk group (black) at pre-SCPC (panel a), and at 14 months (panel b). However, high-risk patients (red) receiving enalapril had lower height z-scores at pre-SCPC, and at 14 months compared to placebo group. n=63, enalapril-treated low risk; n=53, placebo-treated low risk; n=18, enalapril-treated high risk; n=20, placebo-treated high risk. *p<0.05 enalapril vs placebo.
SCPC = Superior cavopulmonary connection, WTZ = weight z-score; HTZ = height z-score; HCZ = head circumference z-score.
RAAS genotype and renal function
Table 4 shows the renal characteristics of enrolled subjects. eGFR at enrollment was 53±15 ml/min/m2 in high-risk and 54±16 ml/min/m2 in low-risk patients (p=0.75). eGFR increased during study follow-up to 98±36 ml/min/m2 (77% of normal) in high-risk, and 105±26 ml/min/m2 (83% of normal) in low-risk patients at 14 months35. This increase from pre-SCPC to 14 months was significant for the low-risk group (change score, 18±33 ml/min/1.73m2, p<0.001) but not for the high-risk group (change score, 12±40 ml/min/1.73m2, p=0.13) (p=0.47 between groups) (Figure 5a). The change in eGFR was independent of enalapril (interaction p=0.71; Figure 5b; Table 4).
Table 4.
Renal function
| eGFR (ml/min/1.73 m2) |
High-risk/ Enalapril |
High-risk/ Placebo |
p* | Low-risk/ Enalapril |
Low-risk/ Placebo |
p* |
|---|---|---|---|---|---|---|
| N† | 18 | 20 | 63 | 53 | ||
| Enrollment | 55±13 | 51±156 | 0.40 | 51±14 | 57±18 | 0.03 |
| Pre-SCPC | 77±24 | 90±31 | 0.19 | 85±26 | 90±22 | 0.39 |
| 14 months | 91±41 | 104±32 | 0.40 | 107±21 | 104±31 | 0.62 |
t-test p-value
N represents sample size at enrollment. N at 14 months was 11 (high-risk/enalapril), 15 (high-risk/placebo), 48 (low-risk/enalapril), and 37 (low-risk/placebo groups).
eGFR = estimated glomerular filtration rate; SCPC = superior cavopulmonary connection
Figure 5.
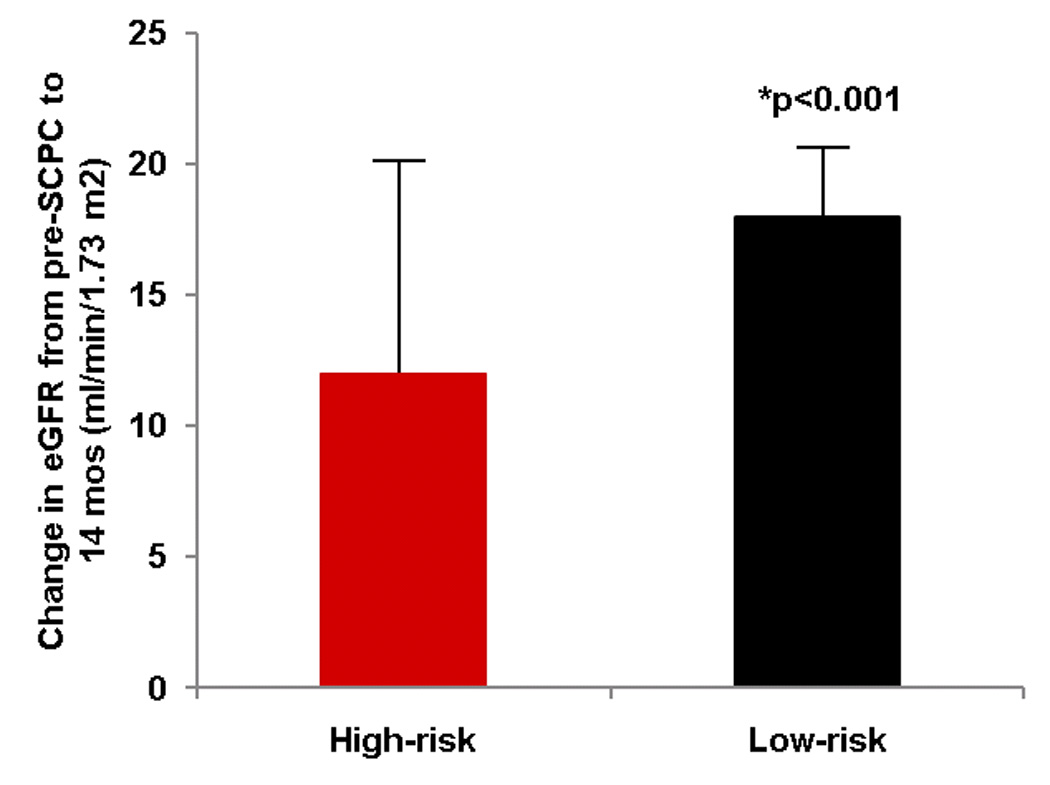
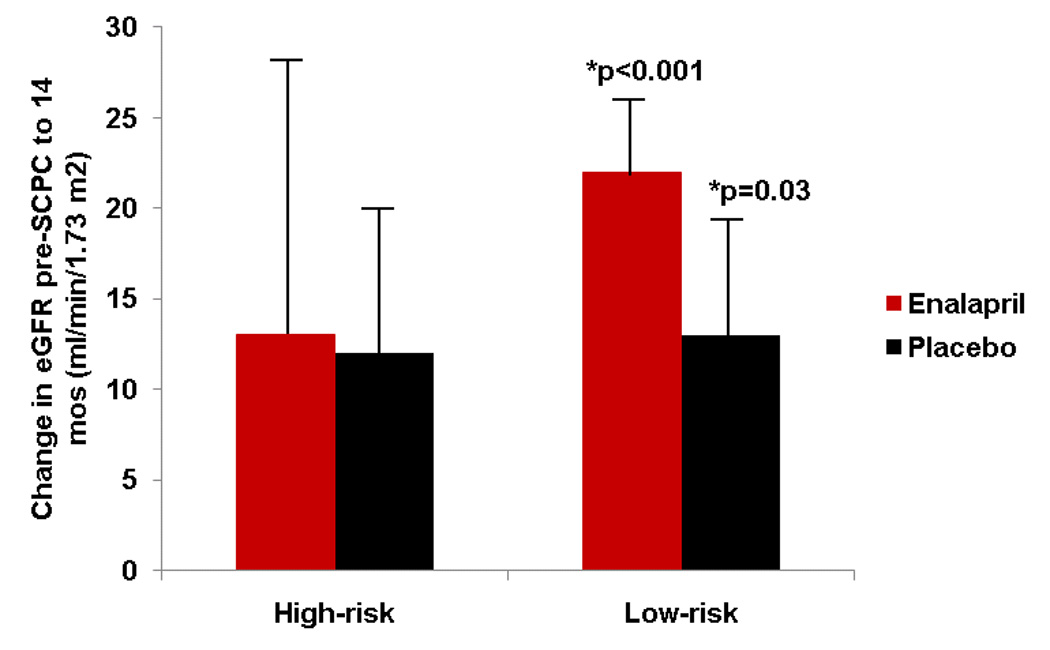
(a) Mean change ± standard error in estimated glomerular filtration rate (eGFR) (14 months minus pre-SCPC). eGFR increased in the low-risk (black, n=85) but not in the high-risk group (red, n=26); *p<0.05 for change score from pre-SCPC. (b) The increase in eGFR in the low-risk group was independent of treatment with enalapril (red) versus placebo (black). High-risk/enalapril = 11; high-risk/placebo 15, low-risk/enalapril = 48; low risk/placebo = 37. *p<0.05 for change score from pre-SCPC.
SCPC = Superior cavopulmonary connection; eGFR = estimated glomerular filtration rate
DISCUSSION
Our study suggests that RAAS-upregulation genotypes in infants with single ventricles are associated with unfavorable remodeling and a deleterious effect of enalapril on growth.
Patients with high-risk RAAS genotypes showed persistent elevation in ventricular mass and volume, less improvement in renal function and persistent impaired somatic growth despite volume unloading surgery. Growth impairment was exacerbated by enalapril treatment in high-risk genotype patients. These findings have important clinical implications for genetic risk stratification and pharmacotherapy in this cohort.
The first important finding is that SCPC surgery was associated with a failure to decrease mass and volume in single ventricle patients with RAAS-upregulation genotypes. Multiple studies have reported an association of individual RAAS genotypes with cardiac hypertrophy although a recent meta-analysis of genome-wide association studies did not identify an association between SNPs in the RAAS pathway and cardiac phenotype.36 Our study was unique since we evaluated the compound effect of SNPs in multiple genes in the same pathway. Also, our study cohort consisted of single ventricle patients during their most vulnerable phase i.e. infancy, when the systemic ventricle is exposed to dynamic shifts in hemodynamic loading conditions which may enhance the influence of variations in RAAS signaling on cardiac remodeling. The association of a higher number of high-risk RAAS genotypes with an incremental increase in ventricular mass at 14 months suggests a gene dosage effect of multiple polymorphisms in the RAAS pathway. Importantly, while the RAAS genotypes have been associated with hypertrophic response to pressure and volume load, this is the first study to report an association of RAAS genotypes with a failure to achieve volume unloading and reverse remodeling following a volume unloading procedure16–18 The failure of effective unloading was not related to a difference in incidence of AVVR, additional sources of pulmonary blood flow, differences in BP or frequency of recurrent coarctation, between the two genotype groups. Also, there was no influence of ventricular morphology on the response of the ventricle to unloading in either risk group. The high-risk group however did fail to significantly increase eGFR post-SCPC. This finding with respect to renal function raises the intriguing possibility that the lack of remodeling in the high-risk cohort may be related in part to persistent volume load and/or elevated systemic vascular resistance due to renal insufficiency. These cardiorenal interactions require further investigation. Other possibilities include a direct pro-hypertrophic effect of tissue RAAS upregulation or to high myocardial oxygen demands in the high-risk group with a resulting failure to decrease stroke volume. High myocardial oxygen consumption has been previously reported in patients with high-risk RAAS genotypes, in particular the ACE DD genotype, especially during conditions with increased metabolic requirements 37, 32, 38, 39.
The second finding of the failure of enalapril to reduce ventricular mass in either risk group is not surprising. A recent study in a rat model of eccentric LV hypertrophy caused by volume overload reported that ACE inhibition did not induce reverse remodeling 40. Other studies have also shown the failure of ACE inhibition to reduce myocardial oxygen consumption in patients with congenital heart disease, and lack of efficacy of ACE inhibition in patients with complex congenital heart defects and volume overloaded ventricles40–42. Together these findings suggest that, unlike pressure-overload hypertrophy, ACE inhibition does not have an anti-hypertrophic effect in volume-overload hypertrophy. Surgical unloading appears to be more effective than conventional pharmacotherapy in promoting reverse remodeling in the volume-loaded ventricle at least in the low-risk genotype group.
The third important finding is that RAAS upregulation genotypes were associated with growth impairment including lower mean weight and height z-scores at enrollment, with persistent impairment in height at age 14 months. This impairment was most significant in the high-risk patients taking enalapril. The mechanism of height impairment in high-risk patients was not assessed in our study. However, other studies report that RAAS activation increases systemic vascular resistance and arterial afterload leading to ventricular diastolic dysfunction and fetal growth restriction 23, 43, 44. Insulin resistance as reported in newborns with an AGT M235T TT genotype is another potential mechanism for growth impairment24. Impaired energy efficiency and increased whole body oxygen consumption as reported in subjects with the ACE DD genotype also contributes to a lower anabolic response in high-risk genotype subjects 45. Together, these studies suggest that RAAS upregulation is associated with impaired physiologic adaptation to increased metabolic demands resulting in growth impairment. The mechanism for the adverse effect of enalapril on height was not assessed although we speculate that this may be related to a failure to increase cardiac output despite afterload reduction by enalapril in a single ventricle physiology. Overall, the failure to see a benefit of enalapril in this cohort compounded by the detrimental effect on growth in the high-risk group argues against the routine use of enalapril in the management of single ventricle lesions.
Limitations
Since this was primarily a pharmacogenetic study nested within a prospective clinical trial, a replication cohort was not available. The study may have been insufficiently powered to detect an association between individual high-risk genotypes and outcomes. Nonetheless significant associations were seen both with linear regression analysis as well with analysis dividing the cohort into two risk groups. This approach highlights the importance of assessing the compound effect of multiple SNPs in a pathway rather than separate analysis of SNPs in a single gene. Since the genetic study was biased towards patients who survived beyond the first few months of life, we were unable to assess the influence of RAAS genotypes on early mortality. Also, in light of the relatively short follow-up after the SCPC surgery, we were unable to assess if persistent ventricular hypertrophy in high-risk patients was associated with adverse outcomes as reported previously, or whether volume unloading surgery should be performed earlier in high-risk patients to achieve maximal benefit before progressive damage from RAAS activation 46, 47.
In conclusion, this is the first prospective pharmacogenetic analysis of ACE inhibition in a congenital heart disease population. We showed an important association of RAAS genotypes with cardiac and renal response to volume unloading surgery. The findings of our study may help in identifying infants with single ventricle lesions who are at risk for persistent elevation in ventricular mass and volume and growth impairment despite volume unloading surgery. The failure of a beneficial effect of enalapril argues for the need to develop alternative approaches that include newer pharmacotherapies and possibly earlier surgical interventions in the high risk cohort to prevent maladaptive ventricular remodeling.
Clinical Summary.
The Pediatric Heart Network conducted a pharmacogenetic study as part of a multicenter, randomized, controlled trial of enalapril versus placebo in single ventricle infants to assess if renin-angiotensin-aldosterone (RAAS)-upregulation genotypes influence the response to enalapril. This represents the first pharmacogenetic study of enalapril in a congenital heart disease population. 154 infants with single ventricle were genotyped and followed till 14 months of age. Patients with RAAS-upregulation genotypes had persistent increase in ventricular mass and volume despite volume unloading surgery i.e. superior cavopulmonary connection (SCPC). Enalapril did not decrease ventricular mass or volume in either genotype group. Patients with high-risk genotypes had lower weight and height at enrollment and the height impairment persisted in high-risk patients who were receiving enalapril while patients receiving placebo normalized their height by 14 months. The high-risk genotype group also showed mild but persistent renal dysfunction. In summary, patients with RAAS-upregulation genotypes failed to show reverse remodeling in response to volume unloading surgery, had persistent growth abnormalities especially with enalapril, and had persistent renal dysfunction. These patients may need earlier SCPC to facilitate reversal of ventricular dilation and hypertrophy before the remodeling becomes irreversible. Since neither enalapril nor surgery showed significant benefit in high-risk genotype patients, there is a need to develop newer therapies in at-risk patients.
ACKNOWLEDGEMENTS
We gratefully acknowledge the contributions of Patricia Lanzano for overall coordination of the Molecular Genetics Core and genomic DNA extraction and to Liyong Deng for performing genotyping for the study.
FUNDING SOURCES
Supported by U01 grants from the National Heart, Lung and Blood Institute (HL068269, HL068270, HL068279, HL068281, HL068285, HL068292, HL068290, HL068288, HL085057) and the FDA Office of Orphan Products Development. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI.
APPENDIX
National Heart, Lung, and Blood Institute: Gail Pearson, Victoria Pemberton, Mario Stylianou, Marsha Mathis
Network Chair: Lynn Mahony, University of Texas Southwestern Medical Center Data Coordinating Center: New England Research Institutes, Lynn Sleeper (PI), Steven Colan, Lisa Virzi, Lisa Wruck*, Victor Zak, David F. Teitel, Leslie Kalish*, Patty Connell* Core Clinical Site Investigators: Children's Hospital Boston, Jane W. Newburger (PI), Roger Breitbart, Jami Levine, Ellen McGrath, Carolyn Dunbar-Masterson; Children's Hospital of New York, Daphne Hsu* (Study Chair), William Hellenbrand (PI), Ashwin Prakash*, Seema Mital*, Darlene Servedio*; Children's Hospital of Philadelphia, Victoria L. Vetter (PI), Chitra Ravishankar, Sarah Tabbutt*, Meryl Cohen, Katherine Lee, Marisa Nolan, Stephanie Piacentino, Michelle Toms; Cincinnati Children's Medical Center, D. Woodrow Benson (PI), Catherine Dent Krawczeski, Lois Bogenschutz, Teresa Barnard, Steven Schwartz*, David Nelson; North Carolina Consortium: Duke University, East Carolina University, Wake Forest University, Page A. W. Anderson (PI) – deceased, Jennifer Li (PI), Wesley Covitz, Kari Crawford, Michael Hines, James Jaggers, Theodore Koutlas, Charlie Sang, Jr, Lori Jo Sutton, Mingfen Xu; Medical University of South Carolina, J. Philip Saul (PI), Andrew Atz, Girish Shirali, Eric M. Graham, Teresa Atz; Primary Children's Medical Center and the University of Utah, Salt Lake City, Utah, L. LuAnn Minich (PI), John A. Hawkins, Richard V. Williams, Linda M. Lambert, Marian E. Shearrow; Hospital for Sick Children, Toronto, Brian McCrindle (PI), Elizabeth Radojewski, Nancy Slater, Svetlana Khaikin, Susan McIntyre.
Auxiliary Sites: Children's Hospital of Wisconsin, Nancy Ghanayem, Kathy Mussatto, Michele Frommelt, Lisa Young-Borkowski; University of Michigan, Albert Rocchini, Laurie Rodgers Augustyniak
Echocardiography Core Laboratory: Children's Hospital Boston: Steven Colan, Renee Margossian
Genetics Core Laboratory: Children's Hospital of New York: Wendy Chung, Liyong Deng, Patricia Lanzano
Protocol Review Committee: Michael Artman, Chair; Judith Massicot-Fisher, Executive Secretary; Timothy Feltes, Julie Johnson, Thomas Klitzner, Jeffrey Krischer, G. Paul Matherne Data and Safety Monitoring Board: John Kugler, Chair; Rae-Ellen Kavey, Executive Secretary; David J. Driscoll, Mark Galantowicz, Sally A. Hunsberger, Thomas J. Knight, Holly Taylor, Catherine L. Webb
*no longer at the institution listed
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
None
REFERENCES
- 1.Steinberger EK, Ferencz C, Loffredo CA. Infants with single ventricle: A population-based epidemiological study. Teratology. 2002;65:106–115. doi: 10.1002/tera.10017. [DOI] [PubMed] [Google Scholar]
- 2.Allgood NL, Alejos J, Drinkwater DC, Laks H, Williams RG. Effectiveness of the bidirectional glenn shunt procedure for volume unloading in the single ventricle patient. Am J Cardiol. 1994;74:834–836. doi: 10.1016/0002-9149(94)90450-2. [DOI] [PubMed] [Google Scholar]
- 3.Gewillig MH, Lundstrom UR, Deanfield JE, Bull C, Franklin RC, Graham TP, Jr, Wyse RK. Impact of fontan operation on left ventricular size and contractility in tricuspid atresia. Circulation. 1990;81:118–127. doi: 10.1161/01.cir.81.1.118. [DOI] [PubMed] [Google Scholar]
- 4.Cheung YF, Penny DJ, Redington AN. Serial assessment of left ventricular diastolic function after fontan procedure. Heart. 2000;83:420–424. doi: 10.1136/heart.83.4.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gentles TL, Mayer JE, Jr, Gauvreau K, Newburger JW, Lock JE, Kupferschmid JP, Burnett J, Jonas RA, Castaneda AR, Wernovsky G. Fontan operation in five hundred consecutive patients: Factors influencing early and late outcome. J Thorac Cardiovasc Surg. 1997;114:376–391. doi: 10.1016/s0022-5223(97)70183-1. [DOI] [PubMed] [Google Scholar]
- 6.Seliem M, Muster AJ, Paul MH, Benson DW., Jr Relation between preoperative left ventricular muscle mass and outcome of the fontan procedure in patients with tricuspid atresia. J Am Coll Cardiol. 1989;14:750–755. doi: 10.1016/0735-1097(89)90121-6. [DOI] [PubMed] [Google Scholar]
- 7.Cohen MI, Bush DM, Perry RJ, Spray TJ, Moshang T, Wernovsky G, Vetter V. Somatic growth failure after the fontan operation. Cardiol Young. 2002;10:447–457. doi: 10.1017/s1047951100008118. [DOI] [PubMed] [Google Scholar]
- 8.Day RW, Denton DM, Jackson WD. Growth of children with a functionally single ventricle following palliation at moderately increased altitude. Cardiol Young. 2000;10:193–200. doi: 10.1017/s1047951100009100. [DOI] [PubMed] [Google Scholar]
- 9.Vogt KN, Manlhiot C, Van Arsdell G, Russell JL, Mital S, McCrindle BW. Somatic growth in children with single ventricle physiology impact of physiologic state. J Am Coll Cardiol. 2007;50:1876–1883. doi: 10.1016/j.jacc.2007.07.050. [DOI] [PubMed] [Google Scholar]
- 10.Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin ii-mediated cardiovascular and renal diseases. Pharmacol Rev. 2000;52:11–34. [PubMed] [Google Scholar]
- 11.Frenneaux M, Stewart RA, Newman CM, Hallidie-Smith KA. Enalapril for severe heart failure in infancy. Arch Dis Child. 1989;64:219–223. doi: 10.1136/adc.64.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mori Y, Nakazawa M, Tomimatsu H, Momma K. Long-term effect of angiotensin-converting enzyme inhibitor in volume overloaded heart during growth: A controlled pilot study. J Am Coll Cardiol. 2000;36:270–275. doi: 10.1016/s0735-1097(00)00673-2. [DOI] [PubMed] [Google Scholar]
- 13.Seguchi M, Nakazawa M, Momma K. Effect of enalapril on infants and children with congestive heart failure. Cardiol Young. 1992;2:14–19. [Google Scholar]
- 14.Shaw NJ, Wilson N, Dickinson DF. Captopril in heart failure secondary to a left to right shunt. Arc Dis Child. 1988;63:360–363. doi: 10.1136/adc.63.4.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu DT, Zak V, Mahony L, Sleeper LA, Atz A, Levine JC, Barker PC, Ravishankar C, McCrindle BW, Williams RV, Altmann K, Ghanayem NS, Margossian R, Chung WK, Border WL, Pearson GD, Stylianou MP, Mital S. Enalapril in infants with single ventricle: Results of a multicenter randomized trial. Circulation. 2010;122:333–340. doi: 10.1161/CIRCULATIONAHA.109.927988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diet F, Graf C, Mahnke N, Wassmer G, Predel HG, Palma-Hohmann I, Rost R, Bohm M. Ace and angiotensinogen gene genotypes and left ventricular mass in athletes. Eur J Clin Invest. 2001;31:836–842. doi: 10.1046/j.1365-2362.2001.00886.x. [DOI] [PubMed] [Google Scholar]
- 17.Kaufman BD, Auerbach S, Reddy S, Manlhiot C, Deng L, Prakash A, Printz BF, Gruber D, Papavassiliou DP, Hsu DT, Sehnert AJ, Chung WK, Mital S. Raas gene polymorphisms influence progression of pediatric hypertrophic cardiomyopathy. Hum Genet. 2007;122:515–523. doi: 10.1007/s00439-007-0429-9. [DOI] [PubMed] [Google Scholar]
- 18.Ortlepp JR, Vosberg HP, Reith S, Ohme F, Mahon NG, Schroder D, Klues HG, Hanrath P, McKenna WJ. Genetic polymorphisms in the renin-angiotensin-aldosterone system associated with expression of left ventricular hypertrophy in hypertrophic cardiomyopathy: A study of five polymorphic genes in a family with a disease causing mutation in the myosin binding protein c gene. Heart. 2002;87:270–275. doi: 10.1136/heart.87.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Semsarian C, Healey MJ, Fatkin D, Giewat M, Duffy C, Seidman CE, Seidman JG. A polymorphic modifier gene alters the hypertrophic response in a murine model of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:2055–2060. doi: 10.1006/jmcc.2001.1466. [DOI] [PubMed] [Google Scholar]
- 20.Hallberg P, Karlsson J, Kurland L, Lind L, Kahan T, Malmqvist K, Ohman KP, Nystrom F, Melhus H. The cyp2c9 genotype predicts the blood pressure response to irbesartan: Results from the swedish irbesartan left ventricular hypertrophy investigation vs atenolol (silvhia) trial. J Hypertens. 2002;20:2089–2093. doi: 10.1097/00004872-200210000-00030. [DOI] [PubMed] [Google Scholar]
- 21.Kurland L, Melhus H, Karlsson J, Kahan T, Malmqvist K, Ohman KP, Nystrom F, Hagg A, Lind L, Trial SILVHIA. Angiotensin converting enzyme gene polymorphism predicts blood pressure response to angiotensin ii receptor type 1 antagonist treatment in hypertensive patients. J Hypertens. 2001;19:1783–1787. doi: 10.1097/00004872-200110000-00012. [DOI] [PubMed] [Google Scholar]
- 22.Kurland L, Melhus H, Karlsson J, Kahan T, Malmqvist K, Ohman P, Nystrom F, Hagg A, Lind L. Polymorphisms in the angiotensinogen and angiotensin ii type 1 receptor gene are related to change in left ventricular mass during antihypertensive treatment: Results from the swedish irbesartan left ventricular hypertrophy investigation versus atenolol (silvhia) trial. J Hypertens. 2002;20:657–663. doi: 10.1097/00004872-200204000-00023. [DOI] [PubMed] [Google Scholar]
- 23.Dotsch J. Renal and extrarenal mechanisms of perinatal programming after intrauterine growth restriction. Hypertens Res. 2009;32:238–241. doi: 10.1038/hr.2009.4. [DOI] [PubMed] [Google Scholar]
- 24.Schlemm L, Haumann HM, Ziegner M, Stirnberg B, Sohn A, Alter M, Pfab T, Kalache KD, Guthmann F, Hocher B. New evidence for the fetal insulin hypothesis: Fetal angiotensinogen m235t polymorphism is associated with birth weight and elevated fetal total glycated hemoglobin at birth. J Hypertens. 2010;28:732–739. doi: 10.1097/HJH.0b013e328336a090. [DOI] [PubMed] [Google Scholar]
- 25.Hsu DT, Mital S, Ravishankar C, Margossian R, Li JS, Sleeper LA, Williams RV, Levine JC, McCrindle BW, Atz AM, Servedio D, Mahony L Pediatric Heart Network I. Rationale and design of a trial of angiotensin-converting enzyme inhibition in infants with single ventricle. Am Heart J. 2009;157:37–45. doi: 10.1016/j.ahj.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perkins MJ, Van Driest SL, Ellsworth EG, Will ML, Gersh BJ, Ommen SR, Ackerman MJ. Gene-specific modifying effects of pro-lvh polymorphisms involving the renin-angiotensin-aldosterone system among 389 unrelated patients with hypertrophic cardiomyopathy. European Heart Journal. 2005;26:2457–2462. doi: 10.1093/eurheartj/ehi438. [DOI] [PubMed] [Google Scholar]
- 27.Auerbach SR, Manlhiot C, Reddy S, Kinnear C, Richmond ME, Gruber D, McCrindle BW, Deng L, Chen JM, Addonizio LJ, Chung WK, Mital S. Recipient genotype is a predictor of allograft cytokine expression and outcomes after pediatric cardiac transplantation. J Am Coll Cardiol. 2009;53:1909–1917. doi: 10.1016/j.jacc.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 28.Grummer-Strawn LM, Reinold C, Krebs NF. Centers for Disease Control and Prevention (CDC). Use of world health organization and cdc growth charts for children aged 0–59 months in the united states. MMWR Recomm Rep. 2010;59:1–15. [PubMed] [Google Scholar]
- 29.Schwartz GJ, Haycock GB, Edelmann CM, Jr, Spitzer A. A simple estimate of glomerular filtration rate in children derived from body length and plasma creatinine. Pediatrics. 1976;58:259–263. [PubMed] [Google Scholar]
- 30.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. CKD-EPI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margossian R, Schwartz ML, Prakash A, Wruck L, Colan SD, Atz AM, Bradley TJ, Fogel MA, Hurwitz LM, Marcus E, Powell AJ, Printz BF, Puchalski MD, Rychik J, Shirali G, Williams R, Yoo SJ, Geva T, Investigators PHN. Comparison of echocardiographic and cardiac magnetic resonance imaging measurements of functional single ventricular volumes, mass, and ejection fraction (from the pediatric heart network fontan cross-sectional study) Am J Cardiol. 2009;104:419–428. doi: 10.1016/j.amjcard.2009.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sluysmans T, Colan SD. Theoretical and empirical derivation of cardiovascular allometric relationships in children. J Appl Physiol. 2005;99:445–457. doi: 10.1152/japplphysiol.01144.2004. [DOI] [PubMed] [Google Scholar]
- 33.Gauderman WJ, Murcray C, Gilliland F, Conti D. Testing association between disease and multiple snps in a candidate gene. Genet Epidemiol. 2007;31:383–395. doi: 10.1002/gepi.20219. [DOI] [PubMed] [Google Scholar]
- 34.Erdfelder E, Faul F, Buchner A. Gpower: A general power analysis program. Behav Res Methods. 1996;28:1–11. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- 35.Heilbron DC, Holliday MA, Al-Dahwi A, Kogan B. Expressing glomerular filtration rate in children. Pediatr Nephrol. 1991;5:5–11. doi: 10.1007/BF00852829. [DOI] [PubMed] [Google Scholar]
- 36.Vasan RS, Glazer NL, Felix JF, Lieb W, Wild PS, Felix SB, Watzinger N, Larson MG, Smith NL, Dehghan A, Groβhennig A, Schillert A, Teumer A, Schmidt R, Kathiresan S, Lumley T, Aulchenko YS, König IR, Zeller T, Homuth G, Struchalin M, Aragam J, Bis JC, Rivadeneira F, Erdmann J, Schnabel RB, Dörr M, Zweiker R, Lind L, Rodeheffer RJ, Greiser KH, Levy D, Haritunians T, Deckers JW, Stritzke J, Lackner KJ, Völker U, Ingelsson E, Kullo I, Haerting J, O’Donnell CJ, Heckbert SR, Stricker BH, Ziegler A, Reffelmann T, Redfield MM, Werdan K, Mitchell GF, Rice K, Arnett DK, Hofman A, Gottdiener JS, Uitterlinden AG, Meitinger T, Blettner M, Friedrich N, Wang TJ, Psaty BM, van Duijn CM, Wichmann H-E, Munzel TF, Kroemer HK, Benjamin EJ, Rotter JI, Witteman JC, Schunkert H, Schmidt H, Völzke H, Blankenberg S. Genetic variants associated with cardiac structure and function. JAMA: The Journal of the American Medical Association. 2009;302:168–178. doi: 10.1001/jama.2009.978-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanazawa H, Hirata K, Yoshikawa J. Effects of captopril administration on pulmonary haemodynamics and tissue oxygenation during exercise in ace gene subtypes in patients with copd: A preliminary study. Thorax. 2003;58:629–631. doi: 10.1136/thorax.58.7.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu CK, Tsai CT, Chang YC, Luo JL, Wang YC, Hwang JJ, Lin JL, Tseng CD, Chiang F. Genetic polymorphisms of the angiotensin ii type 1 receptor gene and diastolic heart failure. J Hypertens. 2009;27:502–507. doi: 10.1097/hjh.0b013e32831fda3a. [DOI] [PubMed] [Google Scholar]
- 39.Henrion D, Benessiano J, Iglarz M, Philip I, Levy B. Genetic determinants of vascular reactivity. Curr Hypertens Rep. 2002;4:41–48. doi: 10.1007/s11906-002-0052-z. [DOI] [PubMed] [Google Scholar]
- 40.Ryan TD, Rothstein EC, Aban I, Tallaj JA, Husain A, Lucchesi PA, Dell'Italia L. Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J Am Coll Cardiol. 2007;49:811–821. doi: 10.1016/j.jacc.2006.06.083. [DOI] [PubMed] [Google Scholar]
- 41.Mital S, Loke KE, Chen JM, Mosca RS, Quaegebeur JM, Addonizio LJ, Hintze T. Mitochondrial respiratory abnormalities in patients with end-stage congenital heart disease. J Heart Lung Transplant. 2004;23:72–79. doi: 10.1016/s1053-2498(03)00095-0. [DOI] [PubMed] [Google Scholar]
- 42.Dore A, Houde C, Chan K-L, Ducharme A, Khairy P, Juneau M, Marcotte F, Mercier L-A. Angiotensin receptor blockade and exercise capacity in adults with systemic right ventricles: A multicenter, randomized, placebo-controlled clinical trial. Circulation. 2005;112:2411–2416. doi: 10.1161/CIRCULATIONAHA.105.543470. [DOI] [PubMed] [Google Scholar]
- 43.Jakoubek V, Bibova J, Herget J, Hampl V. Chronic hypoxia increases fetoplacental vascular resistance and vasoconstrictor reactivity in the rat. Am J Physiol Heart Circ Physiol. 2008;294:H1638–H1644. doi: 10.1152/ajpheart.01120.2007. [DOI] [PubMed] [Google Scholar]
- 44.Tsyvian PB, Markova TV, Mikhailova SV, Hop WCJ, Wladimiroff JW. Left ventricular isovolumic relaxation and renin-angiotensin system in the growth restricted fetus. Eur J Obstet Gynecol Reprod Biol. 2008;140:33–37. doi: 10.1016/j.ejogrb.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 45.Montgomery HE, Clarkson P, Dollery CM, Prasad K, Losi MA, Hemingway H, Statters D, Jubb M, Girvain M, Varnava A, World M, Deanfield J, Talmud P, McEwan JR, McKenna WJ, Humphries S. Association of angiotensin-converting enzyme gene i/d polymorphism with change in left ventricular mass in response to physical training. Circulation. 1997;96:741–747. doi: 10.1161/01.cir.96.3.741. [DOI] [PubMed] [Google Scholar]
- 46.Connor JA, Gauvreau K, Jenkins KJ. Factors associated with increased resource utilization for congenital heart disease. Pediatrics. 2005;116:689–695. doi: 10.1542/peds.2004-2071. [DOI] [PubMed] [Google Scholar]
- 47.Hehir DA, Dominguez TE, Ballweg JA, Ravishankar C, Marino BS, Bird GL, Nicolson SC, Spray TL, Gaynor JW, Tabbutt S. Risk factors for interstage death after stage 1 reconstruction of hypoplastic left heart syndrome and variants. J Thorac Cardiovasc Surg. 2008;136:94–99. doi: 10.1016/j.jtcvs.2007.12.012. [DOI] [PubMed] [Google Scholar]