

Abstract
In order to identify novel targets in pancreatic cancer cells, we utilized high-throughput RNAi (HT-RNAi) to identify genes that, when silenced, would decrease viability of pancreatic cancer cells. The HT-RNAi screen involved reverse transfecting the pancreatic cancer cell line BxPC3 with a siRNA library targeting 572 kinases. From replicate screens, approximately thirty-two kinases were designated as hits, of which twenty-two kinase targets were selected for confirmation and validation. One kinase identified as a hit from this screen was tyrosine kinase non-receptor 1 (TNK1), a kinase previously identified as having tumor suppressor-like properties in embryonic stem cells. Silencing of TNK1 with siRNA showed reduced proliferation in a panel of pancreatic cancer cell lines. Furthermore, we demonstrated that silencing of TNK1 led to increased apoptosis through a caspase-dependent pathway and that targeting TNK1 with siRNA can synergize with gemcitabine treatment. Despite previous reports that TNK1 affects Ras and NFκB signaling, we did not find similar correlations with these pathways in pancreatic cancer cells. Our results suggest that TNK1 in pancreatic cancer cells does not possess the same tumor suppressor properties seen in embryonic cells, but appears to be involved in growth and survival. The application of functional genomics using HT-RNAi screens has allowed us to identify TNK1 as a growth-associated kinase in pancreatic cancer cells.
Keywords: HT-RNAi, TNK1, gemcitabine, pancreatic cancer
Introduction
Pancreatic cancer is one of the most aggressive and lethal cancers known today, with a 5-year survival of only 4%. In 2009, pancreatic cancer was the fourth-leading cause of cancer-related deaths (1). Patients diagnosed with pancreatic cancer typically have a poor prognosis due to a lack of early symptoms, leading to metastatic disease at the time of diagnosis. The treatment options for pancreatic cancer include chemotherapy, surgery and radiation. The most preferred current therapeutic drug to treat pancreatic cancer is gemcitabine, yet the one-year survival of pancreatic cancer patients treated with gemcitabine is about 18%, representing a significant but modest advancement in the quality of life (2). Clearly, novel targets and therapeutic combinations are needed to more effectively treat patients afflicted with this deadly disease.
Previously, our laboratory has reported on the use of a high-throughput RNA interference (HT-RNAi) platform to identify potential sensitizing targets to gemcitabine in pancreatic cancer cells (3) and to cisplatin in ovarian cancer (4). This procedure utilized a library of small interfering RNA (siRNA) to identify functional mediators of the cytotoxic response to chemotherapeutic agents. This HT-RNAi screen is also capable of identifying targets which, when silenced, would cause a decrease in cancer cell viability in the absence of additional therapeutics as previously reported (5). The targets identified by these types of HT-RNAi screens are referred to as Achilles’ Heel (AH) targets. In the current study, we have utilized this HT-RNAi platform to identify novel AH targets in pancreatic cancer, using BxPC3 cells. One novel AH target identified from this screen was tyrosine non-receptor kinase 1 (TNK1).
TNK1 was first identified in human umbilical cells (6) and murine embryonic stem cells as a tumor suppressor that downregulates Ras activity (7). It was further determined that TNK1 knockout mice form spontaneous tumors (8) and TNK1 blocks NFκB activation to facilitate TNFα-induced apoptosis (9). These data collectively suggest that TNK1 functions as a tumor suppressor, at least in embryonic and stem cells. In contrast, a recently published study identified TNK1 as having oncogenic potential based upon a retroviral insertion mutagenesis screen (10). A separate study has also identified an activated version of TNK1 in Hodgkin’s Lymphoma (11). Here we identify TNK1 as a novel AH target in pancreatic cancer and present evidence that TNK1 functions to promote growth and survival in pancreatic cancer cells.
Materials and Methods
Cell Culture and Reagents
The human pancreatic cancer cell lines MiaPaCa-2, BxPC3, PANC-1, AsPC-1, Su.86.86, CAPAN-1, and CAPAN-2 were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). Cells were grown in RPMI-1640 supplemented with 10% FBS, 2 mM L-glutamine, 100 IU/ml penicillin G, and 100 μg/ml streptomycin. The immortalized human pancreatic ductal epithelial cell line HPDE6 was kindly provided by Dr. Ming-Sound Tsao (University of Toronto; Toronto, Ontario, Canada; (12, 13)). HPDE6 cells were maintained in keratinocyte serum-free medium supplemented with epidermal growth factor (0.2 ng/mL) and bovine pituitary extract (30 μg/mL). All media reagents were obtained from Invitrogen (Carlsbad, CA). The cell lines were routinely maintained at 37°C in a humidified 5% CO2 atmosphere and periodically screened for mycoplasma.
Cell line identities were verified by STR profiling (14) using the AmpFISTR Identifiler PCR amplification kit (Applied Biosystems, Foster City, CA). This method simultaneously amplifies 15 STR loci and Amelogenin in a single tube, using 5 dyes, 6-FAM™, JOE™, NED™, PET™, and LIZ™ which are then separated on a 3100 Genetic Analyzer (Applied Biosystems). GeneMapper ID v3.2. AmpFISTR control DNA and the AmpFISTR allelic ladder were run concurrently. Results were compared to published STR sequences from the ATCC. The STR profiling is repeated once a cell line has been passaged more than 6 months after previous STR profiling.
Gemcitabine hydrochloride (Eli Lilly; Indianapolis, IN) was obtained from the Mayo Clinic Pharmacy (Scottsdale, AZ) and stock solutions of 100 mM were prepared by dissolving gemcitabine in phosphate-buffered saline (PBS). Aliquots of gemcitabine were stored at −20°C until use. Short interfering RNAi targeting TNK1 and non-silencing control were obtained from Qiagen (Valencia, CA).
RNAi Screening
High-Throughput RNAi (HT-RNAi) was performed using the validated kinase siRNA library version 1 obtained from Qiagen. Stock siRNA was diluted in siRNA buffer (Qiagen) and 9.3 ng of siRNA was printed onto white Corning 384-well plates (Fisher Scientific; Pittsburgh, PA). Non-silencing and lethal siRNA (Qiagen) were included as negative and positive controls, respectively. Prior to actual RNAi screening, a transfection reagent test was performed by testing commercially available transfection reagents for optimal transfection efficiency of a panel of pancreatic cancer cell lines. SiLentFect reagent (BioRad; Hercules, CA) was selected for siRNA transfection in all assays (Supplemental Table S1). HT-RNAi was done using siRNA reverse transfection of cells as previously described (3). Briefly, diluted siLentFect reagent (BioRad) in OptiMEM (Invitrogen) was added to the wells and allowed to complex with the siRNA. BxPC3 cells were resuspended in growth media without antibiotics at a final concentration of 1000 cells/well. Cell viability was determined by the addition of Cell Titer Glo (Promega, Madison, WI) and relative luminescence units (RLU) were measured using an EnVision plate reader (Perkin-Elmer, Wellesley, MA). Raw RLU data was used to calculate viability relative to control wells. Decreased viability in the lethal positive control wells served as an indicator of successful transfection for each plate. The screening data was normalized using the standard z-score method by correcting the raw data for plate row variation, and then normalizing and pooling data from all assay plates. The assumption is that the majority of the siRNAs are non-hits and the null distribution is normal (15). The criteria for identification of potential hits were z-score values of less than −1.65, which corresponded to a p-value of 0.05, in at least three of the four screens of pancreatic cancer cells. This cutoff was chosen due to the relatively small size and focused nature of the screen. Validation of screening results with a panel of pancreatic cancer cell lines was done in a similar assay format.
Dose response assays
Cells were reverse transfected as described above in 384-well plates and incubated with siRNA (Qiagen) for 24 hours. Gemcitabine was added at a range of concentrations and cells were incubated for a further 72 hours. Cell viability was measured as described above. Drug-dose response curves were generated and IC50 calculated using Prism 5.0 (GraphPad Software; La Jolla, CA).
Apoptotic Activity Assay
Analysis of apoptotic activity was completed using a Caspase-Glo 3/7 Assay System (Promega). All reagents were added according to manufacturer’s instructions. Briefly, BxPC3 cells were reverse transfected with siRNA (Qiagen) on a 384-well plate at a density of 1000 cells/well. Caspase-Glo reagent was added at 24, 48, and 72 hours to lyse cells and permit caspase-induced cleavage of the substrate. Activity was determined by measuring luminescence output as described above.
Western Blot Analysis
Cells were transfected with 16 nM of TNK1 siRNA or non-silencing siRNAs in 6 well plates by reverse transfection. Cells were treated with siRNA for 96 hours and whole cell lysates were prepared using Complete Lysis-M reagent (Roche; Indianapolis, IN). Protein concentration was determined by BCA assay (Pierce; Rockford, IL) and lysates were resolved by SDS-PAGE on a 4-12% resolving gel. Proteins were transferred onto PVDF membranes. Antibodies for TNK1, PARP, GAPDH, p-MEK 1/2, and MEK 1/2 were purchased from Cell Signaling Technology (Danvers, MA). Mouse anti-β-tubulin was purchased from Sigma Aldrich (St. Louis, MO). Secondary HRP-conjugated anti-rabbit and anti-mouse antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc (West Grove, PA). Bound antibodies were detected using SuperSignal West Femto (Pierce) and imaged using an AlphaInnotech Imager.
Immunoprecipitation
Whole cell lysates were immunoprecipitated using bead-bound p-Tyr monoclonal antibody (Cell Signaling) according to manufacturer’s instructions. Protein was eluted from immunobeads, heat denatured, and loaded onto an SDS-PAGE gel. Protein levels were analyzed by western blotting as described above. The anti-TNK1 antibody was purchased from Abgent (San Diego, CA) and the anti-phospho-TNK1 (Y277) and anti-EGFR antibodies were purchased from Cell Signaling.
Quantitative Real-Time PCR
Cells were reverse-transfected with siRNA in 6-well plates and incubated for 24-72 hours. Total RNA was collected using a RNeasy MiniPrep Kit (Qiagen) and concentration was measured using a Nano Drop (Thermo Scientific; Wilmington, DE). cDNA was generated using an iScript cDNA synthesis Kit (Bio-Rad). Primers for TNK1 were purchased from Qiagen. Samples were run in triplicate on a 96-well PCR plate using an Opticon 2 (MJ Research, Waltham, MA). All samples were normalized to levels of GAPDH.
Results
HT-RNAi screening for kinases important in growth of pancreatic cancer cells
In order to identify genes that modulate viability of BxPC3 pancreatic cancer cells, we performed loss-of-function screening using high throughput RNAi. A robust HT-RNAi assay was developed that allowed for high efficiency siRNA transfection of cells by cationic lipids in 384-well plates. The HT-RNAi screen involved reverse-transfecting BxPC3 pancreatic cancer cells with validated library siRNA targeting 572 kinases with 2 siRNA sequences/kinase. Cell viability was assessed using a luminescence-based cell number assay and the data were normalized and analyzed as described in Materials and Methods. Two independent HT-RNAi screens were conducted in order to produce a biological replicate of the results. Data was normalized by z-score analysis and results are shown as the z-score for each kinase siRNA in each screen of the BxPC3 cells as well as previous screens using MiaPaCa-2 cells (Fig. 1A). Hits were designated as having z-scores lower than −1.65 for at least three out of the four assays on the pancreatic cell lines. These criteria identified 32 kinases as significant in growth of both BxPC3 and MiaPaCa-2 cells (Fig. 1B and Supplemental Table S2). Furthermore, comparison of these hits to z-scores from a HT-RNAi kinase screen done on the normal fibroblasts cell line GM05659 showed 22 kinase targets that did not overlap and thus seem to be specific for the pancreatic cancer cells (Table 1). Several of these kinases have previously been associated with growth and survival of pancreatic cancer cells. One kinase identified as a hit in both cell lines was TNK1, which was the focus of subsequent studies due to reports that it had both tumor suppressor and oncogenic properties.
FIGURE 1. High-Throughput siRNA screening of pancreatic cancer cell lines.

A, BxPC3 and MiaPaCa-2 pancreatic cancer cells were reverse transfected with siRNA from the validated kinase siRNA library v.1 (Qiagen) and incubated for 96 hours. Cell viability was determined by the addition of Cell Titer Glo. Each screen was repeated to obtain a biological replicate, yielding a total of four screens. Data was normalized using the standard z-score method by correcting the raw data for plate row variation, and then normalizing and pooling data from all assay plates. The criteria for identification of potential hits were z-score values of less than −1.65, which corresponded to a p-value of 0.05, in at least three of the four screens of pancreatic cancer cells. B, Comparison of the z-scores from the 32 hits from RNAi screening of kinases in pancreatic cancer cells to the normal fibroblast cell line GM05659.
Table 1. List of Achilles’ Heel targets identified by HT-RNAi screening.
A selection of genes that, when knocked down by RNAi, decrease the viability of pancreatic cancer cells.
| Symbol | Gene Name | References* |
|---|---|---|
| AKAP3 | A-kinase anchor protein 110 kDa | |
| AKAP9 | A-kinase anchor protein 350 kDa | |
| ALS2CR7 | cyclin-dependent kinase 15 | |
| CALM1 | calmodulin 1 (phosphorylase kinase, delta) | (16) |
| CDK5R2 | cyclin-dependent kinase 5, regulatory subunit 2 | (17, 18) |
| EPHA5 | ephrin type-A receptor 5 | |
| FLT4 | Fms-like tyrosine kinase 4 | (19) |
| GRK4 | G protein-coupled receptor kinase 4 | |
| HK1 | hexokinase 1 | (20) |
| MAP2K1IP1 | MAPK scaffold protein 1 | |
| MAPK11 | MAP kinase p38 beta | (21-24) |
| NEK3 | NIMA (never in mitosis gene a)-related kinase 3 | |
| PLK3 | polo-like kinase 3 (Drosophila) | |
| PRKCL1 | Protein-kinase C-related kinase 1 | |
| PRKY | protein kinase, Y-linked | |
| PTK9 | twinfilin, actin-binding protein, homolog 1 (Drosophila) | |
| PTK9L | twinfilin, actin-binding protein, homolog 2 (Drosophila) | |
| ROR2 | receptor tyrosine kinase-like orphan receptor 2 | (25) |
| STK10 | serine/threonine kinase 10, Lymphocyte-oriented kinase | (26) |
| TAOK3 | TAO kinase 3 | |
| TNK1 | tyrosine kinase, non-receptor, 1 | |
| TNK2 | tyrosine kinase, non-receptor, 2 |
A selection of references related to the gene target in pancreatic cancer.
Confirmation of gene silencing by TNK1 siRNA
To demonstrate the silencing efficiency of the siRNA targeting TNK1, MiaPaCa-2 and BxPC3 cells were transfected with TNK1 or non-silencing siRNA and incubated at 37°C for 72 hours. For these validation experiments, we utilized three different siRNA sequences of which two of these sequences were identical to the ones used in the initial. Total RNA was collected and cDNA was generated to determine levels of TNK1 mRNA by qRT-PCR. Additionally, cell lysates were analyzed by western blot using an anti-TNK1 antibody. Results using qRT-PCR show that all three TNK1 siRNA sequences tested were able to reduce the TNK1 message levels compared to non-silencing siRNA (Fig. 2A). Furthermore, silencing of TNK1 resulted in decreased TNK1 protein level as demonstrated by the western blot results (Fig. 2B). Silencing of TNK1 by these sequences were further demonstrated by qRT-PCR analysis in MiaPaCa-2, AsPC-1, Su.86.86 (Supplemental Fig. S1) .
FIGURE 2. Validation of TNK1 knockdown following siRNA treatment.
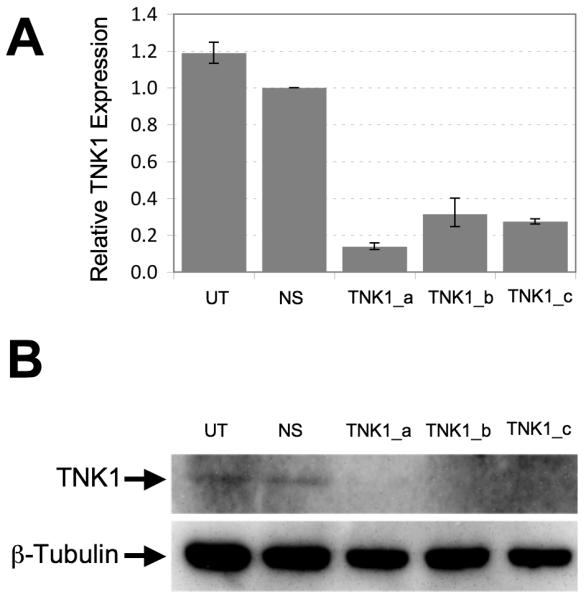
BxPC3 cells were incubated with one of three different siRNA sequences against TNK1, negative control non-silencing siRNA (NS) or left untreated (UT) and allowed to grow for 48 hours prior to RNA purification or 72 hours prior to protein lysate preparation. A, Silencing of TNK1 by siRNA decreased TNK1 expression by qRT-PCR analysis. All samples were normalized against GAPDH. B, Silencing of TNK1 by siRNA decreased TNK1 protein expression in BxPC3 pancreatic cancer cells as determined by Western blot analysis. Anti-β-tubulin is used as a loading control. Final images were cropped to highlight relevant bands.
TNK1 expression in pancreatic cancer cells
Since little is known about TNK1, we first sought to characterize its expression in pancreatic cancer cells. We selected nine common pancreatic cancer cell lines and examined the protein expression levels of TNK1 along with SMAD4, vimentin and E-cadherin by western blotting (Fig. 3A). We further quantified TNK1 protein expression levels using densitometry analysis (Fig. 3B). Only slight differences in expression exist across the panel of cell lines with MiaPaCa-2 exhibiting the lowest expression of TNK1 while HPAC exhibits the highest expression of TNK1. Of note, higher expression levels of TNK1 appear to correlate with mutation or low expression of the SMAD4 gene in this panel of cell lines (i.e. BxPC3, CAPAN-1, HPAC). Conversely, the cell lines in this panel that express lower levels of TNK1 have all been previously characterized as having a wild-type SMAD4 gene (i.e. MiaPaCa-2, Panc-1) (27, 28). Furthermore, cell lines that exhibit high vimentin expression (indicative of mesenchymal phenotype) tended to have lower expression of TNK1 whereas cell lines that have high E-cadherin expression (indicative of epithelial phenotype) appreared to have higher expression of TNK1.
FIGURE 3. Characterization of TNK1 expression in a panel of pancreatic cancer cell lines.

A, Whole cell lysates from nine different pancreatic cancer cell lines were separated by SDS-PAGE and analyzed by western blot analysis. Membranes were probed for expression of TNK1, β-actin (loading control), SMAD4, vimentin, and E-cadherin. Final images were cropped to highlight relevant bands. B, Densitometry analysis was completed to compare levels of TNK1 normalized against the loading control. C, Whole-cell lysates were prepared from TNK1 and SMAD4 siRNA-treated Panc-1 cells, which were then resolved by SDS-PAGE and analyzed for TNK1, SMAD4, and β-tubulin expression relative to untreated (UT) and non-silencing (NS) siRNA controls. D, Densitometry analysis was completed to compare protein levels normalized against the loading control.
To determine if TNK1 knockdown affected SMAD4 expression, we treated Panc-1 cells with TNK1 or non-silencing siRNA and analyzed SMAD4 expression by western blot (Fig. 3C). Conversely, we also treated Panc-1 cells with siRNA against SMAD4 and analyzed for TNK1 expression. A densitometry graph correlating to the western blot is shown for quantitative comparison (Figure 3D). These results show that even though there appears to be a correlation between SMAD4 and TNK1 expressions based on the results of figure 3A, TNK1 knockdown by siRNA does not directly affect SMAD4 expression nor does SMAD4 knockdown directly affect TNK1 expression.
Silencing of TNK1 inhibits growth and induces apoptosis in pancreatic cancer cells
To determine how TNK1 affects pancreatic cancer cell viability, five pancreatic cell lines (BxPC3, MiaPaCa-2, AsPC-1, Panc-1, and Su.86.86) were treated with three different siRNA sequences targeting TNK1. As shown in figures 2A and 2B, these sequences all demonstrate reliable knockdown of the TNK1 message. In BxPC3, MiaPaCa-2, and Su.86.86 cells (Fig. 4A), TNK1 knockdown resulted in a significant decrease in cell viability. This effect was less notable in Panc-1 and AsPC-1 cells, with TNK1 inhibition resulting in an approximately 20% decrease in viability. Treatment of BxPC3 cells with the three TNK1 siRNA resulted in increased apoptosis as demonstrated using an assay for caspase 3/7 activity (Fig. 4B). These results suggest an important role for TNK1 in pancreatic cancer cell growth.
FIGURE 4. TNK1 is an Achilles’ Heel target in pancreatic cancer cells.

A, BxPC3, MiaPaCa-2, AsPC-1, Panc-1, and Su.86.86 cells were treated with three different siRNA sequences targeting TNK1, a Non-silencing control siRNA and a positive control lethal siRNA (not shown). At 96 hours post transfection, cell viability was measured using Cell Titer Glo and expressed as percent of control. B, BxPC3 cells were incubated with TNK1 siRNA or non-silencing control for 24, 48, and 72 hours. Lethal control siRNA was also included (not shown). Apoptotic activity was measured using Caspase-Glo 3/7 reagent. Increased readout indicates an increase in Caspase 3/7 activity. Student’s t-test were completed for statistical analysis (relative to non-silencing control), *p<0.05. C, Whole cell lysates were immunoprecipitated using a bead-bound p-Tyr antibody and analyzed by western blot. A blot for EGFR (a commonly phosphorylated tyrosine kinase) was included as a positive control for the immunoprecipitation. Final images were cropped to highlight relevant bands.
To test for expression of activated TNK1, we first immunoprecipitated whole cell lysates using a bead-bound p-Tyr antibody. Blotting with a TNK1 antibody revealed high levels of phosphorylated TNK1 in BxPC3 and CAPAN-1 cells (Fig. 4C). These samples were also blotted with a phospho-TNK1 antibody specific for the Tyr 277 residue. We note that CAPAN-1, CAPAN-2, and HPAC cells all demonstrate high levels of phosphorylation at this particular residue. In contrast, we did not observe any phosphorylation of TNK1 in the normal pancreatic epithelial cell line HPDE6.
A role for TNK1 in cell response to gemcitabine
To determine the effect of TNK1 knockdown on gemcitabine response in pancreatic cancer cells, we treated BxPC3 cells with TNK1 siRNA or non-silencing siRNA and dosed the transfected cells with gemcitabine (Fig. 5A). The results showed a shift in the IC50 values of the TNK1 siRNA treated cells compared to control siRNA treated cells suggesting potentiation of gemcitabine activity. Using the active concentration of 8 nM, the effect of TNK1 silencing with gemcitabine was examined in a panel of five pancreatic cell lines (Fig. 5B). Results show varying effects of TNK1 silencing on gemcitabine response. Of note, the combination of gemcitabine with TNK1 siRNA resulted in lower cell viability when compared to gemcitabine or TNK1 siRNA alone in all of the cell lines tested.
FIGURE 5. Knockdown of TNK1 shows synergy with gemcitabine.

A, BxPC3 cells were reverse transfected with siRNA against TNK1 (non-silencing and lethal siRNA controls were included) and dosed with a range of gemcitabine concentrations. Cell viability was assessed at 96 hours using Cell Titer Glo. Data was analyzed using GraphPad Prism and the average normalized viability from four replicate wells with standard error was plotted for each treatment dose. The graph is a representative of three independent experiments. B, Cell viability data was further analyzed to directly compare the viability effects of non-silencing siRNA alone, 8 nM gemcitabine alone, or non-silencing siRNA plus 8 nM gemcitabine. Student’s t-test were completed for statistical analysis, *p<0.05 relative to NS, **p<0.05 relative to NS + gem. BxPC3 cells were treated with non-silencing or TNK1 siRNA. The graph is a representative of three independent experiments. C, Silencing of TNK1 potentiates TNFα induced apoptosis of pancreatic cancer cells. BxPC3 cells were treated with non-silencing (NS) or TNK1 siRNA. Samples were incubated with 20 ng/mL TNFα for 12 hours and whole cell lysates were analyzed for cleavage of PARP by western blotting. Protein expression of β-tubulin was completed to ensure appropriate loading of all lanes. Final images were cropped to highlight relevant bands. UT: untreated, NS: non-silencing.
The role of TNK1 in Kras and NFκB pathways
Because previously published studies found a correlation between TNK1 expression and suppression of Kras activity (7), we sought to determine if this relationship holds true in pancreatic cancer cells. Kras gain-of-function (GOF) mutations are often present in pancreatic cancer cells (29), so we examined the ratio of phospho-MEK1/2 (Ser217/221) to total MEK 1/2 (a downstream phosphorylation target of Kras). Following incubation with TNK1 siRNA, we noticed a slight increase in the ratio of p-MEK to total MEK in BxPC3 cells (Supplemental Fig. S2). However, this trend was determined not to be statistically significant, suggesting that TNK1 knockdown does not affect Kras activity in pancreatic cancer cells.
Azoitei et al. noted that overexpression of TNK1 resulted in a dramatic increase in PARP cleavage following treatment with TNFα (9). Therefore, we sought to determine if TNK1 knockdown results in a decrease in TNFα-induced cleavage of PARP. Our results indicate that TNK1 knockdown actually resulted in an increased cleavage of PARP following TNFα induction (Fig. 5C). Additionally, we note that treatment with TNK1 siRNA alone does not result in increased PARP cleavage (Supplemental Fig. S3). These data suggest that TNK1 levels are not closely tied to NFκB activation in pancreatic cancer cells.
Discussion
In this study, we used HT-RNAi screening to identify kinases whose silencing decreased growth of pancreatic cancer cells. Furthermore, we validated the kinase TNK1 as a novel pancreatic cancer target important in cell growth and survival. HT-RNAi screens with a kinase siRNA library on the BxPC3 and MiaPaCa-2 pancreatic cancer cell lines (Fig. 1) identified 32 kinases as being important in cell growth of pancreatic cancer cells (Table 1). Of these, 22 kinases appeared to be specific for the pancreatic cancer cells compared to a previous HT-RNAi kinase dataset from normal fibroblasts. Although several of the kinases have been previously associated with cancer (i.e. p38, CALM1, HK1, STK10), we focused on TNK1 in this study due to previous studies suggesting both tumor suppressor and oncogenic activity (8, 11).
TNK1 was identified as a hit in both the MiaPaCa-2 and BxPC3 siRNA screens and has not been previously associated with pancreatic cancer. Expression analysis of TNK1 in a panel of pancreatic cells showed only slight variations in levels of TNK1 expression with the highest expression seen in HPAC cells (Fig. 2C). While protein expression levels are relatively low overall, we did note the presence of phosphorylated TNK1 in several pancreatic cell lines (Fig. 2A and 2B). Silencing of TNK1 by siRNA reduced the cell viability of several pancreatic cell lines and induced apoptosis (Figs. 4A and B). Furthermore, TNK1 knockdown affected cell survival by potentiating gemcitabine-induced cytotoxicity (Figs. 5A and B).
Though all cell lines demonstrate a statistically significant decrease in viability when treated with TNK1 siRNA, Panc-1 and AsPC-1 were noticeably less affected than the BxPC3, MiaPaCa-2, and Su.86.86 cell lines. While the exact nature of this observation is unclear, we do note that cell lines exhibiting a mesenchymal phenotype tend to demonstrate lower expression of TNK1 than those with epithelial characteristics (Fig. 3A). These data combined with the conflicting studies regarding the role of TNK1 as a tumor suppressor/oncogene (8, 10) provide support the hypothesis that TNK1 exerts context-dependent roles within the cell. Further investigation is needed to determine what factors, if any, play a role in determining whether TNK1 acts as an oncogene or a tumor suppressor.
Previously published results have demonstrated that TNK1 in embryonic cells potentiates Kras activity (7). However, this effect was not noted in BxPC3 cells as silencing of TNK1 did not affect MEK 1/2 phosphorylation (Supplemental Fig. S2). Also, despite reports that TNK1 overexpression enhances PARP cleavage following TNFα induction (9), our data indicates that silencing of TNK1 in BxPC3 cells with siRNA increases PARP cleavage when compared to untreated and non-silencing siRNA controls (Fig. 5C) suggesting a role for TNK1 in apoptosis.
These data collectively suggest an alternative role for TNK1 in pancreatic cancer cells. Knowing that TNK1 knockdown results in a decrease in cell viability, it is unlikely that TNK1 acts as a tumor suppressor, as has been demonstrated in embryonic cells (7, 8). Additionally, this heretofore-unknown role for TNK1 in growth and survival in pancreatic cancer makes it a promising target in the ongoing quest to find novel cancer targets and treatments. Further experimentation is needed to determine if TNK1 plays an oncogenic role in other cancer types and data on expression levels in different tissue types is needed. Very few genes have been characterized as being a tumor suppressor under certain conditions and an oncogene in others (i.e. TGFβ). The addition of TNK1 to this very short list both complicates the view of how cancer develops and presents additional avenues for treating cancer more effectively.
Supplementary Material
Acknowledgements
We wish to acknowledge Tanya Little, Kristen Bisanz, Clifford Whatcott, Haiyong Han, Holly Yin and Donald Chow for their help and support. This work was supported by NIH Project Program P01 CA109552.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA: a cancer journal for clinicians. 2009;59(4):225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Carmichael J, Fink U, Russell RC, et al. Phase II study of gemcitabine in patients with advanced pancreatic cancer. British journal of cancer. 1996;73(1):101–5. doi: 10.1038/bjc.1996.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azorsa DO, Gonzales IM, Basu GD, et al. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J Transl Med. 2009;7:43. doi: 10.1186/1479-5876-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arora S, Bisanz KM, Peralta LA, et al. RNAi screening of the kinome identifies modulators of cisplatin response in ovarian cancer cells. Gynecol Oncol. 2010;118(3):220–7. doi: 10.1016/j.ygyno.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Arora S, Gonzales IM, Hagelstrom RT, et al. RNAi phenotype profiling of kinases identifies potential therapeutic targets in Ewing’s sarcoma. Mol Cancer. 2010;9:218. doi: 10.1186/1476-4598-9-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoehn GT, Stokland T, Amin S, et al. Tnk1: a novel intracellular tyrosine kinase gene isolated from human umbilical cord blood CD34+/Lin−/CD38− stem/progenitor cells. Oncogene. 1996;12(4):903–13. [PubMed] [Google Scholar]
- 7.Hoare K, Hoare S, Smith OM, Kalmaz G, Small D, May W Stratford. Kos1, a nonreceptor tyrosine kinase that suppresses Ras signaling. Oncogene. 2003;22(23):3562–77. doi: 10.1038/sj.onc.1206480. [DOI] [PubMed] [Google Scholar]
- 8.Hoare S, Hoare K, Reinhard MK, Lee YJ, Oh SP, May WS., Jr. Tnk1/Kos1 knockout mice develop spontaneous tumors. Cancer Res. 2008;68(21):8723–32. doi: 10.1158/0008-5472.CAN-08-1467. [DOI] [PubMed] [Google Scholar]
- 9.Azoitei N, Brey A, Busch T, Fulda S, Adler G, Seufferlein T. Thirty-eight-negative kinase 1 (TNK1) facilitates TNFalpha-induced apoptosis by blocking NF-kappaB activation. Oncogene. 2007;26(45):6536–45.l. doi: 10.1038/sj.onc.1210476. [DOI] [PubMed] [Google Scholar]
- 10.Lierman E, Van Miegroet H, Beullens E, Cools J. Identification of protein tyrosine kinases with oncogenic potential using a retroviral insertion mutagenesis screen. Haematologica. 2009;94(10):1440–4. doi: 10.3324/haematol.2009.007328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu TL, Cherry J, Tucker M, Wu J, Reeves C, Polakiewicz RD. Identification of activated Tnk1 kinase in Hodgkin’s lymphoma. Leukemia. 2010 doi: 10.1038/leu.2009.293. [DOI] [PubMed] [Google Scholar]
- 12.Liu N, Furukawa T, Kobari M, Tsao MS. Comparative phenotypic studies of duct epithelial cell lines derived from normal human pancreas and pancreatic carcinoma. Am J Pathol. 1998;153(1):263–9. doi: 10.1016/S0002-9440(10)65567-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouyang H, Mou L, Luk C, et al. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am J Pathol. 2000;157(5):1623–31. doi: 10.1016/S0002-9440(10)64800-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins PJ, Hennessy LK, Leibelt CS, Roby RK, Reeder DJ, Foxall PA. Developmental validation of a single-tube amplification of the 13 CODIS STR loci, D2S1338, D19S433, and amelogenin: the AmpFlSTR Identifiler PCR Amplification Kit. J Forensic Sci. 2004;49(6):1265–77. [PubMed] [Google Scholar]
- 15.Birmingham A, Selfors LM, Forster T, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods. 2009;6(8):569–75. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song D, Chaerkady R, Tan AC, et al. Antitumor activity and molecular effects of the novel heat shock protein 90 inhibitor, IPI-504, in pancreatic cancer. Mol Cancer Ther. 2008;7(10):3275–84. doi: 10.1158/1535-7163.MCT-08-0508. [DOI] [PubMed] [Google Scholar]
- 17.Rosales JL, Lee KY. Extraneuronal roles of cyclin-dependent kinase 5. Bioessays. 2006;28(10):1023–34. doi: 10.1002/bies.20473. [DOI] [PubMed] [Google Scholar]
- 18.Lee CS, Charalambous D. Immunohistochemical localisation of the c-fos oncoprotein in pancreatic cancers. Zentralbl Pathol. 1994;140(3):271–5. [PubMed] [Google Scholar]
- 19.Schneider M, Buchler P, Giese N, et al. Role of lymphangiogenesis and lymphangiogenic factors during pancreatic cancer progression and lymphatic spread. Int J Oncol. 2006;28(4):883–90. [PubMed] [Google Scholar]
- 20.von Forstner C, Egberts JH, Ammerpohl O, et al. Gene expression patterns and tumor uptake of 18F-FDG, 18F-FLT, and 18F-FEC in PET/MRI of an orthotopic mouse xenotransplantation model of pancreatic cancer. J Nucl Med. 2008;49(8):1362–70. doi: 10.2967/jnumed.107.050021. [DOI] [PubMed] [Google Scholar]
- 21.Koizumi K, Tanno S, Nakano Y, et al. Activation of p38 mitogen-activated protein kinase is necessary for gemcitabine-induced cytotoxicity in human pancreatic cancer cells. Anticancer Res. 2005;25(5):3347–53. [PubMed] [Google Scholar]
- 22.Lee KH, Hyun MS, Kim JR. Growth factor-dependent activation of the MAPK pathway in human pancreatic cancer: MEK/ERK and p38 MAP kinase interaction in uPA synthesis. Clin Exp Metastasis. 2003;20(6):499–505. doi: 10.1023/a:1025824816021. [DOI] [PubMed] [Google Scholar]
- 23.Adachi S, Yasuda I, Nakashima M, et al. HSP90 inhibitors induce desensitization of EGF receptor via p38 MAPK-mediated phosphorylation at Ser1046/1047 in human pancreatic cancer cells. Oncol Rep. 2010;23(6):1709–14. doi: 10.3892/or_00000815. [DOI] [PubMed] [Google Scholar]
- 24.Sawai H, Funahashi H, Okada Y, et al. Interleukin-1alpha enhances IL-8 secretion through p38 mitogen-activated protein kinase and reactive oxygen species signaling in human pancreatic cancer cells. Med Sci Monit. 2005;11(10):BR343–50. [PubMed] [Google Scholar]
- 25.Katoh M. Transcriptional mechanisms of WNT5A based on NF-kappaB, Hedgehog, TGFbeta, and Notch signaling cascades. Int J Mol Med. 2009;23(6):763–9. doi: 10.3892/ijmm_00000190. [DOI] [PubMed] [Google Scholar]
- 26.Schreiner B, Baur DM, Fingerle AA, et al. Pattern of secondary genomic changes in pancreatic tumors of Tgf alpha/Trp53+/- transgenic mice. Genes Chromosomes Cancer. 2003;38(3):240–8. doi: 10.1002/gcc.10285. [DOI] [PubMed] [Google Scholar]
- 27.Moore PS, Sipos B, Orlandini S, et al. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 2001;439(6):798–802. doi: 10.1007/s004280100474. [DOI] [PubMed] [Google Scholar]
- 28.Sipos B, Moser S, Kalthoff H, Torok V, Lohr M, Kloppel G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: towards the establishment of an in vitro research platform. Virchows Arch. 2003;442(5):444–52. doi: 10.1007/s00428-003-0784-4. [DOI] [PubMed] [Google Scholar]
- 29.Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16(16):7773–82. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.