

Abstract
Background
Isoflurane and carbon dioxide (CO2) negatively modulate N-methyl-D-aspartate (NMDA) receptors, but via different mechanisms. Isoflurane is a competitive antagonist at the NMDA receptor glycine binding site, whereas CO2 inhibits NMDA receptor current through extracellular acidification. Isoflurane and CO2 exhibit additive minimum alveolar concentration effects in rats, but we hypothesized that they would not additively inhibit NMDA receptor currents in vitro because they act at different molecular sites.
Methods
NMDA receptors were expressed in frog oocytes and studied using two-electrode voltage clamp techniques. A glycine concentration-response for NMDA was measured in the presence and absence of CO2. Concentration-response curves for isoflurane, H+, CO2, and ketamine as a function of NMDA inhibition were measured, and a Hill equation was used to calculate the EC50 for each compound.
Results
Binary drug combinations containing ½ EC50 were additive if NMDA current inhibition was not statistically different from 50%. The ½ EC50 binary drug combinations decreased the percent baseline NMDA receptor current as follows (mean±SD, n=5–6 oocytes each): CO2+H+ (51±5%), CO2+isoflurane (54±5%), H++isoflurane (51±3%), CO2+ketamine (67±8%), H++ketamine (64±2%).
Conclusions
In contrast to our hypothesis, NMDA receptor inhibition by CO2 and isoflurane is additive. Possibly, CO2 acidification modulates a pH-sensitive loop on the NMDA receptor that in turn alters glycine binding affinity on the GluN1 subunit. However, ketamine plus either CO2 or H+ synergistically inhibits NMDA receptor currents. Drugs acting via different mechanisms can thus exhibit additive or synergistic receptor effects. Additivity may not robustly indicate commonality between molecular anesthetic mechanisms.
Introduction
Additive anesthetic interactions have been used to support a common mechanism of anesthetic action, although this inference may not apply when receptor occupancy at sites mediating immobility is much less than the mean effective concentration (EC50) for anesthetics at individual sites.1 In studies in vivo, inhaled anesthetic effects are generally additive, even when the combinations of drugs are postulated to exert their effects via different cell mechanisms.2,3 In contrast, injectable anesthetics in combination with inhaled anesthetics often produce synergistic effects. 4,5
These findings reflect in vitro responses of many anesthetic-sensitive ion channels. Mixtures of halothane and isoflurane produce additive responses on gamma-aminobutyric acid type A (GABAA) and glycine receptors expressed in oocytes.6 Despite marked differences in N-methyl-D-aspartate (NMDA) receptor effects at a minimum alveolar concentration, the volatile anesthetics benzene and isoflurane exhibit additive effects on expressed NMDA receptor currents.6 Nevertheless, the injectable anesthetic ketamine combined with either isoflurane, sevoflurane, or desflurane synergistically inhibits NMDA receptor currents.7
At issue is whether anesthetics acting through different mechanisms must show synergy 8, or conversely, whether it is possible to infer a mechanism of anesthetic action based on an additive interaction. To address this question in vitro, it is essential to study drugs with known effects on an anesthetic-sensitive ion channel that is likely relevant to anesthetic-mediated immobility, such as the NMDA receptor.9 Carbon dioxide (CO2) and hydrogen ions (H+) both negatively modulate NMDA receptors through extracellular acidification.10 Isoflurane does not change solution pH; it competitively inhibits NMDA receptor function at the glycine binding site.11 Ketamine antagonizes NMDA receptors by both open channel block and by closed channel block associated with decreased opening frequency.12 Ketamine must access at least one of these binding sites via the cell membrane,12 and subunit mutations that diminish NMDA receptor sensitivity to volatile anesthetics do not affect responses to ketamine.13 Thus CO2 and H+, isoflurane, and ketamine offer three pharmacologic tools to inhibit NMDA receptors through three different molecular mechanisms. We hypothesized that drug pairs acting via identical mechanisms, such as CO2 + protons, would inhibit NMDA receptor currents additively, and that drugs pairs acting via different molecular sites, such as CO2 + ketamine or CO2 + isoflurane, would inhibit NMDA receptor currents synergistically.
The first study aim was to determine whether anesthetics that inhibit NMDA receptors differently always synergistically inhibit NMDA currents in combination. CO2 and H+ would serve as a positive control for an additive interaction. Ketamine and either CO2 or H+ would serve as a positive control for a synergistic interaction. The inhaled anesthetics CO2 and isoflurane, having different molecular mechanisms, were hypothesized to inhibit NMDA receptors synergistically. As a second study aim, the effects of CO2 on the glutamate and glycine binding sites of NMDA receptors would be determined and compared to known isoflurane interactions at these sites.
Methods
Oocyte Preparation and Receptor Expression
An ovary from a tricaine-anesthetized adult female Xenopus laevis frog was removed surgically according to a protocol approved by the Animal Care and Use Committee at the University of California, Davis. After defolliculation in a 0.2% Type I collagenase solution (Worthington Biochemical, Lakewood, NJ), oocytes were stored in a filtered modified Barth’s solution composed of 88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 20 mM HEPES, 0.82 mM MgSO4, 0.33 mM Ca(NO3)2, 0.41 mM CaCl2, 5 mM sodium pyruvate, gentamycin, penicillin, streptomycin, and corrected to pH=7.4. All salts and antibiotics were A.C.S. grade (Fisher Scientific, Pittsburgh, PA).
The human GluN1 NMDA subunit cloned in a pCDNA plasmid and the rat NMDA GluN2A subunit cloned in a pBSII KS+ plasmid were made available by a generous gift from Dr. Adron Harris (University of Texas, Austin). After plasmid linearization, cRNA was synthesized using a T7 transcription kit (mMessage mMachine, Ambion, Austin, TX). Equal proportions of each subunit cRNA were mixed and diluted with DEPC-treated water to 1 mg/mL total nucleic acid. A microinjection pipette (Nanoject II, Drummond Scientific, Broomall, PA) was used to deliver 9 or 18 nL of the transcript mixture or water (controls). Electrophysiology studies on oocytes were conducted 1 or 2 days later.
Voltage Clamp Protocol
Electrophysiology techniques are similar to published protocols.10,14–16 Oocytes were studied in a 250μL linear-flow perfusion chamber through which syringe pumps (Pump 33, Harvard Apparatus, Holliston, MA) delivered 1.5 ml/min of barium frog Ringer’s solution (BaFR) consisting of 115mM NaCl, 2.5mM KCl, 1.8mM BaCl2, 10mM HEPES, 0.1 mM EGTA, filtered, and corrected to pH=7.4. Syringes and tubing were made only of glass and PTFE to prevent plasticizer contamination.17 A −80mV membrane potential was maintained using a standard two-electrode voltage clamp technique (GeneClamp 500B, Axon Instruments, Union City, CA). After a 5 min baseline measurement during perfusion with BaFR, the perfusate was switched to an agonist solution (BaFREG) composed of BaFR plus 0.1 mM glutamate (E) plus 0.01 mM glycine (G) for 30 seconds followed by a 5 min washout with BaFR. This was repeated 3–4 times to verify constancy of the control agonist response (<10% change in peak current). BaFREG itself produced cell currents ≥ 98% of a maximal response.
The perfusate was then switched to the anesthetic test solution in BaFR for a 2 min washin, followed by a 30 sec exposure to the anesthetic test solution in BaFREG. The anesthetic was then washed out with BaFR for 5 min followed by another 30 sec exposure to BaFREG. Data were only used for analysis if NMDA receptor current responses after washout differed by ≤ 10% from responses before washin of the test solution. Data were recorded by commercially available data acquisition software (Chart, version 5, AD Instruments, Colorado Springs, CO).
Anesthetic Concentration-Response Measurement
All test solutions were prepared in 100ml gastight glass syringes using 50ml of either oxygenated BaFR or oxygenated BaFREG in each syringe; drug concentrations were equal in paired syringes. Carbon dioxide test solutions were prepared by using 99.999% pure CO2 (Matheson Trigas, Newark, CA), and PCO2 was measured using an automated and calibrated gas analyzer with temperature correction to 23°C (ABL5, Radiometer America, Westlake, OH). The pH test solutions were prepared by adding 1M HCl, and pH was measured using a calibrated meter (Accumet XL20, Fischer Scientific, Hampton, NH). Isoflurane solutions were prepared by adding to the solutions 50ml of a desired isoflurane-oxygen mixture collected downstream from 2 precision out-of-circuit isoflurane vaporizers, shaking vigorously for 30sec, expelling all of the headspace gas, and repeating headspace exchanges 9 additional times. The final isoflurane headspace concentration was measured after a 1.3 min retention time by a calibrated gas chromatograph (Clarus 500, Perkin Elmer, Waltham, MA) with a 0.25ml sample loop, 3m SF-96 packed column with Chromasorb WHP support (Perkin Elmer, Waltham, MA), 35ml/min hydrogen flow, 350 ml/min zero air flow, and 150°C oven temperature. Some isoflurane headspace concentrations were also measured using an infrared anesthetic gas analyzer (Andros 4800, LumaSense, Santa Clara, CA) that was calibrated using gas chromatography. A 1/4 atmosphere isoflurane concentration was prepared by direct anesthetic injection into fully oxygenated perfusates within gastight syringes. Solutions containing racemic ketamine (Sigma, St. Louis, MO) were prepared by serial dilution.
NMDA responses were measured as a percent of baseline agonist response. Full drug concentration-response curves for NMDA receptor currents were obtained for CO2, isoflurane, and ketamine. Hill equations were fit to the data using nonlinear regression (v.11, SPSS, Chicago, IL). Since CO2 effects on NMDA receptor current are mediated by pH 10, the responses to H+ were only measured next to the predicted half-maximal response to confirm the mean inhibitory concentration (IC50) using a simple linear regression model, and maximal inhibition efficacy was assumed.
To test for additivity, a solution containing each study compound was prepared at its respective IC50. Equal volumes of solutions pairs were then combined to yield one-half IC50 concentrations for each compound. The effect of binary drug combinations on NMDA receptor currents in voltage-clamped oocytes was measured, and a 50% current inhibition defined an additive drug interaction. Synergy was defined as an interaction yielding >50% inhibition with a P ≤ 0.05 using a t-test.
Glycine and Glutamate Effect Site Measurement
The nature of CO2 interactions at the NMDA receptor’s glycine and glutamate binding sites was determined by measuring whole cell NMDA receptor current responses in oocytes as a function of glycine concentration or glutamate concentration, respectively. Responses were measured in solutions containing either 0 or 17 mmHg of CO2 in perfusate solutions, as determined at 37°C using an automated blood gas analyzer. Glycine concentration-responses were compared to a maximal 0.1 mM glycine concentration response. Glutamate concentration-responses were compared to a maximal 1 mM glutamate concentration response. Hill equations were fit to the data using nonlinear regression, and the maximum current response (Emax), median effective agonist concentration (EC50), Hill coefficient (nH), and respective bootstrap estimates of standard errors18 were calculated. Differences between model parameters in the presence and absence of CO2 were deemed significant when P<0.05 using a t-test.
Competitive inhibition by CO2 at an agonist site was inferred if CO2 significantly increased the agonist EC50 but did not change Emax. Noncompetitive inhibition was inferred if CO2 significantly decreased the agonist Emax but did not change the agonist EC50. If CO2 significantly decreased Emax and increased the agonist EC50, mixed inhibition at the agonist site was inferred.19
Results
NMDA receptor current was inhibited by CO2, H+, isoflurane, and ketamine. A sample whole cell current response to anesthetic exposure is shown in Figure 1, and concentration-response curves for each test agent are shown in Figure 2.
Figure 1.
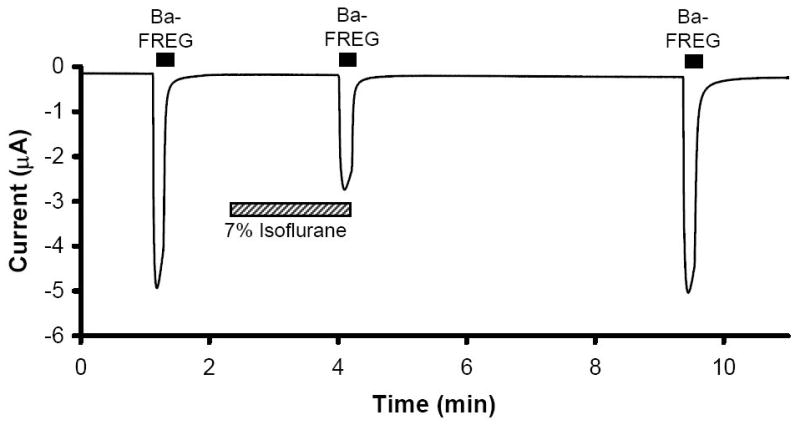
Sample physiograph tracing from a two-electrode voltage clamped oocyte expressing N-methyl-D-aspartate (NMDA) receptors. Currents were stimulated using barium frog Ringer’s solution (Ba-FR) containing glutamate (E) and glycine (G) Isoflurane reversibly inhibited NMDA receptor current.
Figure 2.
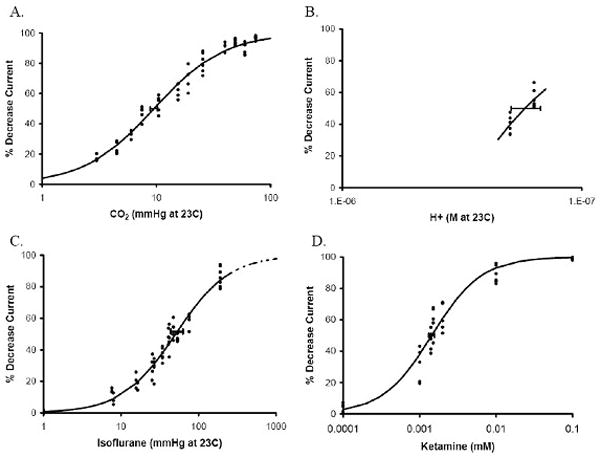
Concentration effects of CO2 (A), pH (B), isoflurane (C) and ketamine (D) on N-methyl-D-aspartate (NMDA) receptor currents measured using two-electrode voltage clamping. Horizontal error bars denote standard deviation at the IC50. Lines in panels A, C, and D are described by Hill equations (parameters given in Table 1). NMDA receptor responses to isoflurane partial pressures >190 mmHg are extrapolated (dotted line).
NMDA receptor responses to binary drug mixtures, with each applied at one-half of its respective IC50, are shown in Table 1. The combination of CO2 and H+ ions, each with a common mechanism of action on NMDA receptor function, inhibited whole cell current by 51%, consistent with a predicted additive interaction (Figure 3). In contrast, the combination of either CO2 or H+ ions with the injectable anesthetic ketamine, which inhibits NMDA receptor function through different mechanisms of action, inhibited whole cell current by 64% to 67%, consistent with a predicted synergistic interaction. However, combinations of either CO2 or H+ with the inhaled anesthetic isoflurane exhibited additive effects (Figure 3). In a separate experiment with 8 oocytes, application of ¼ EC50 CO2 plus ¾ EC50 isoflurane yielded 49 ± 2 % NMDA receptor inhibition, also indicative of additivity.
Table 1.
Maximum N-methyl-D-aspartate (NMDA) receptor inhibition (Imax), median inhibitory concentration (IC50) and Hill coefficient (nH) estimates for agonist response data at 23 °C described by a Hill equation using nonlinear regression analysis. The IC50 for hydrogren ion concentration (H+) was determined by simple linear regression for pH values close to the IC50; a Hill coefficient for pH effects was therefore not determined (n.d.).
| Antagonist | Imax (%) | IC50 | nH |
|---|---|---|---|
| CO2 (mmHg) | 100 ± 2 | 9.5 ± 0.4 | 1.41 ± 0.07 |
| H+ (nM) | 100 | 173 ± 8 | n.d. |
| Isoflurane (mmHg) | 100 ± 5 | 50.6 ± 4.7 | 1.25 ± 0.09 |
| Ketamine (μM) | 100 ± 2 | 1.44 ± 0.06 | 1.34 ± 0.14 |
Figure 3.
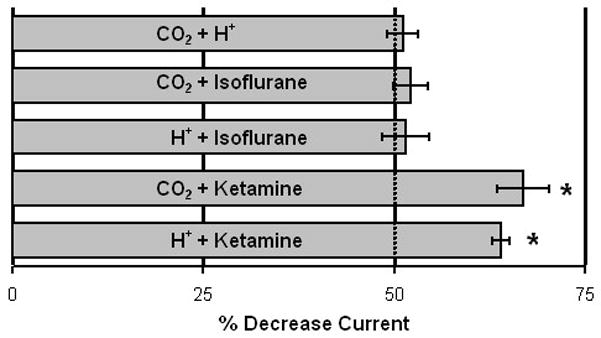
Effects of binary mixtures of N-methyl-D-aspartate (NMDA) receptor inhibitors, each administered at ½ of their respective IC50, on NMDA whole cell currents (n≥6 oocytes tested for each combination). Error bars denote standard errors. Pairs of inhibitors exhibiting additive effects produced a 50% decrease in NMDA receptor current. Ketamine with either CO2 or H+ caused NMDA receptor inhibition that was significantly more than 50%, consistent with a synergistic effect.
NMDA receptor concentration-response curves for the agonists glycine and glutamate, with and without an IC50 of CO2, are shown in Figure 4, and model parameters are given in Table 2. CO2 caused a decrease in the maximum glutamate efficacy (Emax) without changing the glutamate EC50, which is consistent with noncompetitive inhibition by CO2 at the NMDA receptor glutamate binding site. CO2 similarly caused a decrease in the glycine Emax, but it also increased the glycine EC50. This is consistent with mixed inhibition by CO2 at the NMDA receptor glycine binding site. Both glutamate and glycine sigmoid effect models had Hill coefficients >1, indicating cooperative agonist binding. However, only glycine exhibited a change in cooperative NMDA receptor binding with CO2 acidification.
Figure 4.
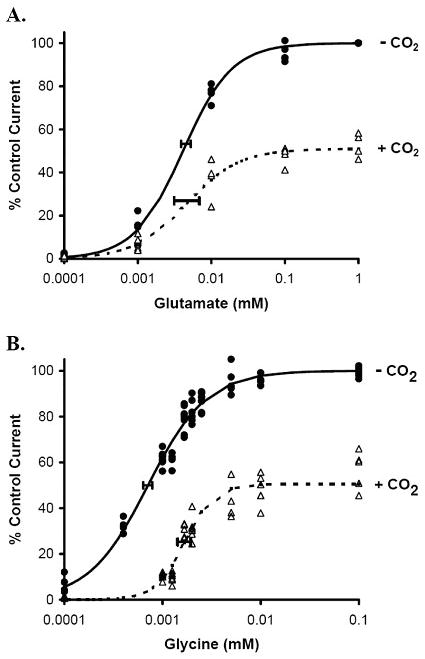
Glutamate (panel A) and glycine (panel B) agonist concentration effect on N-methyl-D-aspartate (NMDA) receptor currents measured using two-electrode voltage clamping in the presence (+CO2) or absence (−CO2) of 9 mmHg carbon dioxide at 23 °C. Lines are described by Hill equations (parameters given in Table 2), and horizontal error bars denote standard deviations at the EC50. In the glutamate concentration-response curve (panel A), CO2 only decreases the maximum control current without affecting the glutamate EC50, consistent with noncompetitive inhibition by CO2 of NMDA receptors at the glutamate binding site. In the glycine concentration-response curve (panel B), CO2 decreases the maximum control current and increases the glycine EC50, consistent with mixed inhibition by CO2 of NMDA receptors at the glycine binding site.
Table 2.
Maximum N-methyl-D-aspartate (NMDA) receptor effect (Emax), median effective concentration (EC50) and Hill coefficient (nH) estimates for NMDA agonist concentration-responses in the presence (9 mmHg) and absence (0 mmHg) of CO2. Statistically significant differences between CO2 treatment parameters for the same agonist (glutamate or glycine) are shaded and boldfaced. A decrease only in the glutamate concentration-response Emax value in the presence of CO2 is consistent with noncompetitive inhibition. A decrease in the glycine concentration-response Emax with an increase in EC50 in the presence of CO2 is consistent with mixed inhibition.
| Agonist | CO2 (23°C) | Emax (%) | EC50 (μM) | nH |
|---|---|---|---|---|
| Glutamate | 0 mmHg | 100 ± 1 | 4.12 ± 0.32 | 1.30 ± 0.09 |
| 9 mmHg | 51 ± 2 | 4.56 ± 0.90 | 1.20 ± 0.20 | |
| Glycine | 0 mmHg | 100 ± 1 | 0.71 ± 0.03 | 1.44 ± 0.07 |
| 9 mmHg | 51 ± 2 | 1.68 ± 0.09 | 2.90 ± 0.48 |
Discussion
CO2 and protons act via an identical mechanism to inhibit NMDA receptor currents in an additive manner.10 CO2 and ketamine 12 act via different mechanisms and combine to synergistically inhibit NMDA receptors. Isoflurane is a competitive antagonist at the glycine binding site of NMDA receptors 11, whereas CO2 is a noncompetitive antagonist at the glutamate binding site and a mixed antagonist at the glycine binding site of NMDA receptors. These inhaled anesthetics, isoflurane and CO2, nonetheless inhibit NMDA currents additively. Thus, the study hypothesis is shown to be incorrect. Two anesthetics can additively modulate receptor function despite known differences in their molecular mechanisms of action.
Site-directed mutagenesis studies in NMDA receptor subunits suggest that different amino acid regions are responsible for glycine binding and pH sensitivity. Based on three-dimensional projections of the NMDA receptor sequence onto the lysine-argenine-ornathine-binding protein structure, the glycine binding cleft is located towards the C-terminal half of the extracellular loop formed between membrane segments M3 and M420,21 on the GluN1 subunit and contains amino acids that confer volatile anesthetic sensitivity to NMDA receptors.13 This site is close to, but distinct from, the extracellular GluN1 regulatory region encoded by exon 5 (S191 to K211) that confers proton sensitivity to NMDA receptors 22, although mutations within the M3-M4 extracellular loop that are proximal to the putative glycine binding site 23,24 or that link it to the M3 segement 24 can modulate pH sensitivity. Amino acids remote from the glycine binding site may also affect the proton IC50, including residues within the M2 pore-forming region of the GluN1 subunit.25 Indeed, certain mutations in the GluN2A subunit, which does not bind glycine at all, can markedly diminish pH sensitivity.24
These molecular actions may explain why, despite their different mechanisms, isoflurane and CO2 combine additively to inhibit NMDA receptor function. Acidification, such as that caused by CO2, decreases the opening frequency of neuronal NMDA channels.26 As a result, there is reduced probability that a channel opens and conducts current at least once before either receptor desensitization or ligand dissociation occurs, and thus acidification decreases Emax for both glutamate and glycine (Figure 4 and Table 2). However, from these remote sites, protons likely also induce allosteric changes at the glycine binding site on GluN1 that decrease glycine affinity and increase the glycine EC50 (Table 2), just as competitive inhibition from isoflurane increases the glycine EC50 for NMDA receptors.11 Perhaps, if negative allosteric modulation is the more important mechanism by which CO2 or H+ inhibits NMDA receptors, then additivity between pH and competitive glycine site inhibitors can become possible.
The concentrations of NMDA receptor antagonist drugs used in this study are not all within clinically relevant ranges for several reasons. First, supra-pharmacologic concentrations of ketamine, isoflurane, and protons were used to obtain complete concentration-response curves in order to more accurately fit a sigmoid curve to the data. In contrast, CO2 was studied at subphysiologic concentrations. This is because oocyte perfusate solutions lack bicarbonate and have a relatively low buffering capacity. Hence, small increases in CO2 partial pressure cause greater perfusate acidification than would be expected in either blood or cerebrospinal fluid.10 Second, if clinically relevant concentrations result in low receptor fractional occupancy, it may be possible to observe additive responses even with two drugs acting via two different targets.1 By testing drug interactions closer to their respective IC50 values, low NMDA receptor occupancy is eliminated as a potential explanation for additivity between drugs with different molecular mechanisms. Third, measuring responses at combinations near the IC50, rather than at either extreme of the sigmoid concentration-response curve, provides the greatest scale over which to observe any potential drug-drug interaction; namely antagonism, additivity, or synergy. However, since only ½ IC50 drug combinations were studied, it is possible that more than one type of interaction might occur using other antagonist drug ratios.
This study has shown that drugs acting on an anesthetic-sensitive ion channel via different molecular mechanisms can combine to act either synergistically, such as with CO2 plus ketamine, or additively, such as with CO2 plus isoflurane. This supports previous work in which anesthetics with suspected dissimilar targets nonetheless additively modulated GABAA, glycine, and NMDA cell receptor currents.6 Consequently, a common molecular mechanism of anesthetic action cannot be reliably inferred from additive in vitro drug interactions. In vivo, inhaled anesthetics generally combine additively to produce a minimum alveolar concentration,3 and this has been taken as evidence for a common site of anesthetic action even though such a site in animals remains elusive.27 However, if in vivo responses reflect in vitro drug effects, then it may be incorrect to infer mechanisms of anesthetic actions on the basis of additivity alone.
Acknowledgments
Funding: This study was supported by funds from the Department of Surgical and Radiological Sciences and by a grant from the National Institutes of Health (GM092821).
Footnotes
Reprints will not be available from the authors.
The authors declare no conflicts of interest
DISCLOSURES
Robert J. Brosnan, D.V.M., Ph.D.
Contribution: This author helped design the study, conduct the study, analyze the data, and write the manuscript
Attestation: Robert J. Brosnan has seen the original study data, reviewed the analysis of the data, approved the final manuscript, and is the author responsible for archiving the study files
Trung L. Pham, B.S.
Contribution: This author helped conduct the study
Attestation: Trung L. Pham has seen the original study data, reviewed the analysis of the data, and approved the final manuscript
Contributor Information
Robert J. Brosnan, University of California, Davis
Trung L. Pham, University of California, Davis
References
- 1.Shafer SL, Hendrickx JF, Flood P, Sonner J, Eger EI., 2nd Additivity versus synergy: a theoretical analysis of implications for anesthetic mechanisms. Anesth Analg. 2008;107:507–24. doi: 10.1213/ane.0b013e31817b7140. [DOI] [PubMed] [Google Scholar]
- 2.Brosnan RJ, Eger EI, 2nd, Laster MJ, Sonner JM. Anesthetic Properties of Carbon Dioxide in the Rat. Anesth Analg. 2007;105:103–6. doi: 10.1213/01.ane.0000265556.69089.78. [DOI] [PubMed] [Google Scholar]
- 3.Eger EI, 2nd, Tang M, Liao M, Laster MJ, Solt K, Flood P, Jenkins A, Raines D, Hendrickx JF, Shafer SL, Yasumasa T, Sonner JM. Inhaled anesthetics do not combine to produce synergistic effects regarding minimum alveolar anesthetic concentration in rats. Anesth Analg. 2008;107:479–85. doi: 10.1213/01.ane.0000295805.70887.65. [DOI] [PubMed] [Google Scholar]
- 4.Solano AM, Pypendop BH, Boscan PL, Ilkiw JE. Effect of intravenous administration of ketamine on the minimum alveolar concentration of isoflurane in anesthetized dogs. Am J Vet Res. 2006;67:21–5. doi: 10.2460/ajvr.67.1.21. [DOI] [PubMed] [Google Scholar]
- 5.Hendrickx JF, Eger EI, 2nd, Sonner JM, Shafer SL. Is synergy the rule? A review of anesthetic interactions producing hypnosis and immobility. Anesth Analg. 2008;107:494–506. doi: 10.1213/ane.0b013e31817b859e. [DOI] [PubMed] [Google Scholar]
- 6.Jenkins A, Lobo IA, Gong D, Trudell JR, Solt K, Harris RA, Eger EI., 2nd General anesthetics have additive actions on three ligand gated ion channels. Anesth Analg. 2008;107:486–93. doi: 10.1213/ane.0b013e31817b70c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hollmann MW, Liu HT, Hoenemann CW, Liu WH, Durieux ME. Modulation of NMDA receptor function by ketamine and magnesium. Part II: interactions with volatile anesthetics. Anesth Analg. 2001;92:1182–91. doi: 10.1097/00000539-200105000-00020. [DOI] [PubMed] [Google Scholar]
- 8.Tallarida RJ. Drug synergism: its detection and applications. J Pharmacol Exp Ther. 2001;298:865–72. [PubMed] [Google Scholar]
- 9.Stabernack C, Sonner JM, Laster M, Zhang Y, Xing Y, Sharma M, Eger EI., 2nd Spinal N-methyl-d-aspartate receptors may contribute to the immobilizing action of isoflurane. Anesth Analg. 2003;96:102–7. doi: 10.1097/00000539-200301000-00022. [DOI] [PubMed] [Google Scholar]
- 10.Brosnan RJ, Pham TL. Carbon dioxide negatively modulates N-methyl-D-aspartate receptors. Br J Anaesth. 2008;101:673–9. doi: 10.1093/bja/aen266. [DOI] [PubMed] [Google Scholar]
- 11.Dickinson R, Peterson BK, Banks P, Simillis C, Martin JC, Valenzuela CA, Maze M, Franks NP. Competitive inhibition at the glycine site of the N-methyl-D-aspartate receptor by the anesthetics xenon and isoflurane: evidence from molecular modeling and electrophysiology. Anesthesiology. 2007;107:756–67. doi: 10.1097/01.anes.0000287061.77674.71. [DOI] [PubMed] [Google Scholar]
- 12.Orser BA, Pennefather PS, MacDonald JF. Multiple mechanisms of ketamine blockade of N-methyl-D-aspartate receptors. Anesthesiology. 1997;86:903–17. doi: 10.1097/00000542-199704000-00021. [DOI] [PubMed] [Google Scholar]
- 13.Ogata J, Shiraishi M, Namba T, Smothers CT, Woodward JJ, Harris RA. Effects of anesthetics on mutant N-methyl-D-aspartate receptors expressed in Xenopus oocytes. J Pharmacol Exp Ther. 2006;318:434–43. doi: 10.1124/jpet.106.101691. [DOI] [PubMed] [Google Scholar]
- 14.Brosnan R, Gong D, Cotten J, Keshavaprasad B, Yost CS, Eger EI, 2nd, Sonner JM. Chirality in anesthesia II: stereoselective modulation of ion channel function by secondary alcohol enantiomers. Anesth Analg. 2006;103:86–91. doi: 10.1213/01.ane.0000221437.87338.af. [DOI] [PubMed] [Google Scholar]
- 15.Brosnan RJ, Yang L, Milutinovic PS, Zhao J, Laster MJ, Eger EI, 2nd, Sonner JM. Ammonia has anesthetic properties. Anesth Analg. 2007;104:1430–3. doi: 10.1213/01.ane.0000264072.97705.0f. [DOI] [PubMed] [Google Scholar]
- 16.Yang L, Zhao J, Milutinovic PS, Brosnan RJ, Eger EI, 2nd, Sonner JM. Anesthetic properties of the ketone bodies beta-hydroxybutyric acid and acetone. Anesth Analg. 2007;105:673–9. doi: 10.1213/01.ane.0000278127.68312.dc. [DOI] [PubMed] [Google Scholar]
- 17.Yang L, Milutinovic PS, Brosnan RJ, Eger EI, 2nd, Sonner JM. The plasticizer di(2-ethylhexyl) phthalate modulates gamma-aminobutyric acid type A and glycine receptor function. Anesth Analg. 2007;105:393–6. doi: 10.1213/01.ane.0000267336.37735.d7. [DOI] [PubMed] [Google Scholar]
- 18.Efron B. Bootstrap methods: another look at the jackknife. Ann Stat. 1979;7:1–26. [Google Scholar]
- 19.Wright JM, Peoples RW. NMDA receptor pharmacology and analysis of patch-clamp recordings. Methods Mol Biol. 1999;128:143–53. doi: 10.1385/1-59259-683-5:143. [DOI] [PubMed] [Google Scholar]
- 20.Kuryatov A, Laube B, Betz H, Kuhse J. Mutational analysis of the glycine-binding site of the NMDA receptor: structural similarity with bacterial amino acid-binding proteins. Neuron. 1994;12:1291–300. doi: 10.1016/0896-6273(94)90445-6. [DOI] [PubMed] [Google Scholar]
- 21.Hirai H, Kirsch J, Laube B, Betz H, Kuhse J. The glycine binding site of the N-methyl-D-aspartate receptor subunit NR1: identification of novel determinants of co-agonist potentiation in the extracellular M3-M4 loop region. Proc Natl Acad Sci U S A. 1996;93:6031–6. doi: 10.1073/pnas.93.12.6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Traynelis SF, Hartley M, Heinemann SF. Control of proton sensitivity of the NMDA receptor by RNA splicing and polyamines. Science. 1995;268:873–6. doi: 10.1126/science.7754371. [DOI] [PubMed] [Google Scholar]
- 23.Sullivan JM, Traynelis SF, Chen HS, Escobar W, Heinemann SF, Lipton SA. Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron. 1994;13:929–36. doi: 10.1016/0896-6273(94)90258-5. [DOI] [PubMed] [Google Scholar]
- 24.Low CM, Lyuboslavsky P, French A, Le P, Wyatte K, Thiel WH, Marchan EM, Igarashi K, Kashiwagi K, Gernert K, Williams K, Traynelis SF, Zheng F. Molecular determinants of proton-sensitive N-methyl-D-aspartate receptor gating. Mol Pharmacol. 2003;63:1212–22. doi: 10.1124/mol.63.6.1212. [DOI] [PubMed] [Google Scholar]
- 25.Kashiwagi K, Pahk AJ, Masuko T, Igarashi K, Williams K. Block and modulation of N-methyl-D-aspartate receptors by polyamines and protons: role of amino acid residues in the transmembrane and pore-forming regions of NR1 and NR2 subunits. Mol Pharmacol. 1997;52:701–13. doi: 10.1124/mol.52.4.701. [DOI] [PubMed] [Google Scholar]
- 26.Banke TG, Dravid SM, Traynelis SF. Protons trap NR1/NR2B NMDA receptors in a nonconducting state. J Neurosci. 2005;25:42–51. doi: 10.1523/JNEUROSCI.3154-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eger EI, 2nd, Raines DE, Shafer SL, Hemmings HC, Jr, Sonner JM. Is a new paradigm needed to explain how inhaled anesthetics produce immobility? Anesth Analg. 2008;107:832–48. doi: 10.1213/ane.0b013e318182aedb. [DOI] [PMC free article] [PubMed] [Google Scholar]