

Abstract
The aggregation kinetics of silver nanoparticles (AgNPs) that were coated with two commonly used capping agents—citrate and polyvinylpyrrolidone (PVP)—were investigated. Time-resolved dynamic light scattering (DLS) was employed to measure the aggregation kinetics of the AgNPs over a range of monovalent and divalent electrolyte concentrations. The aggregation behavior of citrate-coated AgNPs in NaCl was in excellent agreement with the predictions based on Derjaguin–Landau–Verwey–Overbeek (DLVO) theory, and the Hamaker constant of citrate-coated AgNPs in aqueous solutions was derived to be 3.7 × 10-20 J. Divalent electrolytes were more efficient in destabilizing the citrate-coated AgNPs, as indicated by the considerably lower critical coagulation concentrations (2.1 mM CaCl2 and 2.7 mM MgCl2 vs. 47.6 mM NaCl). The PVP-coated AgNPs were significantly more stable than citrate-coated AgNPs in both NaCl and CaCl2, which is likely due to steric repulsion imparted by the large, non-charged polymers. The addition of humic acid resulted in the adsorption of the macromolecules on both citrate- and PVP-coated AgNPs. The adsorption of humic acid induced additional electrosteric repulsion that elevated the stability of both nanoparticles in suspensions containing NaCl or low concentrations of CaCl2. Conversely, enhanced aggregation occurred for both nanoparticles at high CaCl2 concentrations due to interparticle bridging by humic acid clusters.
Introduction
Because of the optical and antimicrobial properties and electrical conductivity of silver nanoparticles (AgNPs) (1-4), these nanomaterials are already finding applications in a wide variety of fields, which includes biomedical, chemical, and electrical engineering. The combination of their antimicrobial activity and relative low cost is likely the key reason for nanosilver-containing products to presently comprise more than 50% of the inventoried consumer products that contain engineered nanoparticles (5). For example, AgNPs are being incorporated into clothes, bandages, and food containers as deodorizers and disinfectants (6). In addition, studies have been conducted to explore the use of AgNPs for drinking water treatment applications (7).
With AgNPs increasingly being manufactured and utilized in consumer products, AgNPs are likely to be released into surface waters and the subsurface. Recent studies have shown that AgNPs can exert toxic effects on mammalian cells (8, 9) and microorganisms (4, 10), even though the mechanisms for cytotoxicity are still not well understood. Thus, the potential release of these engineered nanomaterials into the environment has raised concerns on their impacts on the ecosystem and human health.
Currently, the most commonly used method to synthesize spherical, reasonably monodisperse AgNPs is chemical reduction of silver salts (3). In addition, AgNPs are often modified with capping agents. Since these capping agents are usually negatively charged species or relatively large, hydrophilic polymers, the adsorption of these agents will impart colloidal stability to AgNPs by either enhancing their surface charge or introducing steric or electrosteric repulsion (11). This enhancement in colloidal stability is especially critical for applications which require the dispersion of AgNPs in aqueous solutions, such as the high ionic strength solutions typically encountered in biomedical applications.
The environmental impact of AgNPs is greatly dependent on their mobility and aggregation behavior in the natural and engineered environment. Recently, Li et al. (12) investigated the influence of solution chemistry on the aggregation kinetics of unmodified AgNPs and obtained the critical coagulation concentrations (CCCs) of these nanoparticles through time-resolved dynamic light scattering (DLS). Since the CCC represents the minimum amount of electrolyte needed to completely destabilize the suspension (13), it provides a useful metric of colloidal stability for AgNPs and hence can be used to in the prediction of the fate and transport of AgNPs in natural and engineered systems. To date, no study has been conducted to derive the CCCs of AgNPs that have been modified with capping agents. Furthermore, the impact of natural organic matter (NOM), which is ubiquitous in aquatic systems, on the colloidal stability of modified AgNPs is still not well understood.
The objective of this study is to quantify and compare the aggregation kinetics and colloidal stability of AgNPs that were synthesized through the reduction of a Tollens’ reagent and modified with two commonly used capping agents—citrate and polyvinylpyrrolidone (PVP). Time-resolved DLS measurements were conducted to obtain the aggregation kinetics of both modified AgNPs over a range of monovalent (NaCl) and divalent (MgCl2 and CaCl2) electrolyte concentrations. The Derjaguin–Landau–Verwey–Overbeek (DLVO) theory was used to fit the experimental data in order to derive the Hamaker constant of citrate-coated AgNPs. In addition, the influence of humic acid on the colloidal stability of both citrate- and PVP-coated AgNPs was investigated.
Materials and Methods
Silver Nanoparticle Synthesis and Characterization
All reagents used for the preparation of the citrate- and PVP-coated AgNPs were purchased from Sigma Aldrich. Unmodified AgNPs were first synthesized through the reduction of a Tollens’ reaction using glucose (2, 14). Details for the preparation of the unmodified AgNP suspension are provided in the Supporting Information (SI). This suspension will be referred to as the original AgNP stock suspension. Measurements using inductively coupled plasma mass spectroscopy (ICP-MS) showed that the total and dissolved silver concentrations of a freshly prepared stock suspension were 10.360 mg/L and 0.002 mg/L, respectively. These measurements indicated that the yield of AgNP formation was nearly 100%.
The citrate-coated and PVP-coated AgNP stock suspensions used in this study were prepared by cleaning the original AgNP stock suspension and re-suspending the nanoparticles in citrate and PVP solutions. Details for the preparation of these stock suspensions are provided in the SI. According to the supplier (Sigma), the PVP used to coat the AgNPs has an average MW of 10 kDa. The stock suspensions were contained in tightly capped Pyrex glass bottles and stored in the dark at 4°C. Through ICP-MS analysis, the AgNP and dissolved silver concentrations in the citrate-coated AgNP stock suspension used for the aggregation experiments were determined to be 8.467 mg/L and 0.112 mg/L, respectively. The AgNP and dissolved silver concentrations in the PVP-coated AgNP stock suspension used for the aggregation experiments were 8.247 mg/L and 0.222 mg/L, respectively. The total citrate and PVP concentrations in the citrate- and PVP-coated AgNP stock suspensions were 1 μM and 6.6 mg/L total organic carbon (TOC), respectively.
The absorbance spectra of citrate-and PVP-coated AgNP suspensions were obtained over a range of wavelengths from 300 to 900 nm by using a UV-Vis spectrophotometer (UV-1800, Shimadzu). In addition, the citrate- and PVP-coated AgNPs were examined using a transmission electron microscope (TEM) (Philips CM300 FEG). A drop of AgNP stock suspension was deposited and dried on a specimen copper TEM grid, which was coated with a Lacey carbon film (SPI Supplies, PA), before being observed under the TEM at 300 kV. The elemental composition and crystalline structure of the AgNPs were obtained through energy dispersive X-ray spectroscopy (EDS) and selected area electron diffraction (SAED) analysis, respectively, with the employment of the TEM. The electron diffraction pattern of PVP-coated AgNPs was obtained by using a Philips EM 420 TEM at 120 kV.
Determination of Silver Nanoparticle and Dissolved Silver Concentrations
The AgNP and dissolved silver concentrations of the AgNP stock and diluted suspensions used in this study were obtained through centrifugal ultrafiltration (using 3-kDa centrifugal membrane filters) and ICP-MS. Details are provided in the SI.
Solution Chemistry
ACS-grade electrolyte (NaCl, CaCl2, and MgCl2) stock solutions were prepared and filtered using 0.1-μm alumina syringe filters (Anotop 25, Whatman) before use. Humic acid stock solution was prepared by dissolving Suwannee River humic acid (Standard II, International Humic Substances Society) in DI water. Details are provided in the SI. All experiments and measurements were performed at pH 7.0 ± 0.1 (buffered with 0.15 mM NaHCO3).
Electrophoretic Mobility Measurements
A ZetaPALS analyzer (Brookhaven, NY) was used to measure the electrophoretic mobilities (EPMs) of citrate- and PVP-coated AgNPs over a range of electrolyte concentrations at 25°C. For each solution chemistry, 5–10 measurements were conducted for each of at least 3 samples. The zeta (ζ) potentials were converted from the average EPMs by using the tabulated values that were provided by Ottewill and Shaw (15).
Time-Resolved Dynamic Light Scattering
Time-resolved DLS measurements of aggregating AgNP suspensions were performed using a light scattering unit. This unit comprises an argon laser (Lexel 95, Cambridge laser, CA) with a wavelength of 488 nm, a photomultiplier tube mounted on a goniometer (BI-200SM, Brookhaven, NY), a digital correlator (BI-9000AT, Brookhaven, NY), and a thermostated vat filled with an index-matching cis- and trans-mixture of decahydronaphthalene which was maintained at 25°C for all our measurements. The detailed procedure for the DLS measurements is provided in the SI. For all aggregation experiments, the AgNP suspensions used for DLS measurements had a total volume of 1 mL and a AgNP concentration of 1.129 mg/L of citrate-coated AgNPs or 1.010 mg/L of PVP-coated AgNPs (7.5 times dilution from citrate- or PVP-coated AgNP stock suspensions, respectively). The total citrate and PVP concentrations of the final citrate- and PVP-coated AgNP suspensions were 0.13 μM and 0.88 mg/L TOC, respectively.
All DLS measurements were conducted at a scattering angle of 90°. Each autocorrelation function was accumulated over 15 s and the intensity-weighted hydrodynamic diameter was then derived using second-order cumulant analysis (Brookhaven software). Time-resolved DLS measurements were performed over time periods of between 12 min and 3 h in order to achieve a large enough increase in hydrodynamic diameter for accurate derivation of aggregation kinetics.
Determination of Aggregation Kinetics
The early-stage aggregation kinetics of AgNPs can be obtained from the initial rate of change of hydrodynamic diameter, Dh, with time, t, as measured by time-resolved DLS. In the early aggregation stage, the initial aggregation rate constant, k, is proportional to the initial rate of increase in Dh and inversely proportional to the initial primary AgNP concentration in the suspension, N0 (16, 17):
| (1) |
A linear least squares regression analysis of the initial increase in Dh was conducted to obtain k. For most solution chemistries, this analysis was performed over the time period in which the hydrodynamic diameter increased to 1.3 times of the initial hydrodynamic diameter, Dh,0. At low electrolyte concentrations, however, the hydrodynamic diameter failed to reach 1.3 Dh,0. Under such conditions, the linear regression was performed over a prolonged time period (> 90 min). For all solution chemistries, the y-intercept of the fitted line did not exceed 2 nm in excess of Dh,0.
The attachment efficiency, α, is used to quantify the aggregation kinetics of AgNPs. It is calculated by normalizing the aggregation rate constant obtained in the solution of interest to the rate constant obtained under favorable (non-repulsive) aggregation conditions, kfast (16-18):
| (2) |
The terms with subscript “fast” refer to favorable conditions. To calculate α in the presence of humic acid, kfast was determined using obtained for the same type of AgNPs in the absence of humic acid in the same electrolyte of interest. In such cases, α values exceeding unity indicates that interparticle bridging is taking place (19).
Detection of AgNP Dissolution
The degree of nanoparticle dissolution taking place in our systems was determined since dissolution may influence the mechanism of AgNP aggregation. Specifically, the precipitation of AgCl resulting from the increase in Ag+ concentration due to the dissolution of AgNPs may lead to interparticle bridging between AgNPs, which will in turn enhance the nanoparticle aggregation kinetics (12). In this study, dissolution experiments were conducted by measuring the dissolved silver concentrations of the citrate- and PVP-coated AgNP suspensions prepared at the highest electrolyte concentrations which were used for the aggregation experiments. The measurements were conducted using ICP-MS and the details are provided in the SI.
Results and Discussion
Physicochemical Properties of Citrate- and PVP-Coated AgNPs
The size distribution of the citrate-coated AgNPs (Figure 1a) was determined by using the Digital Micrograph software (Gatan, CA) to measure the diameters of 210 nanoparticles randomly selected from a TEM grid. A representative TEM image of the AgNPs is presented in the insert. The nanoparticles were observed to be spherical with slight angular features. The number average diameter of the citrate-coated AgNPs was 71.3 nm and the standard deviation was 18.9 nm. Through 20 DLS measurements, the average intensity-weighted hydrodynamic diameter of stable citrate-coated AgNPs prepared in DI water was determined to be 72.5 nm, which is very similar to the number average diameter of the nanoparticles. The average intensity-weighted hydrodynamic diameter of PVP-coated AgNPs obtained from 20 DLS measurements was 66.9 nm.
FIGURE 1.
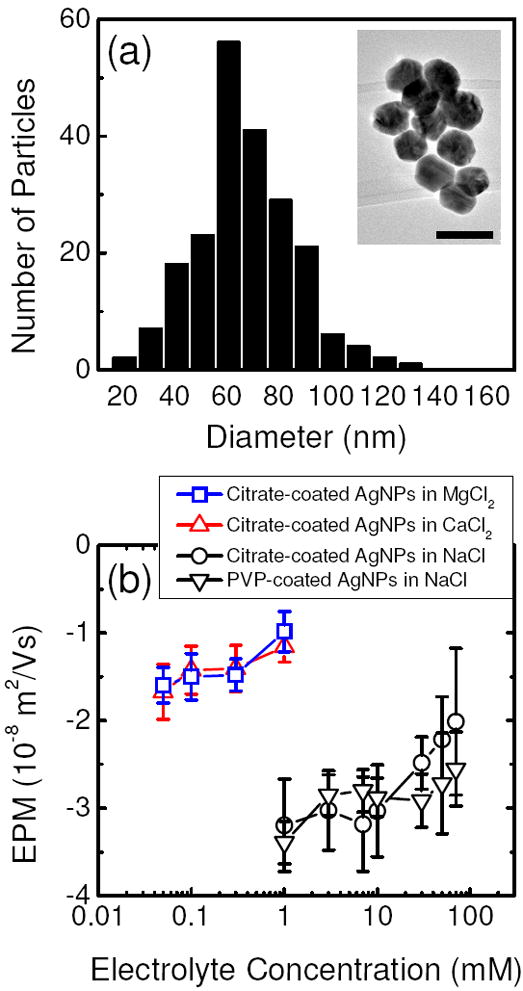
(a) Size distribution of 210 citrate-coated AgNPs. The inset presents a representative TEM image of the citrate-coated AgNPs. The scale bar represents 100 nm. (b) Electrophoretic mobilities (EPMs) of citrate- and PVP-coated AgNPs as a function of electrolyte concentrations at pH 7.0 and 25°C. For all measurements, the citrate and PVP concentrations in the suspension were 0.13 μM and 0.88 mg/L TOC, respectively. Each data point represents the mean EPM. Error bars represent standard deviations.
Absorbance peaks for citrate- and PVP-coated AgNP suspensions were at 431 nm and 418 nm, respectively (Figure S1 in SI). These values are within the reported range for AgNPs with the size of 57–87 nm (12, 20). High resolution TEM imaging of both AgNPs revealed lattice fringes with different alignments within a single nanoparticle, which are indicative of the polycrystalline nature of AgNPs synthesized through the Tollens’ method (Figure S2).
SAED analysis was performed on citrate- and PVP-coated AgNP clusters and the distances between atomic planes (d-spacing) were determined by measuring the diameters of the first four rings on the diffraction images (Figure S3a and Figure S4a). The similarity in the first four d-spacing values between the AgNPs (2.38 Å, 2.05 Å, 1.42 Å, 1.23 Å for citrate-coated AgNPs and 2.40 Å, 2.08 Å, 1.50 Å, 1.23 Å for PVP-coated AgNPs) and silver metal (2.36 Å, 2.04 Å, 1.44 Å, 1.23 Å (21)) confirmed that the nanoparticles have the same crystalline structure as bulk silver.
The elements and their relative abundance of citrate- and PVP-coated AgNPs were obtained by performing TEM-EDS on AgNP samples which were freshly dried on TEM grids. The EDS spectra of randomly selected citrate- and PVP-coated AgNP clusters (Figure S3b and Figure S4b) showed that the nanoparticles were mostly composed of silver and the amount of oxygen of the whole cluster was relatively small. Therefore, the results from both SAED and EDS analyses confirmed that our synthesis method resulted in the production of zero-valent AgNPs.
Electrokinetic Properties of Citrate- and PVP-Coated AgNPs
Figure 1b presents the EPMs of citrate-coated AgNPs measured over a range of monovalent (NaCl) and divalent (CaCl2 and MgCl2) electrolyte concentrations. The measurements showed that citrate-coated AgNPs were negatively charged over the entire range of monovalent and divalent electrolyte concentrations used in this study. Since the pKa values of citric acid are 3.13, 4.72, and 6.33 (22), the carboxylic acid groups of citrate molecules are mostly deprotonated at pH 7 and will thus contribute to the surface charge of citrate-coated AgNPs. In addition, the residual side products from AgNP synthesis could also contribute to the negative surface charge of these nanoparticles. With increasing electrolyte concentrations, the EPMs of citrate-coated AgNPs became less negative due to an increase in charge screening (for NaCl) or charge neutralization (for CaCl2 and MgCl2) effects.
Figure 1b also presents the EPMs of PVP-coated AgNPs over a range of NaCl concentrations. It was observed that PVP-coated AgNPs had similar EPMs as citrate-coated AgNPs. Since PVP polymers are neutral and the total PVP concentration in the suspension was relatively low (0.88 mg/L TOC), the AgNPs may not be completely coated with PVP and the negative surface charge is likely due to the residual side products which were adsorbed on the nanoparticle surface.
Dissolution of Citrate- and PVP-Coated AgNPs at High Electrolyte Concentrations
Because the precipitation of silver salts may enhance the aggregation kinetics of AgNPs, we performed dissolution experiments at the highest electrolyte concentrations that were used in our aggregation experiments to establish the dissolved silver concentrations under these solution chemistries. These dissolution experiments were performed at 455 mM NaCl, 27 mM CaCl2, and 27 mM MgCl2 for citrate-coated AgNPs and at 455 mM NaCl and 27 mM CaCl2 for PVP-coated AgNPs. These are the electrolyte concentrations at which the nanoparticles underwent diffusion-limited aggregation (to be discussed in the following section). Control experiments were performed in the absence of electrolytes. For these experiments, the samples were prepared in the same manner as for the DLS experiments by diluting the citrate- or PVP-coated AgNP stock suspension 7.5 times before the addition of either the electrolyte solution of interest or DI water (for control experiments).
The dissolved silver concentrations of the citrate- and PVP-coated AgNP suspensions prepared in DI water and electrolyte solutions are presented in Figure 2. It should be noted that the initial AgNP and dissolved silver concentrations of citrate- and PVP-coated AgNP suspensions were different because the citrate- and PVP-coated AgNP stock suspensions were prepared separately. The slight increase in dissolved silver concentrations of the citrate- and PVP-coated AgNP suspensions prepared in DI water after 30 min indicated that some dissolution may have occurred. The increase in temperature (from 4 to 25°C) and dissolved oxygen concentration, as well as the decrease in citrate and PVP concentrations, are expected to lead to the slight dissolution of both AgNPs when diluted in DI water (23-25).
FIGURE 2.
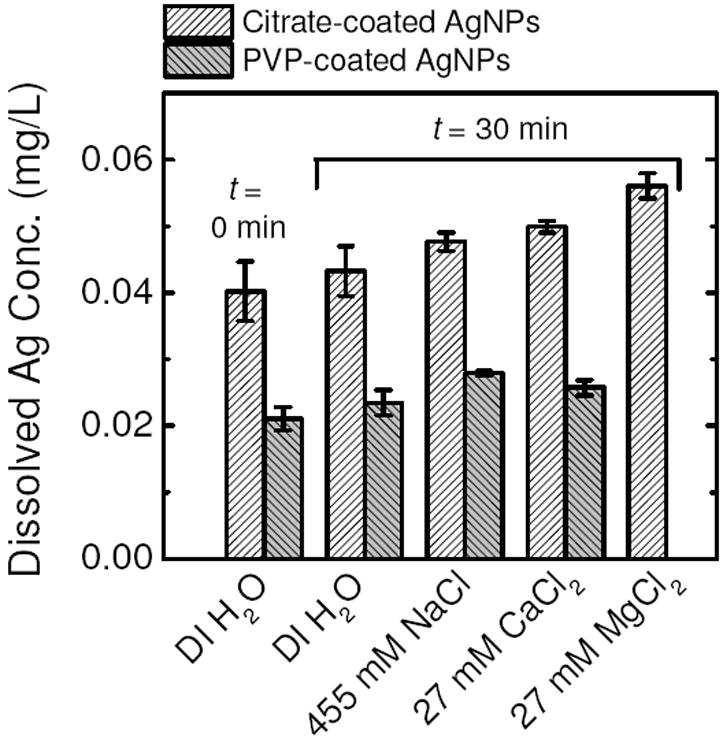
Dissolved silver concentrations at either 0 or 30 min after the preparation of citrate- and PVP-coated AgNP suspensions in different solution chemistries at pH 7.0. The initial AgNP concentrations of the citrate- and PVP-coated AgNP suspensions (at t = 0 min) were 1.074 mg/L and 0.912 mg/L, respectively. Each bar represents the mean measurement of at least 4 samples. Error bars represent standard deviations.
Also shown in Figure 2, citrate- and PVP-coated AgNPs were generally observed to undergo a higher degree of dissolution in all electrolyte solutions than in DI water. These results are consistent to other observations that an increase in ionic strength can enhance AgNP dissolution (12). The presence of chloride ions in the electrolyte solutions will lead to the formation of soluble silver chloride complexes, such as AgCl2− and AgCl32− (24), which can accelerate the dissolution of AgNPs. The measured dissolved silver concentrations in solutions of 455 mM NaCl, 27 mM CaCl2, and 27 mM MgCl2 were used as input parameters for the conservative estimation of the concentrations of dissolved silver species in our aggregation experiments at the same solution chemistries (Visual MINTEQ, Version 3.0). Due to the considerably low dissolved silver concentrations, the simulations confirmed that no precipitation of any silver species (including AgCl) would occur at equilibrium. This result indicates that the minor dissolution of either citrate- or PVP-coated AgNPs in our systems did not lead to precipitation-induced interparticle bridging of AgNPs over the range of NaCl, CaCl2, and MgCl2 concentrations employed in this study.
Aggregation Kinetics of Citrate-Coated AgNPs in Monovalent Electrolyte Solution
The attachment efficiencies of citrate-coated AgNPs are presented as a function of NaCl concentration in Figure 3a. Representative aggregation profiles are presented in Figure S5. The aggregation behavior of citrate-coated AgNPs in NaCl solutions is consistent with the DLVO theory. At low concentrations of NaCl, the increase in NaCl concentration will elevate the degree of charge screening and hence allow for an increase in aggregation kinetics, as reflected by the rise in attachment efficiency. This regime is known as the reaction-limited regime (α < 1). At high NaCl concentrations, the charge of citrate-coated AgNPs is completely screened and the energy barrier between AgNPs is eliminated. Under such conditions, the nanoparticles undergo diffusion-limited aggregation (α = 1). In the diffusion-limited regime, the kinetics of aggregation has reached a maximum and is independent of the NaCl concentration. The CCC, which delineates the reaction- and diffusion-limited regimes, was derived by determining the intersection of the extrapolations through both regimes (17). In NaCl solutions, the CCC of citrate-coated AgNPs was 47.6 mM.
FIGURE 3.
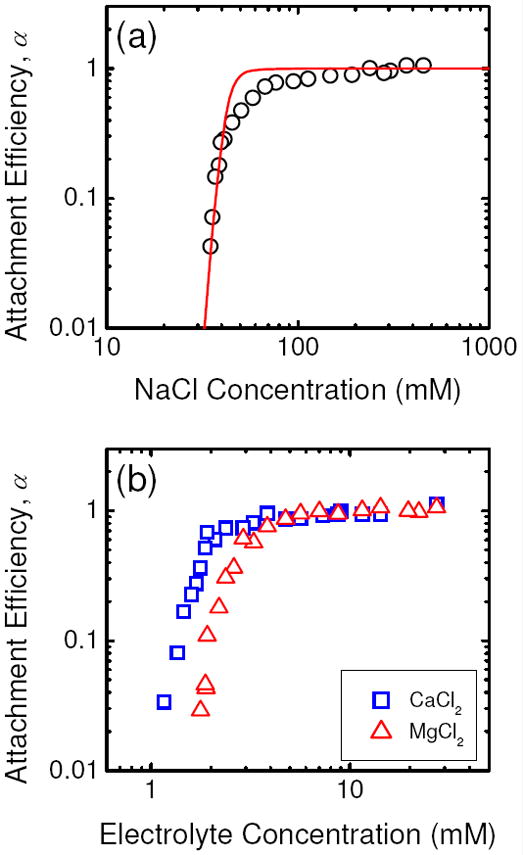
Attachment efficiencies of citrate-coated AgNPs as functions of (a) NaCl and (b) CaCl2 and MgCl2 concentrations at pH 7.0. The solid line in a represents DLVO prediction using a Hamaker constant of 3.7 × 10-20 J. For all experiments, the citrate concentration in the suspension was 0.13 μM.
Recently, Li et al. (12) employed time-resolved DLS to investigate the aggregation kinetics of unmodified AgNPs that were synthesized through the reduction of a Tollens’ reagent using d-maltose. In their study, significant AgNP dissolution appeared to have taken place concurrently with nanoparticle aggregation. Li and coworkers detected an immediate and drastic decrease in hydrodynamic diameter (up to 50% decrease) when electrolyte stock solutions (NaCl, NaNO3, or CaCl2) were introduced into the AgNP suspensions. Through TEM imaging, they also observed interconnected AgNPs within AgNP aggregates which were formed in the presence of NaCl, which they attributed to interparticle bridging by AgCl precipitates (12). In contrast, the degree of dissolution of the citrate- and PVP-coated AgNPs used in this study was too low to result in AgCl precipitation. The considerably higher degree of nanoparticle dissolution in Li et al.’s study (12) is possibly due to the absence of capping agent in their systems. In addition, citrate can serve as a reducing agent which will inhibit oxidative dissolution of the citrate-coated AgNPs in our study. Hence, a comparison of the aggregation behavior of AgNPs in both studies demonstrates that both the methods for nanoparticle preparation and presence of capping agents have significant impacts on the colloidal stability and mechanisms of aggregation of AgNPs.
Comparing Citrate-Coated AgNP Aggregation Kinetics with DLVO Theory
The aggregation kinetics of citrate-coated AgNPs obtained through time-resolved DLS was compared with DLVO theory. The attachment efficiency of aggregating spherical colloidal particles can be calculated by using the following expression which accounts for colloidal and hydrodynamic interactions (17, 26)
| (3) |
where h is the separation distance between two particles, a is the particle radius (35.65 nm, based on the number average diameter of the citrate-coated AgNPs), k is the Boltzmann constant, and T is the absolute temperature (298.15 K). The van der Waals attraction, VA(h), was calculated using the expression proposed by Gregory (27) that accounts for the electromagnetic retardation effect. The total interaction energy between two particles, VT(h), is the sum of the van der Waals attraction, VA(h), and electrical double layer interaction, VR(h). The linear superposition approximation expression was used to calculate the electrical double layer interaction (28). The function β(h) is used to correct for the hydrodynamic interactions between two approaching particles (17, 29). Details on the calculation of DLVO interactions can be found in the SI.
In this study, ζ potentials, which were converted from the EPMs, were used instead of surface potentials for the calculation of VR(h) (30). Logarithmic regression analysis was performed on the ζ potentials to derive the relationship between ζ potential and NaCl concentration. This relationship was then used for the calculation of VR(h). Using the Hamaker constant for citrate-coated AgNPs in aqueous solutions, which is part of the expression for VA(h), as the single fitting parameter, the experimentally obtained attachment efficiencies were fitted with the theoretical calculations for attachment efficiencies (eq 3).
The solid line in Figure 3a represents the best-fitting theoretical attachment efficiencies derived using a Hamaker constant of 3.7 × 10-20 J for citrate-coated AgNPs in aqueous solutions. This value falls within the range of values (2.9 ± 0.8) × 10-20 J suggested by Fornasiero and Grieser (31). Furthermore, Figure 3a shows that the experimentally obtained attachment efficiencies are in remarkable agreement with DLVO predictions. One of the key assumptions of DLVO theory is that the charge on the particle surface is uniformly distributed (30). Previously, discrepancies between experimental results and theoretical predictions had been observed for other types of particles and these discrepancies were attributed to the heterogeneity of surface charge (30). In contrast, the good agreement between the experimental results and theoretical prediction in this study may be an indication of a uniform charge distribution on the citrate-coated AgNP surface.
Aggregation Kinetics of Citrate-Coated AgNPs in Divalent Electrolyte Solutions
The aggregation kinetics of citrate-coated AgNPs in CaCl2 and MgCl2 electrolytes are presented in Figure 3b. Similar to the inverse stability profile obtained in the presence of NaCl (Figure 3a), the presence of the reaction- and diffusion-limited regimes showed that the aggregation kinetics of citrate-coated AgNPs in CaCl2 and MgCl2 electrolytes are controlled by electrostatic interactions. The CCCs of citrate-coated AgNPs in the presence of CaCl2 and MgCl2 were 2.1 mM and 2.7 mM, respectively. These values are much smaller than the CCC obtained in NaCl, since Ca2+ and Mg2+ ions can neutralize the surface charge of citrate-coated AgNPs through specific interactions with the carboxyl groups of the adsorbed citrate molecules. The results from the aggregation experiments corroborate with the EPM measurements which showed that mobilities of citrate-coated nanoparticles were considerably less negative in the presence of CaCl2 and MgCl2 than in NaCl (Figure 1b). In addition, the CCC of citrate-coated AgNPs in CaCl2 is lower than that in MgCl2. This disparity in CCCs is likely due to Ca2+ ions having a higher propensity to form complexes with citrate compared with Mg2+ ions, as evident from the higher stability constant of monodentate Ca-citrate complexes compared to monodentate Mg-citrate complexes at 0 mM ionic strength and 25°C (101.4 vs. 101.0) (32). Therefore, Ca2+ ions are expected to be more efficient than Mg2+ ions in neutralizing the surface charge of citrate-coated AgNPs.
Comparing Aggregation Kinetics of PVP-Coated AgNPs with Citrate-Coated AgNPs
Figure 4 presents the inverse stability profiles of PVP- and citrate-coated AgNPs in NaCl and CaCl2 electrolytes. The total PVP concentration in the PVP-coated AgNP suspensions was 0.88 mg/L TOC, which is calculated to be equivalent to 0.14 μM PVP by using a PVP MW of 10 kDa. Therefore, the molar concentration of PVP in the suspensions is comparable to that of citrate in the citrate-coated AgNP suspensions (0.13 μM). When similar concentrations of capping agents were used, the PVP-coated AgNPs were more stable than citrate-coated AgNPs in both electrolytes, as indicated by the considerably higher CCCs of PVP-coated AgNPs. In the presence of NaCl, the CCC of PVP-coated AgNPs was 111.5 mM, compared to 47.6 mM for the citrate-coated AgNPs. In the presence of CaCl2, the CCCs of PVP- and citrate-coated AgNPs were 4.9 mM and 2.1 mM, respectively.
FIGURE 4.
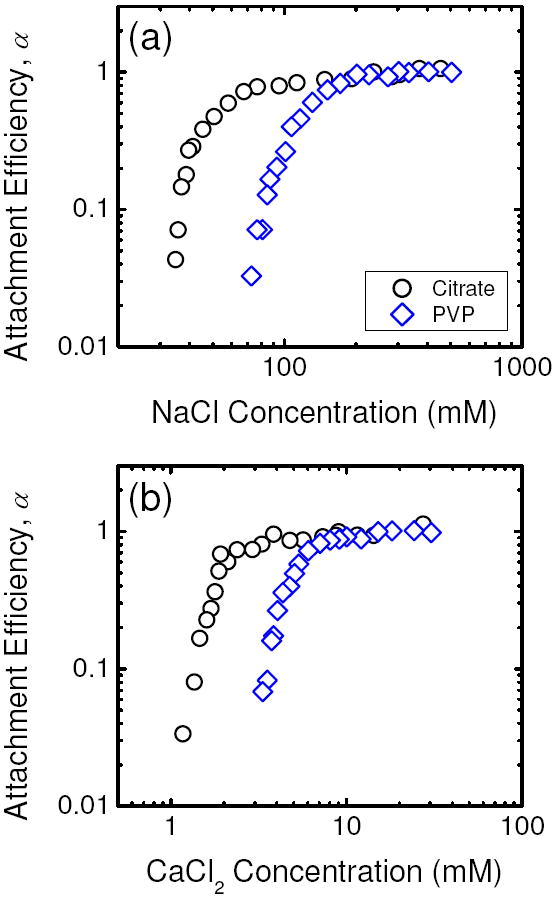
Attachment efficiencies of citrate- and PVP-coated AgNPs as functions of (a) NaCl and (b) CaCl2 concentrations at pH 7.0. The attachment efficiencies of citrate-coated AgNPs in a and b are reproduced from Figures 3a and b, respectively. For citrate-coated AgNPs, the citrate concentration in the suspension was 0.13 μM. For PVP-coated AgNPs, the PVP concentration in the suspension was 0.88 mg/L TOC (equivalent to 0.14 μM PVP).
Because PVP is an amphiphilic, non-charged polymer, the adsorption of PVP on AgNPs is likely to result in steric repulsion. Since the PVP concentration in the suspension was relatively low (0.88 mg/L TOC), it is expected that the AgNPs were only partially coated with PVP. Thus, sufficient space was likely to be available for PVP polymers to lie relatively flat on the nanoparticle surface with some polymer segments protruding into the bulk solution (33). However, the existence of the reaction- and diffusion-limited regimes in the inverse stability profiles in both NaCl and CaCl2 indicated that electrostatic interaction still played a significant role in controlling the stability of PVP-coated AgNPs. Trace amount of residual side products from the AgNP synthesis process on the nanoparticle surface are likely the origin of the negative surface charge of the PVP-coated AgNPs.
At higher PVP concentrations, the PVP coverage will be increased and significant sections of adsorbed PVP polymers will be expected to extend into the bulk solution (33), thus leading to steric repulsion to become the major contributor to the stability of PVP-coated AgNPs. In the study of Badawy et al. (11), the stability of AgNPs in PVP solutions with a concentration of 2,500 mg/L, which is much higher than that used in this study, were investigated. The authors found that the ζ potential, which was close to neutral (ca. −7 mV), and hydrodynamic diameter of PVP-coated AgNPs remained constant over a wide range of pH conditions. Their observation confirmed that steric repulsion was the dominant contributor to the high stability of the AgNPs and that the contribution by electrostatic interactions was minor at elevated PVP concentrations.
Influence of Humic Acid on Aggregation Kinetics of Citrate- and PVP-Coated AgNPs
The aggregation kinetics of citrate-coated AgNPs in the absence and presence of humic acid are presented as functions of NaCl concentrations in Figure 5a. The presence of humic acid (1 mg/L TOC) increased the CCC from 47.6 mM to 72.1 mM NaCl. The adsorption of humic acid macromolecules, which contains negatively charged carboxyl groups, on the surface of citrate-coated AgNPs is very likely to result in electrosteric repulsion and thus increase the stability of the citrate-coated AgNPs. Although both citrate-coated AgNPs and humic acid are negatively charged at pH 7.0, humic acid may still adsorb on the modified nanoparticles through hydrophobic interaction. It is also possible that the nanoparticles were not completely coated with citrate and there were still available surface sites for humic acid adsorption to take place.
FIGURE 5.
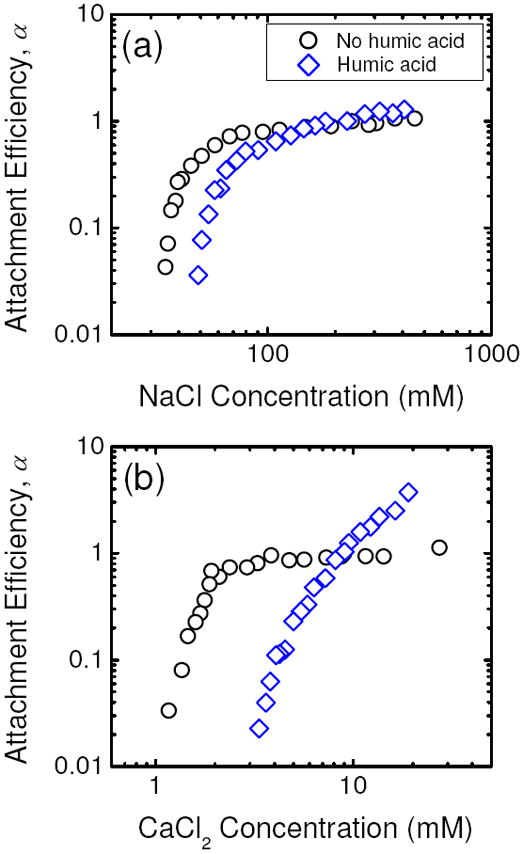
Attachment efficiencies of citrate-coated AgNPs in the absence and in the presence of humic acid (1 mg/L TOC) as functions of (a) NaCl and (b) CaCl2 concentrations at pH 7.0. The attachment efficiencies of citrate-coated AgNPs in the absence of humic acid in a and b are reproduced from Figures 3a and b, respectively. For all experiments, the citrate concentration in the suspension was 0.13 μM.
The aggregation behavior of citrate-coated AgNPs in the presence of humic acid and CaCl2 is dissimilar from that in NaCl, as shown in Figure 5b. Specifically, no distinct reaction- and diffusion-limited regimes were observed in the inverse stability profile. When the concentration of CaCl2 was smaller than 9 mM, the citrate-coated AgNPs were more stable in the presence than in the absence of humic acid due to electrosteric stabilization. At CaCl2 concentrations above 9 mM, the attachment efficiencies were higher than unity and increased with increasing CaCl2 concentration. Similarly, enhanced aggregation of fullerene (19), gold (34), and silicon (35) nanoparticles was previously observed in the presence of humic acid at high CaCl2 concentrations. Chen and Elimelech (19) hypothesized that the enhanced aggregation was due to interparticle bridging of nanoparticles by humic acid aggregates which were created from the complex formation between humic acid macromolecules and Ca2+ ions.
PVP-coated AgNPs exhibited similar aggregation behavior in the presence of humic acid as that of citrate-coated AgNPs in both NaCl and CaCl2 electrolytes (Figure S6). In NaCl solutions, the presence of humic acid increased the CCC of PVP-coated AgNPs from 111.5 mM to 155.1 mM. In the presence of CaCl2, the PVP-coated AgNPs were more stable in the presence than in the absence of humic acid below 18 mM. At CaCl2 concentrations above 18 mM, enhanced aggregation of PVP-coated AgNPs occurred in the presence of humic acid. However, even at the highest CaCl2 concentration employed (30 mM), the enhancement in aggregation kinetics of PVP-coated AgNPs (α = 1.6) was not as dramatic as that of citrate-coated AgNPs (α = 3.7 at 19 mM CaCl2). The smaller enhancement in aggregation kinetics may be an indication that the adsorbed PVP can cause steric hindrance to interparticle bridging by the humic acid clusters.
Environmental Implications
The aggregation kinetics of citrate-coated AgNPs is shown to be in excellent agreement with DLVO theory. The Hamaker constant of citrate-coated AgNPs derived from this study is a critical parameter for the modeling and prediction of the fate and transport of these nanoparticles in aquatic systems (36). PVP-coated AgNPs are significantly more stable than citrate-coated AgNPs, likely due to the steric repulsion imparted by the adsorbed PVP molecules. Since the CCCs for both AgNPs are much higher than typical monovalent and divalent salt concentrations in freshwater systems, both AgNPs are expected to be highly mobile in these environments. While humic acid elevates the colloidal stability of both citrate- and PVP-coated AgNPs in NaCl and at low concentrations of CaCl2, it enhances the aggregation kinetics at high CaCl2 concentrations. Similar studies should be conducted to establish the effects of other key components of NOM, such as fulvic acids and polysaccharides, on the aggregation kinetics of modified AgNPs. In addition, further research is required to assess the long-term reversibility of the adsorption of citrate and PVP on AgNPs, which will have important implications for the aggregation behavior and persistence of these nanoparticles in aquatic systems.
Supplementary Material
Acknowledgments
K.A.H. was funded by the Vietnam Education Foundation and the Johns Hopkins University (JHU) NIEHS Center in Urban Environmental Health (P30ES03819). We acknowledge Dr. Kenneth Livi from the Department of Earth and Planetary Sciences (JHU) for performing the TEM, EDS, and SAED analyses of the AgNPs.
Footnotes
Supporting Information Available
Additional figures and details for Materials and Methods and Results and Discussion are presented. This material is available free of charge via the Internet at http://pubs.acs.org.
Literature Cited
- 1.McFarland AD, Van Duyne RP. Single silver nanoparticles as real-time optical sensors with zeptomole sensitivity. Nano Lett. 2003;3(8):1057–1062. [Google Scholar]
- 2.Panáček A, Kvítek L, Prucek R, Kolár M, Večcřová R, Pizúrová N, Sharma VK, Nevěčná Tj, Zbořil R. Silver colloid nanoparticles: synthesis, characterization, and their antibacterial activity. J Phys Chem B. 2006;110:16248–16253. doi: 10.1021/jp063826h. [DOI] [PubMed] [Google Scholar]
- 3.Sharma VK, Yngard RA, Lin Y. Silver nanoparticles: green synthesis and their antimicrobial activities. Advances in Colloid and Interface Science. 2009;145(1-2):83–96. doi: 10.1016/j.cis.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Sondi I, Salopek-Sondi B. Silver nanoparticles as antimicrobial agent: a case study on E-coli as a model for Gram-negative bacteria. J Colloid Interf Sci. 2004;275(1):177–182. doi: 10.1016/j.jcis.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 5.The Project on Emerging Nanotechnologies. http://www.nanotechproject.org/inventories/consumer/analysis_draft/
- 6.Benn TM, Westerhoff P. Nanoparticle silver released into water from commercially available sock fabrics. Environ Sci Technol. 2008;42(11):4133–4139. doi: 10.1021/es7032718. [DOI] [PubMed] [Google Scholar]
- 7.Li Q, Mahendra S, Lyon DY, Brunet L, Liga MV, Li D, Alvarez PJJ. Antimicrobial nanomaterials for water disinfection and microbial control: Potential applications and implications. Water Research. 2008;42(18):4591–4602. doi: 10.1016/j.watres.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 8.AshaRani PV, Mun GLK, Hande MP, Valiyaveettil S. Cytotoxicity and Genotoxicity of Silver Nanoparticles in Human Cells. ACS Nano. 2009;3(2):279–290. doi: 10.1021/nn800596w. [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Choi JE, Choi J, Chung KH, Park K, Yi J, Ryu DY. Oxidative stress-dependent toxicity of silver nanoparticles in human hepatoma cells. Toxicology in Vitro. 2009;23(6):1076–1084. doi: 10.1016/j.tiv.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Choi O, Hu ZQ. Size dependent and reactive oxygen species related nanosilver toxicity to nitrifying bacteria. Environ Sci Technol. 2008;42(12):4583–4588. doi: 10.1021/es703238h. [DOI] [PubMed] [Google Scholar]
- 11.El Badawy AM, Luxton TP, Silva RG, Scheckel KG, Suidan MT, Tolaymat TM. Impact of Environmental Conditions (pH, Ionic Strength, and Electrolyte Type) on the Surface Charge and Aggregation of Silver Nanoparticles Suspensions. Environ Sci Technol. 2010;44(4):1260–1266. doi: 10.1021/es902240k. [DOI] [PubMed] [Google Scholar]
- 12.Li X, Lenhart JJ, Walker HW. Dissolution-Accompanied Aggregation Kinetics of Silver Nanoparticles. Langmuir. 2010;26(22):16690–16698. doi: 10.1021/la101768n. [DOI] [PubMed] [Google Scholar]
- 13.Chen KL, Smith BA, Ball WP, Fairbrother DH. Assessing the colloidal properties of engineered nanoparticles in water: case studies from fullerene C60 nanoparticles and carbon nanotubes. Environmental Chemistry. 2010;7(1):10–27. [Google Scholar]
- 14.Yin Y, Li Z-Y, Zhong Z, Gates B, Xia Y, Venkateswaran S. Synthesis and characterization of stable aqueous dispersions of silver nanoparticles through the Tollens process. J Matter Chem. 2002;12:552–527. [Google Scholar]
- 15.Ottewill RH, Shaw JN. Electrophoretic Studies On Polystyrene Latices. J Electroanal Chem. 1972 Jun;37:133–142. [Google Scholar]
- 16.Holthoff H, Egelhaaf S, Borkovec M, Schurtenberger P, Sticher H. Coagulation rate measurements of colloidal particles by simultaneous static and dynamic light scattering. Langmuir. 1996;12(23):5541–5549. [Google Scholar]
- 17.Chen KL, Elimelech M. Aggregation and Deposition Kinetics of Fullerene (C60) Nanoparticles. Langmuir. 2006;22(26):10994–11001. doi: 10.1021/la062072v. [DOI] [PubMed] [Google Scholar]
- 18.Chen KL, Mylon SE, Elimelech M. Aggregation kinetics of alginate-coated hematite nanoparticles in monovalent and divalent electrolytes. Environ Sci Technol. 2006;40(5):1516–1523. doi: 10.1021/es0518068. [DOI] [PubMed] [Google Scholar]
- 19.Chen KL, Elimelech M. Influence of humic acid on the aggregation kinetics of fullerene (C60) nanoparticles in monovalent and divalent electrolyte solutions. J Colloid Interf Sci. 2007;309(1):126–134. doi: 10.1016/j.jcis.2007.01.074. [DOI] [PubMed] [Google Scholar]
- 20.Kvítek L, Prucek R, Panáek A, Novotný R, Hrbáč J, Zbořil R. The influence of complexing agent concentration on particle size in the process of SERS active silver colloid synthesis. J Mater Chem. 2005;15:1099–1105. [Google Scholar]
- 21.FIZ Karlsruhe. ICSD Web, Database #44387. http://icsd.fiz-karlsruhe.de/
- 22.Benjamin MM. Water Chemistry. Boston: McGraw-Hill; 2002. [Google Scholar]
- 23.Liu J, Sonshine DA, Shervani S, Hurt RH. Controlled release of biologically active silver from nanosilver surfaces. ACS Nano. 2010;4(11):6903–13. doi: 10.1021/nn102272n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu JY, Hurt RH. Ion Release Kinetics and Particle Persistence in Aqueous Nano-Silver Colloids. Environ Sci Technol. 2010;44(6):2169–2175. doi: 10.1021/es9035557. [DOI] [PubMed] [Google Scholar]
- 25.Kittler S, Greulich C, Diendorf J, Koller M, Epple M. Toxicity of Silver Nanoparticles Increases during Storage Because of Slow Dissolution under Release of Silver Ions. Chemistry of Materials. 2010;22(16):4548–4554. [Google Scholar]
- 26.McGown DNL, Parfitt GD. Improved Theoretical Calculation of Stability Ratio for Colloidal Systems. Journal of Physical Chemistry. 1967;71(2):449–&. [Google Scholar]
- 27.Gregory J. Approximate Expressions for Retarded van der Waals Interaction. Journal of Colloid and Interface Science. 1981;83(1):138–145. [Google Scholar]
- 28.Gregory J. Interaction of Unequal Double-Layers at Constant Charge. Journal of Colloid and Interface Science. 1975;51(1):44–51. [Google Scholar]
- 29.Honig EP, Roeberse Gj, Wiersema PH. Effect of Hydrodynamic Interaction on Coagulation Rate of Hydrophobic Colloids. Journal of Colloid and Interface Science. 1971;36(1):97–109. [Google Scholar]
- 30.Elimelech M, Gregory J, Jia X, Williams RA. Particle deposition and aggregation: measurement, modelling, and simulation. Butterworth-Heinemann: Oxford; Boston: 1995. p. xv.p. 441. [Google Scholar]
- 31.Fornasiero D, Grieser F. The Kinetics of Electrolyte Induced Aggregation of Carey Lea Silver Colloids. J Colloid Interf Sci. 1991;141(1):168–179. [Google Scholar]
- 32.Martell AE, Smith RM, Motekaitis RJ. NIST Critically Selected Stability Constants of Metal Complexes Database. National Institute of Science and Technology; Gaithersburg, MD: 2004. [Google Scholar]
- 33.Robinson S, Williams PA. Inhibition of protein adsorption onto silica by polyvinylpyrrolidone. Langmuir. 2002;18(23):8743–8748. [Google Scholar]
- 34.Stankus DP, Lohse SE, Hutchison JE, Nason JA. Environ Sci Technol. ASAP; 2010. Interactions between Natural Organic Matter and Gold Nanoparticles Stabilized with Different Organic Capping Agents. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Wazne M, Chou T, Xiao R, Xu S. Influence of Ca2+ and Suwannee River Humic Acid on aggregation of silicon nanoparticles in aqueous media. Water Research. 2011;45(1):105–112. doi: 10.1016/j.watres.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 36.Petosa AR, Jaisi DP, Quevedo IR, Elimelech M, Tufenkji N. Aggregation and Deposition of Engineered Nanomaterials in Aquatic Environments: Role of Physicochemical Interactions. Environ Sci Technol. 2010;44(17):6532–6549. doi: 10.1021/es100598h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.