

Abstract
We describe an infant presenting with contractures of the fingers, a large ventricular septal defect (VSD), and severe pulmonary artery dilatation. He had clinical and echocardiographic features of both neonatal or infantile Marfan syndrome (MFS) and congenital contractural arachnodactyly. After surgical VSD closure, the aortic root developed progressive dilatation while the size of pulmonary artery returned to normal limits. Eventually the diagnosis of MFS was confirmed by DNA analysis.
1. Introduction
In infancy the diagnosis of Marfan syndrome may be difficult, and clinical presentation and prognosis are variable. We present a special case of infantile Marfan syndrome with a congenital heart disease.
2. Clinical Presentation
Our patient is a boy born to healthy parents (father is 37 years old, mother is 29 years old), who lives in Surinam. The pregnancy was uneventful, and he was born at a normal term. He had two healthy sisters of 6 and 9 years old. Family history was noncontributory. At the age of 4 months he developed heart failure and echocardiographic examination showed a large ventricular septal defect (VSD) and atrial septal defect (ASD) secundum type. The patient was sent to our institution for cardiac surgery at the age of 8 months. Physical examination showed a tall and dystrophic boy with a height of 75.0 cm (+1.5 SDS), weight of 5.5 kg (<−2.5 SDS), and an arm span of 72.0 cm (ratio arm span/height is 0.96). His target height is 187 cm (+0.4 SDS). He had no distinguished facial anomalies with a normal palate and normal ears. He had a pectus carinatum, arachnodactyly, and contractures of both proximal interphalanges of the third and fourth digits. He was dyspnoeic and tachypnoeic, and cardiac examination showed a hyperdynamic precordium, fixed splitting of the second heart sound, and no cardiac murmurs. Echocardiography showed a secundum-type ASD and a 17 mm muscular inlet VSD with a large left- to-right shunt causing pulmonary flow hypertension. The right atrium and right ventricle were dilated. Both mitral valve (MV) and tricuspid valve (TV) had a mild prolapse with a normal function. The pulmonary valve (PV), trunk, and branches were severely dilated with the diameter of the main pulmonary artery of 27.5 mm (Table 1 and Figure 1). The tricuspid aortic valve (AoV) showed normal function, and the aortic root diameter was dilated (Table 1). Surgical correction included primary closure of the ASD and closure of the VSD with a Gore-Tex patch. The postoperative course was uneventful with prompt recovery. Based on the clinical and echocardiographic features the differential diagnosis consisted of congenital contractural arachnodactyly (CCA) and neonatal or infantile Marfan syndrome (MFS). At that time no further gene testing was performed. After recovery the patient went home to Surinam, where the followup continued up to an age of 6 years.
Table 1.
Aortic root diameters and pulmonary artery diameters during followup.
| Age (yr) |
Height cm (SDS) |
Weight (SDS) |
BSA (m2) |
Aortic root mm (z-score) |
PV mm (z-score) |
MPA mm (z-score) |
|---|---|---|---|---|---|---|
| 0.7 | 75.0 (+1.5) | 5.5 (<−2.5) | 0.34 | 22.0 (+8.6) | 22.0 (+6.2) | 27.5 (+8) |
| 0.8 | 75.1 (+1.5) | 5.6 (<−2.5) | 0.34 | 23.4 (+9.2) | 17.4 (+4.0) | 21.2 (+6) |
| 3.7 | 114.7 (>+2.5) | 16.0 (<−2.5) | 0.73 | 32.3 (+8.4) | 20.0 (+1.9) | 21.0 (+2) |
| 5.7 | 129.0 (+2.5) | 18.0 (<−2.5) | 0.83 | 38.7 (+9.9) | 19.0 (+0.9) | 19.1 (+1) |
BSA: body surface area; PV: pulmonary valve; MPA: main pulmonary artery. z-score according to Daubeney ([1], 284/id).
Figure 1.
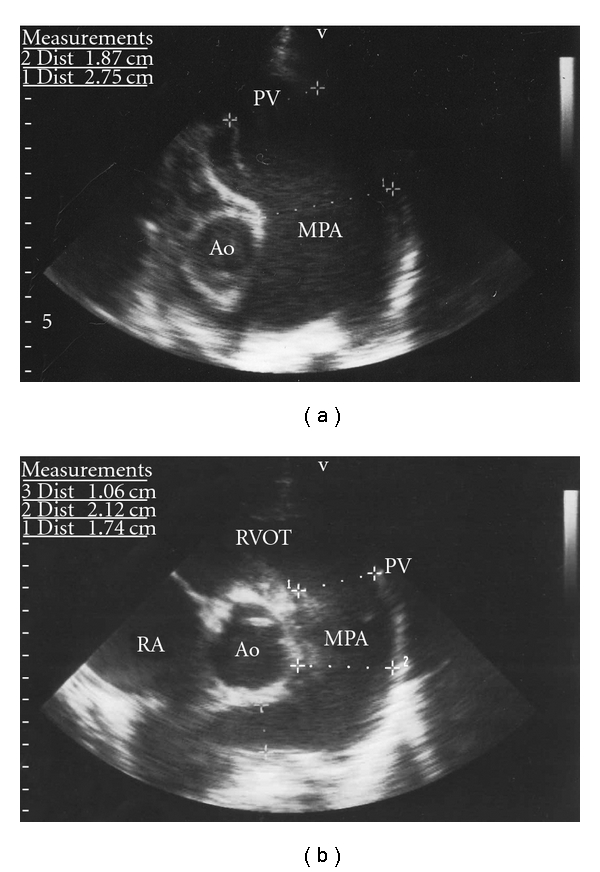
Parasternal short-axis view of the pulmonary valve (PV) and main pulmonary artery (MPA) of the patient preoperatively (a) and directly postoperatively (b). The severe dilatation of the PV (diameter 22 mm) and MPA (diameter 27 mm) diminished directly postoperatively, almost to normal limits (17.4 mm and 21.2 mm, resp.). RA: right atrium; RVOT: right ventricle outflow tract; Ao: aorta.
During followup his height was excessive (SDS > 2.5), the contractures of the fingers were progressive, and he was wearing glasses because of myopia. Psychomotor development was normal. Interestingly, echocardiographic examinations showed significant decrease of diameters of the pulmonary valve and main pulmonary artery directly after operation which remained within normal limits during followup (Table 1, Figure 1). However, the diameter of the aortic root increased from 22 to 23.4 mm directly after operation and with progressive dilatation to 38.7 mm during followup (Table 1, Figure 2). Thus far, aortic valve function remained normal. The mild prolapse of the mitral valve remained stable with trivial mitral regurgitation. Because of progressive aortic root dilatation beta-blocking therapy was started at the age of 5 years.
Figure 2.
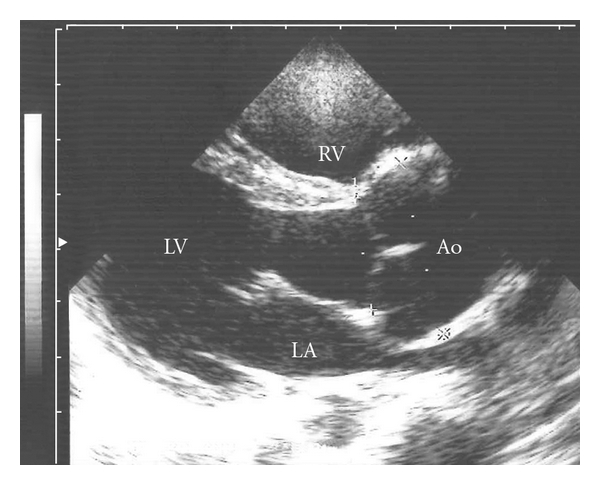
Parasternal long-axis view of left ventricle (LV) shows severe dilatation of the aortic root at the age of almost 4 years. Diameter of the aortic root is 32.2 mm. LA: left atrium; LV: left ventricle; RV: right ventricle; Ao: aorta.
At that time-gene testing was performed to further differentiate between infantile MFS and CCA. Using denaturing high-performance liquid chromatography (DHPLC), multiplex ligation-dependent probe amplification (MLPA kit P065/P066 v3), and DNA sequence-analysis in 2008, we found a frameshift mutation encoding a stop-mutation in the FBN1 gene c.3396delA (p.Glu1133ArgfsX29) confirming the diagnosis of infantile Marfan syndrome.
3. Discussion
In infancy, the diagnosis of MFS can be differentiated in neonatal MFS, severe infantile MFS or early-onset form of MFS, and infants with a positive family mutation without major features at that moment. Differentiation is important because treatment and prognosis will be different, but this may be difficult [2–6]. Marfan syndrome is an autosomal dominant inherited disorder of connective tissue in which ocular, skeletal, cardiovascular, integumentary, pulmonary, and neurological features may be present in a highly variable degree [7–9]. The diagnosis is still a clinical one, based on fulfillment of diagnostic criteria [10]. It is caused by missense mutations in FBN1 gene [11] on chromosome 15q21.1, the gene encoding fibrillin-1, a principle component of extracellular matrix microfibrils. A Marfan-like phenotype can be caused by mutations in the TGFBR2 gene on chromosome 3, encoding TGF-beta receptor 2 [12]. In the classic form of Marfan syndrome, isolated occurrence is in 25–35% of the patients. Fibrillin indirectly controls TGF-beta activation, and dysregulation of TGF-beta may play a role in the pathophysiology of Marfan syndrome [13–23]. Main cardiovascular features are aortic root dilatation and/or aorta dissection and MV prolapse. Prognosis is mainly determined by progressive aortic root dilatation, potentially resulting in dissection and rupture. Prophylactic aortic root repair has raised the life expectancy by 30 years or more [24, 25].
Neonatal Marfan syndrome is a different group of patients with 50% mortality before the first year of life caused by heart failure. The definition of neonatal Marfan syndrome is an estimated diagnosis before 3 months of age, congenital pulmonary emphysema, severe atrioventricular valve (TV and MV) regurgitation in combination with congenital arachnodactyly, contractures, megalocornea, iridodonesis, rocker bottom feet, crumpled ears, and loose skin [3, 26, 27]. Especially congenital pulmonary emphysema and MV and/or TV insufficiency are very common (specific) in neonatal Marfan syndrome [3, 28]. The mutations are located in the central region of the FBN1 gene (exons 24–32) and all are de novo mutations. However, mutations of patients with the classic form of Marfan syndrome have also been found in this region [4, 6, 26, 28–36].
Clinically our patient had severe infantile Marfan syndrome, early diagnosed because of the symptoms of a large ASD and VSD. We found a severely dilated pulmonary artery before operation, due to a large left-to-right shunt and pulmonary flow hypertension. Although pulmonary artery dilatation is a common finding in patients of all ages with MFS [37, 38], this case illustrates that the main pulmonary artery can dilate extremely due to increased pulmonary artery pressure and flow which appeared to be reversible after normalization of pulmonary artery pressure. In contrast, the aortic root showed progressive dilatation after shunt closure and improvement of left ventricular cardiac output. Congenital heart disease in MFS is rare and only a few cases have been described in literature [2, 4, 39–52]. Patent ductus arteriosus and atrial septal defect have been described in 4–6% of the patients [2, 4, 39, 41, 44, 47, 51] and may need surgical or interventional closure. The use of septal occluders should be considered with caution in the Marfan syndrome, because of risk of penetration [47]. An isolated VSD, without complex heart defect, as in our patient, has been reported in only 5 cases (infants and adults) in literature over the last 40 years [39–41, 46, 50, 52]. Coarctation of the aorta was reported once in a 10-year-old girl [45]. Three cases with complex congenital heart disease were described [42, 48, 49]. Congenital heart disease appears to be more common in Beals-Hecht syndrome or congenital contractural arachnodactyly (CCA), a dominantly inherited disorder of connective tissue characterized by multiple contractures, arachnodactyly, dolichostenomelia, scoliosis, and external ear anomalies, with mutations in FBN2 gene [53, 54]. Skeletal features are almost the same as in Marfan syndrome, and dilatation of the aortic root and mitral valve prolapse, although less common, can also occur in CCA. The congenital heart defects described in CCA are very similar to those in MFS, including ASD, VSD, patent ductus arteriosus, coarctation, or interruption of the aorta [55, 56].
4. Conclusion
This special case of infantile MFS with congenital heart disease demonstrates severe dilatation of both great arteries with reversibility of pulmonary artery dilatation after normalization of the pulmonary artery pressure. It further illustrates that the clinical phenotypes of MFS and CCA show significant overlap and that genetic testing was required for final differentiation.
Abbreviations
- AoV:
Aortic valve
- ASD:
Atrial septal defect
- CCA:
Congenital contractural arachnodactyly
- FBN1:
Fibrillin-1
- FBN2:
Fibrillin-2
- MFS:
Marfan syndrome
- MPA:
Main pulmonary artery
- MV:
Mitral valve
- PV:
Pulmonary valve
- SDS:
Standard deviation score
- TV:
Tricuspid valve
- TGFBR2:
TGF-beta receptor 2
- TGF:
Tissue growth factor
- VSD:
Ventricular septal defect.
References
- 1.Daubeney PE, Blackstone EH, Weintraub RG, Slavik Z, Scanlon J, Webber SA. Relationship of the dimension of cardiac structures to body size: an echocardiographic study in normal infants and children. Cardiology in the Young. 1999;9(4):402–410. doi: 10.1017/s1047951100005217. [DOI] [PubMed] [Google Scholar]
- 2.Geva T, Sanders SP, Diogenes MS, Rockenmacher S, Van Praagh R. Two-dimensional and Doppler echocardiographic and pathologic characteristics of the infantile Marfan syndrome. The American Journal of Cardiology. 1990;65(18):1230–1237. doi: 10.1016/0002-9149(90)90979-b. [DOI] [PubMed] [Google Scholar]
- 3.Hennekam RC. Severe infantile Marfan syndrome versus neonatal Marfan syndrome. American Journal of Medical Genetics. 2005;139(1):1–15. doi: 10.1002/ajmg.a.30979. [DOI] [PubMed] [Google Scholar]
- 4.Morse RP, Rockenmacher S, Pyeritz RE, et al. Diagnosis and management of infantile Marfan syndrome. Pediatrics. 1990;86(6):888–895. [PubMed] [Google Scholar]
- 5.Tiecke F, Katzke S, Booms P, et al. Classic, atypically severe and neonatal Marfan syndrome: twelve mutations and genotype-phenotype correlations in FBN1 exons 24–40. European Journal of Human Genetics. 2001;9(1):13–21. doi: 10.1038/sj.ejhg.5200582. [DOI] [PubMed] [Google Scholar]
- 6.Wang M, Wang JY, Cisler J, et al. Three novel fibrillin mutations in exons 25 and 27: classic versus neonatal Marfan syndrome. Human Mutation. 1997;9(4):359–362. doi: 10.1002/(SICI)1098-1004(1997)9:4<359::AID-HUMU10>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 7.Mckusick VA. The cardiovascular aspects of Marfan’s syndrome: a heritable disorder of connective tissue. Circulation. 1955;11(3):321–342. doi: 10.1161/01.cir.11.3.321. [DOI] [PubMed] [Google Scholar]
- 8.Pyeritz RE. The Marfan syndrome. Annual Review of Medicine. 2000;51:481–510. doi: 10.1146/annurev.med.51.1.481. [DOI] [PubMed] [Google Scholar]
- 9.Pyeritz RE, Mckusick VA. The Marfan syndrome: diagnosis and management. New England Journal of Medicine. 1979;300(14):772–777. doi: 10.1056/NEJM197904053001406. [DOI] [PubMed] [Google Scholar]
- 10.De Paepe A, Devereux RB, Dietz HC, Hennekam RCM, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. American Journal of Medical Genetics. 1996;62(4):417–426. doi: 10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 11.Lee B, Godfrey M, Vitale E, et al. Linkage of Marfan syndrome and a phenotypically related disorder to two different fibrillin genes. Nature. 1991;352(6333):330–334. doi: 10.1038/352330a0. [DOI] [PubMed] [Google Scholar]
- 12.Mizuguchi T, Collod-Beroud G, Akiyama T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nature Genetics. 2004;36(8):855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byers PH. Determination of the molecular basis of Marfan syndrome: a growth industry. Journal of Clinical Investigation. 2004;114(2):161–163. doi: 10.1172/JCI22399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dallas SL, Keene DR, Bruder SP, et al. Role of the latent transforming growth factor beta binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. Journal of Bone and Mineral Research. 2000;15(1):68–81. doi: 10.1359/jbmr.2000.15.1.68. [DOI] [PubMed] [Google Scholar]
- 15.De Crescenzo G, Grothe S, Zwaagstra J, Tsang M, O’Connor-McCourt MD. Real-time monitoring of the interactions of transforming growth factor-beta (TGF-beta) isoforms with latency-associated protein and the ectodomains of the TGF-beta Type II and III receptors reveals different kinetic models and stoichiometries of binding. Journal of Biological Chemistry. 2001;276(32):29632–29643. doi: 10.1074/jbc.M009765200. [DOI] [PubMed] [Google Scholar]
- 16.Isogai Z, Ono RN, Ushiro S, et al. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. Journal of Biological Chemistry. 2003;278(4):2750–2757. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- 17.Kaartinen V, Warburton D. Fibrillin controls TGF-beta activation. Nature Genetics. 2003;33(3):331–332. doi: 10.1038/ng0303-331. [DOI] [PubMed] [Google Scholar]
- 18.Kanzaki T, Olofsson A, Moren A, et al. TGF-beta 1 binding protein: a component of the large latent complex of TGF-beta 1 with multiple repeat sequences. Cell. 1990;61(6):1051–1061. doi: 10.1016/0092-8674(90)90069-q. [DOI] [PubMed] [Google Scholar]
- 19.Miyazono K, Olofsson A, Colosetti P, Heldin CH. A role of the latent TGF-beta 1-binding protein in the assembly and secretion of TGF-beta 1. EMBO Journal. 1991;10(5):1091–1101. doi: 10.1002/j.1460-2075.1991.tb08049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nature Genetics. 2003;33(3):407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 21.Saharinen J, Hyytiainen M, Taipale J, Keski-Oja J. Latent transforming growth factor-beta binding proteins (LTBPs)—sructural extracellular matrix proteins for targeting TGF-beta action. Cytokine and Growth Factor Reviews. 1999;10(2):99–117. doi: 10.1016/s1359-6101(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 22.Saharinen J, Keski-Oja J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Molecular Biology of the Cell. 2000;11(8):2691–2704. doi: 10.1091/mbc.11.8.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saharinen J, Taipale J, Keski-Oja J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO Journal. 1996;15(2):245–253. [PMC free article] [PubMed] [Google Scholar]
- 24.Pyeritz RE. Marfan syndrome: 30 years of research equals 30 years of additional life expectancy. Heart. 2009;95(3):173–175. doi: 10.1136/hrt.2008.160515. [DOI] [PubMed] [Google Scholar]
- 25.Silverman DI, Burton KJ, Gray J, et al. Life expectancy in the Marfan syndrome. The American Journal of Cardiology. 1995;75(2):157–160. doi: 10.1016/s0002-9149(00)80066-1. [DOI] [PubMed] [Google Scholar]
- 26.Booms P, Cisler J, Mathews KR, et al. Novel exon skipping mutation in the fibrillin-1 gene: two ‘hot spots’ for the neonatal Marfan syndrome. Clinical Genetics. 1999;55(2):110–117. doi: 10.1034/j.1399-0004.1999.550207.x. [DOI] [PubMed] [Google Scholar]
- 27.Revencu N, Quenum G, Detaille T, Verellen G, De Paepe A, Verellen-Dumoulin C. Congenital diaphragmatic eventration and bilateral uretero-hydronephrosis in a patient with neonatal Marfan syndrome caused by a mutation in exon 25 of the FBN1 gene and review of the literature. European Journal of Pediatrics. 2004;163(1):33–37. doi: 10.1007/s00431-003-1330-8. [DOI] [PubMed] [Google Scholar]
- 28.Heide HT, Schrander-Stumpel CT, Pals G, Delhaas T. Neonatal Marfan syndrome: clinical report and review of the literature. Clinical Dysmorphology. 2005;14(2):81–84. [PubMed] [Google Scholar]
- 29.Abdel-Massih T, Goldenberg A, Vouhe P, et al. Marfan syndrome in the newborn and infant less than 4 months: a series of 9 patients. Archives des Maladies du Coeur et des Vaisseaux. 2002;95(5):469–472. [PubMed] [Google Scholar]
- 30.Buntinx IM, Willems PJ, Spitaels SE, Van Reempst PJ, De Paepe AM, Dumon JE. Neonatal Marfan syndrome with congenital arachnodactyly, flexion contractures, and severe cardiac valve insufficiency. Journal of Medical Genetics. 1991;28(4):267–273. doi: 10.1136/jmg.28.4.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elcioglu NH, Akalin F, Elcioglu M, Comeglio P, Child AH. Neonatal Marfan syndrome caused by an exon 25 mutation of the Fibrillin-1 gene. Genetic Counseling. 2004;15(2):219–225. [PubMed] [Google Scholar]
- 32.Gruber MA, Graham TP, Jr., Engel E, Smith C. Marfan syndrome with contractural arachnodactyly and severe mitral regurgitation in a premature infant. Journal of Pediatrics. 1978;93(1):80–82. doi: 10.1016/s0022-3476(78)80608-8. [DOI] [PubMed] [Google Scholar]
- 33.Jacobs AM, Toudjarska I, Racine A, Tsipouras P, Kilpatrick MW, Shanske A. A recurring FBN1 gene mutation in neonatal Marfan syndrome. Archives of Pediatrics and Adolescent Medicine. 2002;156(11):1081–1085. doi: 10.1001/archpedi.156.11.1081. [DOI] [PubMed] [Google Scholar]
- 34.Ramaswamy P, Lytrivi ID, Nguyen K, Gelb BD. Neonatal Marfan syndrome: in utero presentation with aortic and pulmonary artery dilatation and successful repair of an acute flail mitral valve leaflet in infancy. Pediatric Cardiology. 2006;27(6):763–765. doi: 10.1007/s00246-006-1378-0. [DOI] [PubMed] [Google Scholar]
- 35.Tekin M, Cengiz FB, Ayberkin E, et al. Familial neonatal Marfan syndrome due to parental mosaicism of a missense mutation in the FBN1 gene. American Journal of Medical Genetics. 2007;143(8):875–880. doi: 10.1002/ajmg.a.31660. [DOI] [PubMed] [Google Scholar]
- 36.Wang M, Kishnani P, Decker-Phillips M, Kahler SG, Chen YT, Godfrey M. Double mutant fibrillin-1 (FBN1) allele in a patient with neonatal Marfan syndrome. Journal of Medical Genetics. 1996;33(9):760–763. doi: 10.1136/jmg.33.9.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De BJ, Loeys B, Devos D, Dietz H, De SJ, De PA. A critical analysis of minor cardiovascular criteria in the diagnostic evaluation of patients with Marfan syndrome. Genetics in Medicine. 2006;8(7):401–408. doi: 10.1097/01.gim.0000223550.41849.e3. [DOI] [PubMed] [Google Scholar]
- 38.Nollen GJ, van Schijndel KE, Timmermans J, et al. Pulmonary artery root dilatation in Marfan syndrome: quantitative assessment of an unknown criterion. Heart. 2002;87(5):470–471. doi: 10.1136/heart.87.5.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bauer M, Siniawski H, Bauer U, et al. Ventricular septal aneurysm in a patient with Marfan syndrome. Zeitschrift fur Kardiologie. 2000;89(8):702–705. doi: 10.1007/s003920070199. [DOI] [PubMed] [Google Scholar]
- 40.Bilgrami NL, Tewari SG, Aslam M, Khan ZA. Marfan syndrome with microcornea, aphakia and ventricular septal defect. Case report. Indian Heart Journal. 1981;33(2):78–80. [PubMed] [Google Scholar]
- 41.Bolande RP, Tucker AS. Pulmonary emphysema and other cardiorespiratory lesions as part of the Marfan abiotrophy. Pediatrics. 1964;33:356–366. [PubMed] [Google Scholar]
- 42.Childers RW, McCrea PC. Absence of the pulmonary valve. A case occuring in the Marfan syndrome. Circulation. 1964;29:598–603. doi: 10.1161/01.cir.29.4.598. [DOI] [PubMed] [Google Scholar]
- 43.Cronin CC, Harris AM. Atrial fibrillation and interatrial septal aneurysm in a patient with Marfan’s syndrome. International Journal of Cardiology. 1992;34(1):115–117. doi: 10.1016/0167-5273(92)90095-k. [DOI] [PubMed] [Google Scholar]
- 44.Figueiredo S, Martins E, Lima MR, Alvares S. Cardiovascular manifestations in Marfan syndrome. Revista Portuguesa de Cardiologia. 2001;20(12):1203–1218. [PubMed] [Google Scholar]
- 45.Headley RN, Carpenter HM, Sawyer CG. Unusual features of Marfan’s syndrome including two postmortem studies. The American Journal of Cardiology. 1963;11(2):259–266. doi: 10.1016/0002-9149(63)90068-7. [DOI] [PubMed] [Google Scholar]
- 46.Karoly P, Gyorgy B, Kornél B. Isolated ventricular septal defect in Marfan’s syndrome. Orvosi Hetilap. 1969;110(23):1325–1328. [PubMed] [Google Scholar]
- 47.Loeffelbein F, Schlensak C, Dittrich S. Penetration of left and right atrial wall and aortic root by an Amplatzer atrial septal occluder in a nine year old boy with Marfan syndrome: case report. Journal of Cardiothoracic Surgery. 2008;3, article 25(1) doi: 10.1186/1749-8090-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Metelka R, Dusek J, Vomacka J. Dextrocardia and Marfan's syndrome. Acta Universitatis Palackianae Olomucensis Facultatis Medicae. 1992;133:43–45. [PubMed] [Google Scholar]
- 49.Nathan D, Kraus J, Deutsch V, Neufeld HN. Dilatation of the aorta and pulmonary artery with aortic and pulmonary insufficiency in the presence of a ventricular septal defect and infundibular pulmonic stenosis. Report of a case of forme fruste of the Marfan syndrome. Diseases of the Chest. 1966;50(2):199–205. doi: 10.1378/chest.50.2.199. [DOI] [PubMed] [Google Scholar]
- 50.Roberts WC, Honig HS. The spectrum of cardiovascular disease in the Marfan syndrome: a clinico-morphologic study of 18 necropsy patients and comparison to 151 previously reported necropsy patients. American Heart Journal. 1982;104(1):115–135. doi: 10.1016/0002-8703(82)90650-0. [DOI] [PubMed] [Google Scholar]
- 51.Stuart AG, Williams A. Marfan’s syndrome and the heart. Archives of Disease in Childhood. 2007;92(4):351–356. doi: 10.1136/adc.2006.097469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yajnik VH, Kshatriya PK, Vaidya MH, Vassa NT. Marfan’s syndrome, ventricular septal defect and basilar impression in one patient. Journal of the Indian Medical Association. 1975;64(9):242–244. [PubMed] [Google Scholar]
- 53.Beals RK, Hecht F. Congenital contractural arachnodactyly. A heritable disorder of connective tissue. Journal of Bone and Joint Surgery. 1971;53(5):987–993. [PubMed] [Google Scholar]
- 54.Callewaert BL, Loeys BL, Ficcadenti A, et al. Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: report of 14 novel mutations and review of the literature. Human Mutation. 2009;30(3):334–341. doi: 10.1002/humu.20854. [DOI] [PubMed] [Google Scholar]
- 55.Anderson RA, Koch S, Camerini-Otero RD. Cardiovascular findings in congenital contraction arachnodactyly: report of an affected kindred. American Journal of Medical Genetics. 1984;18(2):265–271. doi: 10.1002/ajmg.1320180210. [DOI] [PubMed] [Google Scholar]
- 56.Macnab AJ, D’Orsogna L, Cole DE, Baguley PE, Adderley RJ, Patterson MW. Cardiac anomalies complicating congenital contractural arachnodactyly. Archives of Disease in Childhood. 1991;66(10):1143–1146. doi: 10.1136/adc.66.10_spec_no.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]