

Abstract
Interactions between mucosal surfaces and microbial microbiota are key to host defense, health, and disease. These surfaces are exposed to high numbers of microbes and must be capable of distinguishing between those that are beneficial or avirulent and those that will invade and cause disease. Our understanding of the mechanisms involved in these discriminatory processes has recently begun to expand as new studies bring to light the importance of epithelial cells and novel immune cell subsets such as Th17 T cells in these processes. Elucidating how these mechanisms function will improve our understanding of many diverse diseases and improve our ability to treat patients suffering from these conditions. In our voyage to discover these mechanisms, mucosal interactions with opportunistic commensal organisms such as the fungus Candida albicans provide insights that are invaluable. Here, we review current knowledge of the interactions between C. albicans and epithelial surfaces and how this may shape our understanding of microbial-mucosal interactions.
1. Introduction
Fungal diseases became recognised as being of clinical importance in the second half of the last century largely due to a combination of rising numbers of patients with immunodeficiency illnesses such as HIV infections, advances in medical treatments such as cancer therapy and transplantation, and improvements in general life expectancies. The incidence of fungal infections has increased dramatically over the past two to three decades and this trend will inevitably continue into the 21st century, particularly as further improvements are made in health care for immunocompromised patients. Thus, these infections will become an increasingly pressing problem with ever mounting cost pressures on national health facilities.
Candida species are the most common fungal pathogens of humans and the causative agents of oral, gastrointestinal, and vaginal candidiasis, giving rise to severe morbidity in millions of individuals worldwide. Vaginal candidiasis alone affects ~75% of women at least once during fertile age [1, 2], equating to ~30 million infection episodes/year. Candida infections are also the most common oral manifestation of HIV infection, with 50% of HIV+ patients and 90% AIDS patients suffering from oral candidiasis [3–5]. With ~4 million cases of HIV/year, this equates to ~2 million oral candidiasis cases/year. Indeed, one of the biggest killers of the immunocompromised population is fungal infection. Candida species also cause mucosal diseases in the elderly and edentulous individuals, such as Candida-associated denture stomatitis. Furthermore, depending on the study, Candida infections are also the 3rd or 4th most common hospital-acquired bloodstream infection, making Candida species as medically important as many mainstream bacterial infections including Enterococci (E. coli) and Pseudomonas spp [6, 7]. In the USA, yearly healthcare costs for systemic fungal infections are ~$2.6 billion, of which Candida infections account for ~$1.8 billion [8]. European Union healthcare costs are estimated to be similar. Furthermore, when taking into account mucosal infections, true healthcare costs are likely to be far higher, although precise figures are scarce. Therefore, Candida pathogens carry an immense health burden and represent a major socioeconomic challenge for worldwide communities.
Therefore, it is important to understand the mechanisms involved in host-Candida interactions, particularly those involved in initiating immune responses and in discriminating between the commensal and pathogenic forms of this fungus. As such, the interactions of Candida with cells of the host immune system have been widely studied with several key reports indicating how Candida and other fungal species are detected by macrophages, dendritic cells, and neutrophils. However, given that the vast majority of Candida infections occur at mucosal surfaces, recent interest has turned towards investigating the interactions between Candida and epithelial cells (ECs) and how this might elicit protective immunity.
2. C. albicans and Mucosal Surfaces
With the increases in our knowledge of the human microbiome and the interplay with its host, it has become increasingly evident that there exists a highly specialised set of interactions between host organism, residential microbiota, and pathogenic microbes. These interactions lead to either a degree of mutualism in the case of resident, commensal microbes, or breaches in the epithelial barrier followed by disease pathology and immune activation in the case of pathogenic microbes. Exactly how the host discriminates between commensal and pathogenic microbes is not well understood but is key to our understanding of health and disease. Of particular importance is the identification of host mechanisms that discriminate between the commensal and pathogenic states of “opportunistic” microbes, such as the fungus Candida albicans, as this will provide valuable insights into how we can manipulate host immunity to control such infections. In particular, understanding these mechanisms will allow us to understand and potentially manipulate immunological events that allow chronic infection by this fungus.
Candida species commonly reside as commensal organisms, being part of the normal microbiome in the gut, oral cavity, or vagina in approximately 50% of the population. Although normally these fungi cause no pathology, if there are changes in the local environment, such as alterations in normal microbiota or compromised local immune defences, then these fungi can become pathogenic. As such, they cause mucosal disease in a significant proportion of immunosuppressed patients and women of fertile age [2] with the majority of these individuals experiencing superficial mucosal candidiasis such as thrush. How the host is able to recognise this shift and respond to control these infections is not well understood but has become the subject of growing research interest. Many putative virulence factors have been proposed as playing roles in C. albicans infections [9], but of these the most studied and widely accepted is hypha formation leading to invasion [10, 11]. What role, if any, these factors play in host discriminatory responses has, until recently, been unclear, but studies are beginning to show an important role for ECs within mucosal surfaces in this process. Further, the mechanisms that the fungus uses to allow evasion of acute immune responses, resulting in chronic infections, are only now beginning to be understood. In this paper, we will outline current understanding of the interactions of C. albicans with mucosal surfaces and discuss the important role ECs play in this process.
3. Immune Recognition of C. albicans
The discovery of Toll-like receptors (TLRs) as pattern recognition receptors (PRRs) in the late 1990s lead to a change in our understanding of how pathogens are recognised by the immune system. We now understand that as well as recognition of specific antigens by T-cells (through the T cell receptor) in the adaptive immune response, conserved pathogen associated molecular patterns (PAMPs) are recognised by PRRs as part of the innate immune response. Since the discovery of TLRs, there has been a rapid expansion of characterised PRRs that now includes a panoply of different families and individual molecules. These include the main families of the TLRs, CLRs (C-type lectin receptors), NLRs (Nacht-like receptors), and RLRs (RIG-like receptors) as well as a host of other, individual receptor molecules [12, 13]. Different locations and cell types throughout the body also show differences in the PRRs they express, and, since each PRR recognises individual PAMPs, this may result in differing sensitivities of various body locales to the multitude of microbes that we encounter.
In the case of fungi and particularly C. albicans, the predominant immune cell types involved in combating mucosal infection are neutrophils [14]. Recognition of fungal cells by these cells has been the subject of the majority of antifungal immunity research during the last ten years, culminating in the discovery of a new PRR, Dectin-1 (β-1,3 glucan) [15, 16], and identification of a role for several other PRRs involved in recognition of different cell wall polysaccharides of this pathogen, including TLR2 (phospholipomannan), TLR4 (O-mannan), and mannose receptor (N-mannan) [17–19] (Table 1). These PRRs have been shown to work both independently and in conjunction with one another. For example, Dectin-1 and TLR2 play a role in the recognition of fungal yeasts, each being responsible for separate actions with Dectin-1 inducing phagocytosis, whilst TLR2 activation induces cytokine production [20, 21]. Each can act independently, but together they produce a synergistic response. Although these are the main receptors used by macrophages and neutrophils, other receptors have also been identified, including Dectin-2 [22], mincle [23], DC-SIGN [24, 25], and galectin-3 [26]. The role of these receptors is currently not fully established and is thus a focus of research by different groups; however, Dectin-2 and DC-SIGN have recently been suggested to play an important role in the recognition of high mannose structures [27] and galectin-3 in the recognition of β-1,2 mannosides [26] (Table 1).
Table 1.
Pattern recognition receptors that sense fungal-associated PAMPs.
| Family | Receptor | PAMP | References |
|---|---|---|---|
| TLRs | TLR2 | Phospholipomannan | [19] |
| TLR3 | Double-stranded RNA | [28] | |
| TLR4 | Mannan | [29] | |
| O-linked Mannan residues | [21] | ||
| TLR9 | CpG DNA | [30] | |
|
| |||
| CLRs | Dectin-1 | β-1,3-glucan | [16] |
| Dectin-2 | High-mannose structures | [31] | |
| α-mannans | [22] | ||
| Mannose receptor | Mannan | [32] | |
| MINCLE | Unknown | [23] | |
| Galectin-3 | β-1,2-Mannosides | [26] | |
| DC-SIGN | High-mannose structures | [24] | |
|
| |||
| NLRs | NLRP3 | Unknown | [33] |
|
| |||
| Others | Cdw17 | Unknown | [34] |
4. Epithelial Recognition of Candida albicans and Interaction with Immune Cells
Despite our knowledge of the PRR-mediated interactions between myeloid cells and C. albicans, the relative importance of these interactions in the detection of this fungus at mucosal surfaces is unclear. This is because, when C. albicans colonises a host, there is little evidence that the fungus interacts directly with neutrophils or macrophages in the first instance, rather the initial interaction is with ECs. Therefore, the key question is: do ECs utilise the same PRRs as myeloid cells in the recognition of C. albicans? ECs are known to express a range of PRRs such as TLRs, Dectin-1 and galectins along with their coreceptors and adaptors [35–37]. Expression of TLR2 and TLR5, in particular, is expressed at high levels by oral ECs, which is notable given that these receptors have been associated with epithelial growth, survival, and repair [38, 39]. Interestingly, TLR4 is expressed at extremely low levels on oral ECs [35], implying that ECs may be refractory to initial stimulation by TLR4 ligands such as lipopolysaccharide (LPS) and thus Gram-negative bacteria. The exact composition of the PRRs utilised by ECs to recognise C. albicans upon initial infection is currently unknown. Recently, we demonstrated that TLR2, TLR4, and Dectin-1 do not appear to be involved in activating epithelial immunity as blockade or inhibition of these receptors did not affect the EC cytokine response to C. albicans [40]. This is supported by another study showing a lack of TLR4 involvement in the induction of GM-CSF by C. glabrata [41]. Furthermore, although the fungal PAMPs inducing cytokine responses in myeloid cells are well described, including mannans and β-glucans, we found that none of these PAMPs or the other polysaccharide constituent of the fungal cell wall, chitin, induced cytokine responses in oral ECs [40]. This was also recently demonstrated for skin keratinocytes [42]. Together, these studies suggest that ECs may utilize different receptors for immune activation and/or target different fungal moieties than myeloid cells, indicating that epithelial fungal detection mechanisms may differ from myeloid cell detection mechanisms.
Despite our knowledge of immune cell- and epithelial-fungal interactions, the relative importance of each interaction in the context of a mucosal infection is unclear. One would, however, expect a high level of immunological cross-talk between the ECs, Candida, and local immune cells in order to either maintain homeostasis (commensal state) or to elicit a protective immune response (pathogenic state). To date, this complex but highly interesting area has largely been ignored, but recently it has become evident that these three-way interactions are critical for host defence. One of the ground-breaking studies in this area was undertaken by Weindl et al. [37] using a three-dimensional organotypic oral epithelial model. Such models permit the direct analysis of pathogen-epithelial interactions that are not complicated by nonepithelial factors, and, although these models are not direct mimics of the in vivo environment, they can be supplemented with immune cells to investigate more complex cell-cell interactions that are applicable to the in vivo situation. Using this model, the authors found that when applied alone, C. albicans induced a chemoattractive and proinflammatory cytokine “effector response” but failed to significantly modulate TLR1-10 expression. However, addition of PMNs to the Candida-infection model strongly upregulated epithelial TLR4 expression (~100-fold) and protected against C. albicans infection. No significant alterations in TLR1-10 expression or protection were observed when PMNs were added in the absence of C. albicans. Interestingly, C. albicans-induced cell damage was abolished irrespective of whether the PMNs were applied directly to the epithelium or were separated by a membrane. This demonstrated that (i) PMN-dependent protection against C. albicans infection was independent of PMN migration or direct cell-cell contact with the oral epithelium, and (ii) three-way communication between Candida, EC, and immune cell was essential for TLR4 upregulation and subsequent protection. Further studies demonstrated that Candida invasion and cell injury could be restored by TLR4 blockade or “knockdown” of TLR4 using siRNA, even in the presence of PMNs, demonstrating a direct role of epithelial TLR4 in antifungal protective responses. This data is of specific interest as it demonstrates that although epithelial TLR4 is not required for the initial activation of ECs [40], it is required for subsequent epithelial protection in the presence of immune cells. This is the first description of such a PMN-dependent TLR4-mediated protective mechanism at epithelial surfaces and may provide significant insights into how fungal infections are managed and controlled in mucosal tissues. It also demonstrates that far from being bystanders during infections, ECs play an active and integral role in mucosal protection against pathogens.
5. Epithelial Signalling Detection Mechanisms
Binding of PRRs by their PAMPs is only the first step in the process of recognition. Ligation of PRRs results in activation of a collection of different intracellular signalling pathways which in turn lead to alterations in gene transcription and ultimately changes in protein expression profiles. In myeloid cells, several different signalling pathways are activated by PRR ligation, including the MAPK pathways and the NF-κB pathway (Figure 1). The events leading to the activation of these pathways depend on the triggering receptor. For example, activation of TLRs such as TLR2 and TLR4 leads to interaction of their cytoplasmic TIR (Toll/IL-1 receptor) domain with several different adaptor molecules including MYD88, MAL, TRAM, and TRIF. This results in the assembly of a complex of proteins including IRAK1, 2, and 4 with TRAF6, leading to activation of the MAPK and NF-κB signalling pathways [43], as well as transcription through activation of IRF-3, IRF-5, and IRF-7 [44]. Activation of Dectin-1 and other CLRs leads to activation of similar pathways, although through differing early mechanisms. In contrast to the TLR receptors, CLR ligation results in activation of a CARD9-MALT1-Bcl10 protein complex. This occurs after phosphorylation of an ITAM-like domain in the cytoplasmic domain of the CLR (as with Dectin-1) or co-receptor (e.g., FcRγ for Dectin-2) results in phosphorylation of SYK [15, 45]. Subsequent to these events is the activation of both NF-κB and MAPK signalling and downstream transcriptional events such as activation of the MAPK transcription factor AP-1 heterodimer. Activation of the MAPK pathway also leads to activation of a group of phosphatases known as the dual specificity phosphatases (DUSPs) including MKP1. These phosphatases act to dephosphorylate and thus deactivate the MAPK proteins, ERK1/2, p38, and JNK [46]. As these phosphatases are activated by the MAPK proteins as a result of MAPK signalling, they form part of a negative feedback loop to regulate the activity of these pathways with each DUSP being specific for a different MAPK protein.
Figure 1.
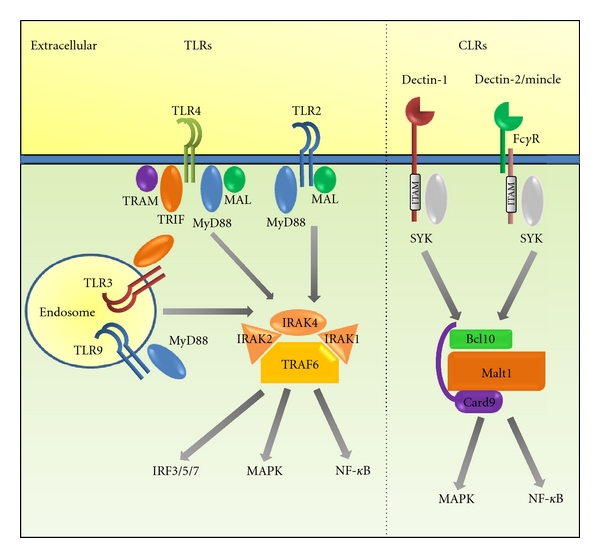
Signal pathway activation by the main TLR and CLR receptors that detect Candida. Signalling through TLRs proceeds mainly via TRAF6 with a variety of adaptor proteins acting as intermediaries between receptor and TRAF6. Foremost among these is MyD88 which is utilised by all known TLRs except TLR3. As well as MyD88, there are other adaptor molecules, including TRIF, MAL, and TRAM, with the different TLRs using different combinations of these adaptors. Activation of these adaptors leads to activation of IRAK1, 2, and 4 followed by ubiquitination of TRAF6 which leads to subsequent activation of downstream signalling pathways. Signalling through CLRs utilises cytoplasmic ITAM domains to interact with the SYK adaptor molecule, activating the Card-9-Bcl10-Malt1 protein complex. Some CLRs, such as Dectin-1, include a modified ITAM domain in their cytoplasmic domain. Others, such as Dectin-2, associate with other receptor molecules, notably the FcRγ and DAP12 proteins, which possess the ITAM domain that transduces the signal into the cell. In all cases, the net effect is to activate the MAPK and NF-κB pathways, leading to upregulation of specific gene transcription. In addition to this, TLRs are also known to activate transcription via members of the IRF family, including IRF3, IRF5, and IRF7.
The exact roles of these pathways in normal, healthy responses to Candida vary depending on the celltype and the context of the stimulation. Different cell types express varying levels of the relevant transcription factors, resulting in alternate transcriptional profiles being activated. Equally, the combination of PRRs on the surface of different cells will result in alternate mechanisms being activated. Currently, almost all studies investigating cellular responses to Candida infection have been carried out using myeloid or lymphoid cells. However, given that most pathogens effect through a mucosal surface, detailed analysis of the responses of other cell types that comprise these surfaces, particularly ECs, may identify novel and unusual mechanisms for host-microbe interactions. Such studies may also identify mechanisms that enable a host to distinguish between commensal and pathogenic microbes in general or between the commensal and pathogenic state of individual organisms, including C. albicans.
Several previous investigations have identified NF-κB and MAPK signalling as the main response mechanisms to bacterial infection in ECs [47, 48]. Further studies have identified NF-κB as an important pathway in EC responses to Candida infections [18, 49]. In our own recent study, we analysed the responses of oral ECs over time to Candida infection and identified a unique mechanism that enables these cells to discriminate between the yeast and hyphal form of C. albicans, which we believe has strong correlations as to whether the fungus is viewed by the host as “pathogenic” or “commensal” [40]. We confirmed that the NF-κB pathway is important in oral EC responses to this fungus. However, we identified MAPK signalling through all three pathways as the mechanism by which oral ECs identify when this fungus becomes invasive and pathogenic. Whilst NF-κB signalling increases linearly over time, MAPK activation shows a bi-phasic response in which a transient, early response through ERK1/2 and JNK signalling induces c-Jun activity in response to the presence of Candida (yeast or hyphae). A second prolonged, late response induced in response to hyphae drives further ERK1/2 and p38 signalling, activating MAPK regulation via the MAPK phosphatase MKP1 and induces c-Fos activity resulting in production of cytokines. Of particular importance is that this second MAPK response is only induced when a sufficient fungal hyphal burden is present, demonstrating that a threshold level of activation needs to be reached prior to epithelial immune activation (Figure 2). This MAPK-based discriminatory pathway may, therefore, provide a mechanism for epithelial tissues to remain quiescent in the presence of low-fungal burdens whilst responding specifically and strongly to hyphae when burdens increase. We propose that this mechanism may comprise a “danger response” pathway, which may be critical in identifying when this normally commensal fungus has become pathogenic. Failure or deficiencies in this “danger response” mechanism could lead to a potentially chronic infection that is inefficiently managed by the host, as no “danger” signal would be elicited to recruit an immune response. Thus, it appears that ECs are instrumental in discriminating between the commensal and pathogenic states of opportunistic pathogens and that this discrimination is communicated via the MAPK pathway.
Figure 2.
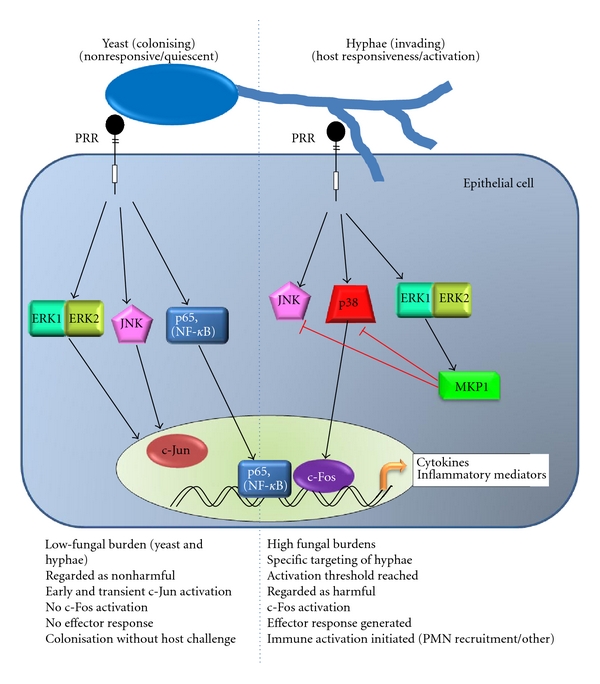
Epithelial cell recognition of C. albicans. Epithelial cells recognise C. albicans via a two-step process [40]. Initial recognition of yeast by surface PRRs results in prolonged activation of NF-kB and an early transient activation of MAPK signalling leading to activation of c-Jun through the ERK1/2 and JNK pathways. When the burden of hyphae passes a threshold, recognition of these hyphae triggers a second, prolonged activation of MAPK signalling. This results in activation of MKP1 through the ERK1/2 pathway and c-Fos via p38 signalling. NF-κB and c-Fos then play essential roles in the transcription of cytokines secreted by the epithelial cells, whilst MKP1 acts as a negative regulator to control the activation of JNK and p38 signalling.
6. Candida-Induced EC Cytokine Responses
Recognition of Candida by host cells leads to activation of a cytokine response profile. For myeloid cells, this profile is fairly well documented and includes release of IL-12, IL-1α/β and TNFα along with other proinflammatory cytokines [17]. Although less well defined, the EC effector response has to some extent been described. We and others have shown that infected ECs produce cytokines and chemokines with a proinflammatory profile [37, 40, 50–52]. Among these are included IL-1α/β, IL-6, G-CSF, GM-CSF, and TNFα as well as the chemokines RANTES, IL-8, and CCL20. In contrast to myeloid and lymphoid cells, however, ECs do not produce IL-12, IFNγ, IL-4 or IL-13. What direct effect these cytokines have on epithelial protection is unclear, although, given that these cytokines act upon both lymphoid and myeloid cells it is likely that they are involved in activating and recruiting these cells into the mucosal layer. For example, IL-8 will recruit neutrophils to the epithelium, subsequently inducing neutrophil-dependent mucosal defence against C. albicans [37].
As well as cytokines and chemokines, Candida infection of ECs results in an increase in expression of matrix metalloproteases (MMPs) [53] which will play a role in remodelling of the epithelium and modulating the barrier function. Infection also results in the upregulation of various antimicrobial peptides such as β-defensins and LL-37 [54, 55]. These antimicrobial peptides have anticandidacidal activity and play a significant role in combating infections and invasion as well as initiating other immune responses [56, 57].
7. Mucosal Surface Immune Responses to C. albicans
The secretion of cytokines and chemokines by ECs in response to Candida invasion will result in the recruitment, differentiation, and activation of a variety of immune cells, including neutrophils, dendritic cells and T cells. The role of neutrophils in anti-Candida mucosal immunity appears to be twofold. As described earlier, neutrophils can induce EC-mediated protection against C. albicans infections through upregulation of TLR4 [37]. Neutrophils can also directly kill Candida cells through ingestion and killing, degranulation, or through the recently discovered Neutrophils extracellular Traps (NETs). NETs occur as a specialised form of neutrophils cell death and comprise a web of chromatin “fibres” coated with serine proteases, antimicrobial proteins, and other neutrophils contents which capture and kill C. albicans on various surfaces [58, 59].
As well as IL-8-recruited neutrophils, secreted CCL20 will recruit the Th17 T cell subset [60]. These cells secrete IL-17 and IL-22 and have been associated with anti-Candida immunity [61]. Recently, dendritic cell recognition of fungi through Dectin-1 and Dectin-2 has been shown to play an instrumental role in driving development of Th17 cells [62], indicating a relationship between fungal infections and this important T cell subset. The exact role of these cells in anti-Candida immunity is not fully understood, with evidence to suggest both a positive [63, 64] and negative [65] role, although it is becoming clear that cytokines secreted by these cells, most notably IL-17 [66–68] and IL-22 [69], play a significant role in antifungal immunity. IL-17 acts on ECs and neutrophils, functioning as a bridge between the adaptive and innate immune responses. Its effects on ECs include induction of antimicrobial peptides, MMPs, and other inflammatory mediators. The role of L-17 in anti-Candida immunity is controversial with evidence to indicate that it both increases [65] and reduces [67, 68] C. albicans burdens after infection through various routes. Interestingly, infection of peripheral blood mononuclear cells by live C. albicans results in inhibition of IL-17 secretion resulting from the effects of C. albicans-released 5-hydroxytryptophan metabolites [70] with a concurrent suppression of immunity to the fungus, suggesting that IL-17 may be important in coordinating immune responses to the fungus. IL-22 has similar effects to IL-17 on ECs but has been suggested to control yeast cell growth, as well as controlling epithelial layer integrity during infection [69], thus helping to control cell numbers and invasion of the epithelium during an infection event. The importance of the Th17 response in mucosal immunity to Candida spp infections is underlined by several recent studies linking defects in the Th17 response and production of IL-17 to cases of chronic mucocutaneous candidiasis (CMC) [67, 71]. This link is further supported by the finding that in cases of autoimmunity with neutralising antibodies to Th17 cytokines (IL-17A, IL-17F, and IL-22), there is an increased incidence of CMC [72]. Despite this, the report that IL-17 may play a deleterious role in anti-Candida immunity [65] suggests that the situation may be far more complex in vivo.
As well as driving innate immunity and neutrophil responses, Th17 cells have also been shown to drive antibody responses at mucosal surfaces, in particular secretory IgA (sIgA). In mice, Th17 cells induce an influx of CD19+ B cells and boost levels of sIgA as well as epithelial expression of polymeric IgA receptor [73]. Increases in secreted IgA at mucosal surfaces have previously been reported [74], indicating that this may be another mechanism by which Th17 responses mediate protection against mucosal candidiasis, especially since sIgA antibodies can inhibit the adherence of C. albicans to epithelial cells [75].
8. Conclusions
Until recently, our understanding of the events and mechanisms involved in host mucosal responses to fungal cells was elementary. In particular, the role of ECs in these events was considered to be relatively unimportant. Recent advances in our knowledge of immunity have resulted in the identification of a novel subset of T cells that produce cytokines targeting ECs, thus forming a direct link involved in maintaining the mucosal barrier. Equally, the discovery of an EC-driven mechanism for protection against Candida infection demonstrates that these cells communicate with immune cells and play a role in combating fungal infections. The elucidation of an epithelial-specific mechanism for identifying the pathogenic state of C. albicans has confirmed the importance of ECs in mediating protective mucosal mechanisms and in discriminating commensal microbes from pathogens. The identification of this role for ECs may open new avenues of research for treatments for use in immunocompromised patients or for those with chronic mucosal infections.
References
- 1.Sobel JD. Pathogenesis and epidemiology of vulvovaginal candidiasis. Annals of the New York Academy of Sciences. 1988;544:547–557. doi: 10.1111/j.1749-6632.1988.tb40450.x. [DOI] [PubMed] [Google Scholar]
- 2.Sobel JD. Pathogenesis and treatment of recurrent vulvovaginal candidiasis. Clinical Infectious Diseases. 1992;14(supplement 1):S148–S153. doi: 10.1093/clinids/14.supplement_1.s148. [DOI] [PubMed] [Google Scholar]
- 3.Korting HC, Ollert M, Georgii A, Froschl M. In vitro susceptibilities and biotypes of Candida albicans isolates from the oral cavities of patients infected with human immunodeficiency virus. Journal of Clinical Microbiology. 1988;26(12):2626–2631. doi: 10.1128/jcm.26.12.2626-2631.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palmer GD, Robinson PG, Challacombe SJ, et al. Aetiological factors for oral manifestations of HIV. Oral Diseases. 1996;2(3):193–197. doi: 10.1111/j.1601-0825.1996.tb00223.x. [DOI] [PubMed] [Google Scholar]
- 5.Phelan JA, Saltzman BR, Friedland GH, Klein RS. Oral findings in patients with acquired immunodeficiency syndrome. Oral Surgery, Oral Medicine, Oral Pathology. 1987;64(1):50–56. doi: 10.1016/0030-4220(87)90116-2. [DOI] [PubMed] [Google Scholar]
- 6.Giusiano G, Mangiaterra M, Saito V, Rojas F, Gomez V, Diaz MC. Fluconazole and itraconazole resistance of yeasts isolated from the bloodstream and catheters of hospitalized pediatric patients. Chemotherapy. 2006;52(5):254–259. doi: 10.1159/000094867. [DOI] [PubMed] [Google Scholar]
- 7.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clinical Infectious Diseases. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 8.Wilson LS, Reyes CM, Stolpman M, Speckman J, Allen K, Beney J. The direct cost and incidence of systemic fungal infections. Value in Health. 2002;5(1):26–34. doi: 10.1046/j.1524-4733.2002.51108.x. [DOI] [PubMed] [Google Scholar]
- 9.Naglik JR, Challacombe SJ, Hube B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiology and Molecular Biology Reviews. 2003;67(3):400–428. doi: 10.1128/MMBR.67.3.400-428.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calderone RA, Fonzi WA. Virulence factors of Candida albicans . Trends in Microbiology. 2001;9(7):327–335. doi: 10.1016/s0966-842x(01)02094-7. [DOI] [PubMed] [Google Scholar]
- 11.Zakikhany K, Naglik JR, Schmidt-westhausen A, Holland G, Schaller M, Hube B. In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cellular Microbiology. 2007;9(12):2938–2954. doi: 10.1111/j.1462-5822.2007.01009.x. [DOI] [PubMed] [Google Scholar]
- 12.Hollmig ST, Ariizumi K, Cruz PD. Recognition of non-self-polysaccharides by C-type lectin receptors Dectin-1 and Dectin-2. Glycobiology. 2009;19(6):568–575. doi: 10.1093/glycob/cwp032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumagai Y, Takeuchi O, Akira S. Pathogen recognition by innate receptors. Journal of Infection and Chemotherapy. 2008;14(2):86–92. doi: 10.1007/s10156-008-0596-1. [DOI] [PubMed] [Google Scholar]
- 14.Fidel PL., Jr. Distinct protective host defenses against oral and vaginal candidiasis. Medical Mycology. 2002;40(4):359–375. [PubMed] [Google Scholar]
- 15.Brown GD. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nature Reviews Immunology. 2006;6(1):33–43. doi: 10.1038/nri1745. [DOI] [PubMed] [Google Scholar]
- 16.Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature. 2001;413(6851):36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 17.Netea MG, Brown GD, Kullberg BJ, Gow NA. An integrated model of the recognition of Candida albicans by the innate immune system. Nature Reviews Microbiology. 2008;6(1):67–78. doi: 10.1038/nrmicro1815. [DOI] [PubMed] [Google Scholar]
- 18.Roeder A, Kirschning CJ, Rupec RA, Schaller M, Weindl G, Korting HC. Toll-like receptors as key mediators in innate antifungal immunity. Medical Mycology. 2004;42(6):485–498. doi: 10.1080/13693780400011112. [DOI] [PubMed] [Google Scholar]
- 19.Jouault T, Ibata-Ombetta S, Takeuchi O, et al. Candida albicans phospholipomannan is sensed through toll-like receptors. Journal of Infectious Diseases. 2003;188(1):165–172. doi: 10.1086/375784. [DOI] [PubMed] [Google Scholar]
- 20.Dennehy KM, Willment JA, Williams DL, Brown GD. Reciprocal regulation of IL-23 and IL-12 following co-activation of Dectin-1 and TLR signaling pathways. European Journal of Immunology. 2009;39(5):1379–1386. doi: 10.1002/eji.200838543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Netea MG, Gow NA, Munro CA, et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. Journal of Clinical Investigation. 2006;116(6):1642–1650. doi: 10.1172/JCI27114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sato K, Yang XL, Yudate T, et al. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. Journal of Biological Chemistry. 2006;281(50):38854–38866. doi: 10.1074/jbc.M606542200. [DOI] [PubMed] [Google Scholar]
- 23.Wells CA, Salvage JA, Li X, et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans . Journal of Immunology. 2008;180(11):7404–7413. doi: 10.4049/jimmunol.180.11.7404. [DOI] [PubMed] [Google Scholar]
- 24.Cambi A, Gijzen K, de Vries JM, et al. The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. European Journal of Immunology. 2003;33(2):532–538. doi: 10.1002/immu.200310029. [DOI] [PubMed] [Google Scholar]
- 25.Taylor PR, Brown GD, Herre J, Williams DL, Willment JA, Gordon S. The role of SIGNR1 and the beta-glucan receptor (Dectin-1) in the nonopsonic recognition of yeast by specific macrophages. Journal of Immunology. 2004;172(2):1157–1162. doi: 10.4049/jimmunol.172.2.1157. [DOI] [PubMed] [Google Scholar]
- 26.Jouault T, El Abed-El Behi M, Martínez-Esparza M, et al. Specific recognition of Candida albicans by macrophages requires galectin-3 to discriminate Saccharomyces cerevisiae and needs association with TLR2 for signaling. Journal of Immunology. 2006;177(7):4679–4687. doi: 10.4049/jimmunol.177.7.4679. [DOI] [PubMed] [Google Scholar]
- 27.Netea MG, Marodi L. Innate immune mechanisms for recognition and uptake of Candida species. Trends in Immunology. 2010;31(9):346–353. doi: 10.1016/j.it.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Muller V, Viemann D, Schmidt M, et al. Candida albicans triggers activation of distinct signaling pathways to establish a proinflammatory gene expression program in primary human endothelial cells. Journal of Immunology. 2007;179(12):8435–8445. doi: 10.4049/jimmunol.179.12.8435. [DOI] [PubMed] [Google Scholar]
- 29.Tada H, Nemoto E, Shimauchi H, et al. Saccharomyces cerevisiae- and Candida albicans-derived mannan induced production of tumor necrosis factor alpha by human monocytes in a CD14- and Toll-like receptor 4-dependent manner. Microbiology and Immunology. 2002;46(7):503–512. doi: 10.1111/j.1348-0421.2002.tb02727.x. [DOI] [PubMed] [Google Scholar]
- 30.Miyazato A, Nakamura K, Yamamoto N, et al. Toll-like receptor 9-dependent activation of myeloid dendritic cells by Deoxynucleic acids from Candida albicans . Infection and Immunity. 2009;77(7):3056–3064. doi: 10.1128/IAI.00840-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGreal EP, Rosas M, Brown GD, et al. The carbohydrate-recognition domain of Dectin-2 is a C-type lectin with specificity for high mannose. Glycobiology. 2006;16(5):422–430. doi: 10.1093/glycob/cwj077. [DOI] [PubMed] [Google Scholar]
- 32.Ezekowitz RA, Sastry K, Bailly P, Warner A. Molecular characterization of the human macrophage mannose receptor: demonstration of multiple carbohydrate recognition-like domains and phagocytosis of yeasts in Cos-1 cells. Journal of Experimental Medicine. 1990;172(6):1785–1794. doi: 10.1084/jem.172.6.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gross O, Poeck H, Bscheider M, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459(7245):433–436. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 34.Jimenez-Lucho V, Ginsburg V, Krivan HC. Cryptococcus neoformans, Candida albicans, and other fungi bind specifically to the glycosphingolipid lactosylceramide (GAl beta 1-4 Glc beta 1-1Cer), a possible adhesion receptor for yeasts. Infection and Immunity. 1990;58(7):2085–2090. doi: 10.1128/iai.58.7.2085-2090.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Backhed F, Hornef M. Toll-like receptor 4-mediated signaling by epithelial surfaces: necessity or threat? Microbes and Infection. 2003;5(11):951–959. doi: 10.1016/s1286-4579(03)00189-8. [DOI] [PubMed] [Google Scholar]
- 36.Hornef MW, Bogdan C. The role of epithelial Toll-like receptor expression in host defense and microbial tolerance. Journal of Endotoxin Research. 2005;11(2):124–128. doi: 10.1179/096805105X35224. [DOI] [PubMed] [Google Scholar]
- 37.Weindl G, Naglik JR, Kaesler S, et al. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. Journal of Clinical Investigation. 2007;117(12):3664–3672. doi: 10.1172/JCI28115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rhee SH, Im E, Riegler M, Kokkotou E, O’Brien M, Pothoulakis C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagenllin in colonic inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(38):13610–13615. doi: 10.1073/pnas.0502174102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaykhiev R, Behr J, Bals R. Microbial patterns signaling via toll-like receptors 2 and 5 contribute to epithelial repair, growth and survival. PLoS One. 2008;3(1) doi: 10.1371/journal.pone.0001393. Article ID e1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moyes DL, Runglall M, Murciano C, et al. A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host and Microbe. 2010;8(3):225–235. doi: 10.1016/j.chom.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li L, Dongari-Bagtzoglou A. Epithelial GM-CSF induction by Candida glabrata . Journal of Dental Research. 2009;88(8):746–751. doi: 10.1177/0022034509341266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Koning HD, Rodijk-Olthuis D, van Vlijmen-Willems IM, et al. A comprehensive analysis of pattern recognition receptors in normal and inflamed human epidermis: upregulation of Dectin-1 in psoriasis. Journal of Investigative Dermatology. 2010;130(11):2611–2620. doi: 10.1038/jid.2010.196. [DOI] [PubMed] [Google Scholar]
- 43.O’Neill LAJ, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Reviews Immunology. 2007;7(5):353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 44.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nature Reviews Immunology. 2006;6(9):644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 45.Kerrigan AM, Brown GD. Syk-coupled C-type lectin receptors that mediate cellular activation via single tyrosine based activation motifs. Immunological Reviews. 2010;234(1):335–352. doi: 10.1111/j.0105-2896.2009.00882.x. [DOI] [PubMed] [Google Scholar]
- 46.Liu Y, Shepherd EG, Nelin LD. MAPK phosphatases—regulating the immune response. Nature Reviews Immunology. 2007;7(3):202–212. doi: 10.1038/nri2035. [DOI] [PubMed] [Google Scholar]
- 47.Handfield M, Baker HV, Lamont RJ. Beyond good and evil in the oral cavity: insights into host-microbe relationships derived from transcriptional profiling of gingival cells. Journal of Dental Research. 2008;87(3):203–223. doi: 10.1177/154405910808700302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kinane DF, Galicia JC, Gorr SU, Stathopoulou PG, Benakanakere M. P. gingivalis interactions with epithelial cells. Frontiers in Bioscience. 2008;13(3):966–984. doi: 10.2741/2736. [DOI] [PubMed] [Google Scholar]
- 49.Pivarcsi A, Bodai L, Rethi B, et al. Expression and function of Toll-like receptors 2 and 4 in human keratinocytes. International Immunology. 2003;15(6):721–730. doi: 10.1093/intimm/dxg068. [DOI] [PubMed] [Google Scholar]
- 50.Dongari-Bagtzoglou A, Fidel PL., Jr. The host cytokine responses and protective immunity in oropharyngeal candidiasis. Journal of Dental Research. 2005;84(11):966–977. doi: 10.1177/154405910508401101. [DOI] [PubMed] [Google Scholar]
- 51.Schaller M, Boeld U, Oberbauer S, Hamm G, Hube B, Korting HC. Polymorphonuclear leukocytes (PMNs) induce protective Th1-type cytokine epithelial responses in an in vitro model of oral candidosis. Microbiology. 2004;150(9):2807–2813. doi: 10.1099/mic.0.27169-0. [DOI] [PubMed] [Google Scholar]
- 52.Villar CC, Kashleva H, Mitchell AP, Dongari-Bagtzoglou A. Invasive phenotype of Candida albicans affects the host proinflammatory response to infection. Infection and Immunity. 2005;73(8):4588–4595. doi: 10.1128/IAI.73.8.4588-4595.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Claveau I, Mostefaoui Y, Rouabhia M. Basement membrane protein and matrix metalloproteinase deregulation in engineered human oral mucosa following infection with Candida albicans . Matrix Biology. 2004;23(7):477–486. doi: 10.1016/j.matbio.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 54.Schneider JJ, Unholzer A, Schaller M, Schafer-Korting M, Korting HC. Human defensins. Journal of Molecular Medicine. 2005;83(8):587–595. doi: 10.1007/s00109-005-0657-1. [DOI] [PubMed] [Google Scholar]
- 55.Li M, Chen Q, Tang R, Shen Y, Liu WD. The expression of beta-defensin-2, 3 and LL-37 induced by Candida albicans phospholipomannan in human keratinocytes. Journal of Dermatological Science. 2011;61(1):72–75. doi: 10.1016/j.jdermsci.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 56.Lopez-Garcia B, Lee PH, Yamasaki K, Gallo RL. Anti-fungal activity of cathelicidins and their potential role in Candida albicans skin infection. Journal of Investigative Dermatology. 2005;125(1):108–115. doi: 10.1111/j.0022-202X.2005.23713.x. [DOI] [PubMed] [Google Scholar]
- 57.Vylkova S, Nayyar N, Li W, Edgerton M. Human beta-defensins kill Candida albicans in an energy-dependent and salt-sensitive manner without causing membrane disruption. Antimicrobial Agents and Chemotherapy. 2007;51(1):154–161. doi: 10.1128/AAC.00478-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Urban CF, Ermert D, Schmid M, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans . PLoS Pathogens. 2009;5(10) doi: 10.1371/journal.ppat.1000639. Article ID e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans and hyphal forms. Cellular Microbiology. 2006;8(4):668–676. doi: 10.1111/j.1462-5822.2005.00659.x. [DOI] [PubMed] [Google Scholar]
- 60.Ghannam S, Dejou C, Pedretti N, et al. CCL20 and beta-defensin-2 induce arrest of human Th17 cells on inflamed endothelium in vitro under flow conditions. Journal of Immunology. 2011;186(3):1411–1420. doi: 10.4049/jimmunol.1000597. [DOI] [PubMed] [Google Scholar]
- 61.Conti HR, Gaffen SL. Host responses to Candida albicans: Th17 cells and mucosal candidiasis. Microbes and Infection. 2010;12(7):518–527. doi: 10.1016/j.micinf.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gringhuis SI, Wevers BA, Kaptein TM, et al. Selective C-Rel activation via Malt1 controls anti-fungal TH-17 immunity by Dectin-1 and Dectin-2. PLoS Pathogens. 2011;7(1) doi: 10.1371/journal.ppat.1001259. Article ID e1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Conti HR, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. Journal of Experimental Medicine. 2009;206(2):299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin L, Ibrahim AS, Xu X, et al. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathogens. 2009;5(12) doi: 10.1371/journal.ppat.1000703. Article ID e1000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zelante T, De Luca A, Bonifazi P, et al. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. European Journal of Immunology. 2007;37(10):2695–2706. doi: 10.1002/eji.200737409. [DOI] [PubMed] [Google Scholar]
- 66.Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans . Journal of Immunology. 2010;185(9):5453–5462. doi: 10.4049/jimmunol.1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eyerich K, Foerster S, Rombold S, et al. Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. Journal of Investigative Dermatology. 2008;128(11):2640–2645. doi: 10.1038/jid.2008.139. [DOI] [PubMed] [Google Scholar]
- 68.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. Journal of Infectious Diseases. 2004;190(3):624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 69.De Luca A, Zelante T, D’Angelo C, et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunology. 2010;3(4):361–373. doi: 10.1038/mi.2010.22. [DOI] [PubMed] [Google Scholar]
- 70.Cheng SC, van de Veerdonk F, Smeekens S, et al. Candida albicans dampens host defense by downregulating IL-17 production. Journal of Immunology. 2010;185(4):2450–2457. doi: 10.4049/jimmunol.1000756. [DOI] [PubMed] [Google Scholar]
- 71.Ng WF, von Delwig A, Carmichael AJ, et al. Impaired TH17 responses in patients with chronic mucocutaneous candidiasis with and without autoimmune polyendocrinopathy-candidiasis- ectodermal dystrophy. Journal of Allergy and Clinical Immunology. 2010;126(5):1006–1015.e4. doi: 10.1016/j.jaci.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 72.Kisand K, Boe Wolff AS, Podkrajsek KT, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. Journal of Experimental Medicine. 2010;207(2):299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jaffar Z, Ferrini ME, Herritt LA, Roberts K. Cutting edge: lung mucosal Th17-mediated responses induce polymeric Ig receptor expression by the airway epithelium and elevate secretory IgA levels. Journal of Immunology. 2009;182(8):4507–4511. doi: 10.4049/jimmunol.0900237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Coogan MM, Sweet SP, Challacombe SJ. Immunoglobulin A (IgA), IgA1, and IgA2 antibodies to Candida albicans in whole and parotid saliva in human immunodeficiency virus infection and AIDS. Infection and Immunity. 1994;62(3):892–896. doi: 10.1128/iai.62.3.892-896.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Epstein JB, Kimura LH, Menard TW, Truelove EL, Pearsall NN. Effects of specific antibodies on the interaction between the fungus Candida albicans and human oral mucosa. Archives of Oral Biology. 1982;27(6):469–474. doi: 10.1016/0003-9969(82)90086-3. [DOI] [PubMed] [Google Scholar]