

Abstract
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) is an inherited form of cardiomyopathy with low penetrance and variable expressivity. Dominant mutations and rare polymorphisms in desmosome genes are frequently identified. We reasoned that individuals with earlier onset disease would have more frequent desmosome gene mutations and rare polymorphisms. Three groups were compared: Young with symptoms attributable to ARVD/C or a diagnosis of ARVD/C at age of 21 years or earlier, Middle with first symptoms or diagnosis age of 22–49 years, and Late with first symptoms or diagnosis at age of 50 or more years. deoxyribonucleic acid (DNA) sequence analysis was performed on five cardiac desmosome genes, and the presence of mutations and rare missense polymorphisms was compared among the three groups. In the entire Young cohort, 20 (67%) had one or more cardiac desmosome gene mutations. The prevalence of cardiac desmosome gene mutations was similar in the Middle (48%) and Late (53%) cohorts (P=0.23). Similar numbers of individuals in each cohort had more than one desmosome gene mutation, although the numbers are too small for statistical comparisons. The prevalence of certain rare missense DNA variants was not different among the cohorts (P=0.71), yet these rare missense alleles were more prevalent in the overall study cohort of 112 ARVD/C participants compared to 100 race-matched controls (P=0.027). The presence of these variants did not associate with the age of onset of ARVD/C or ventricular tachycardia. These findings highlight the complex interplay of environmental and genetic factors contributing to this condition.
Keywords: Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C), Genetics, Sudden cardiac death, Desmosome
Introduction
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) is an inherited form of cardiomyopathy affecting up to 1 in 5,000 individuals in the USA [1, 2]. Although the median age at presentation was 26 years in one study, this included a broad range of ages extending from 2 to 70 years [3]. Factors influencing the age of onset are not well understood.
Over the past several years, it has become clear that many cases of ARVD/C are caused by mutations in genes encoding elements of the cardiac desmosome (PKP2, DSG2, JUP, DSC2, and DSP) [4–8]. Its transmission is typically autosomal dominant, but recessive variants of ARVD/C have also been described [9, 10]. In rare cases, affected individuals have been reported with three distinct desmosome gene mutations, although the absence of a functional assay for novel desmosome variants leads to difficulty in classification [11].
Determination of the age of onset is complicated by a “latent period” during which overt evidence of disease may be lacking, and sudden cardiac death may be the first symptom [12]. Children under the age of 11 years account for less than 2% of ARVD/C [13]. Very little is known about the genotypic characteristics of this subset of ARVD/C individuals. In addition, incomplete penetrance and variable expressivity affect the interpretation of family screening for ARVD/C [14, 15]. Similarly, late onset disease is rare but well described [12, 16]. Genetic factors influencing low penetrance and later onset disease are also poorly understood.
The aim of our study is: (1) To investigate the prevalence of desmosome mutations and multiple mutations (compound and digenic heterozygosity) in young people with ARVD/C compared to more typical age of onset and to those with later onset disease, and (2) to determine whether low-frequency amino acid substitutions that are found among individuals with ARVD/C are similarly prevalent among controls.
Methods
The study population consists of 112 unrelated participants with definite and probable ARVD/C from the Johns Hopkins ARVD registry. Three specific subgroups were identified based on the age of onset of symptoms or, if asymptomatic, the age of diagnosis of definite or probable ARVD/C. The Young cohort consisted of 19 definite ARVD/C and 11 probable ARVD/C probands, age≤ 21 years. The Middle cohort consisted of 56 individuals with definite ARVD/C and 11 with probable ARVD/C, age between 22 and 49 years of age. The Late cohort included 15 probands with definite ARVD/C, age≥50 years. The Middle cohort overlaps with a prior report by our group, but the Young and Late cohorts were expanded by ten (Young) and six (Late) to increase the representation of these groups [11]. Only one member of any family is included in this analysis; family members who provided medical records, blood for deoxyribonucleic acid (DNA) testing, and consent for participation were assessed for cosegregation of mutations with ARVD/C, but they are not included in the three cohorts in order to prevent skewed representation of families with high penetrance.
This study was approved by the Johns Hopkins School of Medicine Institution Review Board and written informed consent was obtained from all study subjects. Clinical history and medical records were obtained at enrollment and at yearly intervals. Family history was determined by interviewing participants or family members. The age of onset of symptoms was defined as the age at which those individuals experienced symptoms attributable to ARVD/C. Symptoms include palpitations, syncope, ventricular tachycardia, sudden cardiac death (SCD), chest discomfort, and symptoms related to right heart failure. All participants underwent a thorough physical examination.
Further testing included 12-lead echocardiogram (ECG; n=112), exercise testing using standard protocols, signal-averaged electrocardiogram (SAECG; n=95), 24-h Holter monitoring (n=78), imaging studies such as echocardiography or cardiac magnetic resonance imaging (MRI; n=112), and right ventricular endomyocardial biopsy (n=39). The presence or absence of ventricular tachycardia or ventricular ectopics and its morphology was determined with the standard 12-lead ECG, the results of 24-h Holter, and exercise testing. SAECG in subjects without preexisting right bundle branch block was considered positive for late potentials if any two of the following were present: (1) filtered QRS duration ≥ 114 ms, (2) low-amplitude signal duration≥38 ms, and (3) RMS20 μV. RV dysfunction and structural abnormalities were determined with imaging studies. Histological evidence of fibrofatty replacement of myocytes was obtained from endomyocardial biopsies of the right ventricular free wall.
The diagnosis of ARVD/C was established based on the criteria set in 1994 by the Task Force of the Working Group of Myocardial and Pericardial Disease of the European Society of Cardiology and the International Society and Federation of Cardiology [17]. The diagnosis of definite ARVD/C is established by the presence of two major criteria, one major and two minor criteria, or four minor criteria. The diagnosis of probable ARVD/C is established by the presence one major and one minor criterion or three minor criteria. The criteria must come from different categories. (1) Structural alteration of the heart (Major: Severe dilatation and reduction of right ventricular ejection fraction with no significant LV impairment; localized right ventricular aneurysms; severe segmental dilatation of the right ventricle. Minor: Mild global right ventricular dilatation and/or dysfunction with normal left ventricle; mild segmental dilatation of the right ventricle; regional right ventricular hypokinesia). (2) Tissue characterization of walls (Major: Fibrofatty replacement of myocardium on endomyocardial biopsy). (3) Repolarization abnormalities (Minor: Inverted T waves in right precordial leads in people aged >12 years, in the absence of right bundle branch block). (4) Depolarization/conduction abnormalities (Major: epsilon waves or localized prolongation >110 ms of the QRS complex in right precordial leads; Minor: late potentials on signal averaged ECG). (5) Arrhythmias (Minor: left bundle branch block type ventricular tachycardia, sustained or non-sustained on ECG, Holter, or exercise testing; frequent ventricular extrasystoles >1000/24 h) [6]. Family history [Major: familial disease confirmed at necropsy or surgery. Minor: familial history of premature sudden death (<35 years) due to suspected ARVD/C, family members independently diagnosed with ARVD/C] [17].
Mutation Analysis
Each participant had genomic DNA extracted from leukocytes present in whole blood using QIAmp DNA blood maxi kits (Qiagen, Valencia, CA). Intronic PKP2, DSG2, DSC2, DSP, and JUP primers flanking each exon were used in polymerase chain aeaction (PCR) amplification. The resultant PCR products were purified using the QIAquick PCR Purificatin Kit (Qiagen) followed by bidirectional sequence analysis using the ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA) and chromatograms analyzed with Sequencer 4.1 software. Mutations were confirmed by restriction fragment length polymorphisms when possible or by Taqman genotyping assays (Applied Biosystems).
A novel variant in the DNA sequence was defined as a mutation when it does not occur in 400 unrelated, healthy, race-matched, control chromosomes (NIGMS Human Genetic Cell Repository, Coriell Institute for Medical Research), when it alters one or more conserved amino acids, and when it segregates with disease in the family if that information is available. All other novel variants are classified as polymorphisms. Sequence variants that were previously reported as mutations were considered mutations unless we could show otherwise. Novel DNA variants that alter a restriction site were tested for their presence in controls using restriction enzyme digest. If the sequence variants did not alter any restriction sites, the presence among controls was determined by the use of Taqman genotyping assays (Applied Biosystems). Novel missense protein variants were analyzed by ClustalW (MacVector, Cary, NC), Polyphen (http://genetics.bwh.harvard.edu/pph/), and MUpro (http://www.ics.uci.edu/~baldig/mutation.html) for conservation, stability, and likelihood for pathogenic effect.
Low-frequency amino acid substitutions are defined as missense variants that were found in 200 unrelated, healthy, race-matched, control chromosomes (NIGMS Human Genetic Cell Repository, Coriell Institute for Medical Research) at a frequency of less than 5%, or were previously reported as single-nucleotide polymorphisms at a similar low-frequency. The ARVD/C Genetic Variants Database was reviewed for all mutations and low-frequency missense alleles (www.arvcdatabase.info).
Statistical Analysis
Continuous variables were expressed as mean ± SD and compared by unpaired t test when comparing two groups and analysis of variance (ANOVA) in cases of multiple groups. Categorical variables were expressed as frequency (%) and compared by chi-squared test in case of large samples and by Fisher’s exact test in case of small sample size.
A low-frequency amino acid substitution score was calculated by assigning a score of one for the presence of each missense variant. Participants were then categorized based on this score. Categorical variables were compared based on the score category. For the survival analysis, individuals were categorized into three groups based on the presence or absence of low-frequency amino acid substitution and known desmosome gene mutations. Kaplan–Meier analysis and log–rank test was used to compare arrhythmia-free survival at the age of evaluation as a function of the three groups. Analyses were performed using STATA statistical software (College Station, TX) and a p value< 0.05 was considered statistically significant.
Results
Demographics and Presentation
The demographics and presenting symptoms of individuals in each cohort are summarized in Table 1. The Young cohort consisted of 19 people with definite ARVD/C with a mean age of symptom onset or a diagnosis of ARVD/C at 15.5±4.1 years (min=2, max=20 years). Sixteen were symptomatic before the age of 21 years, and the remaining three were diagnosed due to a family history of ARVD/C prior to symptoms. The 11 individuals with probable ARVD/C in the Young cohort had a mean age of 16.5± 3 years (min=12, max=21). Nine were symptomatic before the age of 21 years, and two were screened and diagnosed due to a family history of ARVD/C prior to symptoms.
Table 1.
Demographics, presentations, clinical data, and genotypic characteristics
| Characteristics | Young cohort with definite ARVD/C (N=19) | Young cohort with probable ARVD/C (N=11) | Middle cohort with definite ARVD/C (N=56) | Middle cohort with probable ARVD/C, (N=11) | Late cohort with definite ARVD/C (N=15) | P values |
|---|---|---|---|---|---|---|
| Male | 12 | 7 | 29 | 6 | 12 | 0.08 |
| Age at symptom onset | 15.5±4.1 (min=2, max=20) | 16.5±3.0 (min=12, max 21) | 34.3±8.1 (min=22, max 48) | 36.2±4.7 (min=29, max=46) | 56.3±7.0 (min=50, max=76) | |
| ARVD/C symptoms: n (%) | 0.57 | |||||
| Palpitations | 8 (42%) | 6 (54%) | 19 (34%) | 5 (45%) | 4 (27%) | |
| Syncope | 4 (21%) | 1 (9%) | 14 (25%) | 1 (9%) | 3 (20%) | |
| VT | 3 (16%) | 1 (9%) | 14 (25%) | 3 (27%) | 3 (20%) | |
| SCD | 0 (0%) | 0 (0%) | 1 (2%) | 0 (0%) | 0 (0%) | |
| Others (asymptomatic, atrial fibrillation, atypical chest pain, shortness of breath) | 4 (21%) | 3 (27%) | 8 (14%) | 2 (18%) | 5 (33%) | |
| Family history | 0.34 | |||||
| ARVD/C on autopsy | 6 (32%) | 2 (18%) | 13 (23%) | 1 (9%) | 5 (33%) | |
| SCD expected to be caused by ARVD/C | 5 (26%) | 0 (0%) | 5 (9%) | 0 (0%) | 2 (13%) | |
| ARVD/C based on current criteria | 5 (26%) | 4 (36%) | 13 (23%) | 0 (0%) | 1 (7%) | |
| Any family history (major or minor criterion) | 9 (47%) | 6 (54%) | 21 (37%) | 2 (18%) | 5 (33%) | |
| Repolarization abnormalities | 0.56 | |||||
| T wave inversion V1–V3 [n (%), N] | 13 (81%), N=16 | 8 (80%), N=10 | 51 (93%), N=55 | 4, (36%), N=11 | 10 (71%), N=14 | |
| Depolarization abnormalities: [n (%), N] | ||||||
| Epsilon waves in V1–V3 | 0 (0%), N=19 | 0 (0%), N=11 | 11 (20%), N=56 | 0 (0%), N=11 | 1 (7%), N=14 | 0.03 |
| QRS prolongation in V1–V3 | 7 (37%), N=19 | 2 (18%), N=11 | 18 (33%), N=55 | 0 (0%), N=11 | 5 (36%), N=14 | 0.65 |
| Late potentials on SAECG | 10 (56%), N=18 | 1 (10%), N=10 | 34 (72%), N=47 | 5 (55%), N=9 | 9 (90%), N=10 | 0.006 |
| Arrhythmias [n (%), N] | ||||||
| LBBB VT on Holter or ECG | 11(58%), N=19 | 5 (45%), N=11 | 32 (58%), N=55 | 7 (63%), N=11 | 8 (53%), N=15 | 0.76 |
| >1000 PVCs on Holter | 15 (94%), N=16 | 5 (50%), N=10 | 21 (57%), N=37 | 3 (33%), N=9 | 4 (67%), N=6 | 0.11 |
| Minor dysfunction and structural alterations, [n (%), N] | 9 (47%), N=19 | 9 (82%), N=11 | 32 (57%), N=56 | 6 (55%), N=11 | 7 (47%), N=15 | 0.71 |
| Major dysfunction and structural alterations, [n (%), N] | 10 (53%), N=19 | 0 (0%), N=11 | 23 (41%), N=56 | 1 (9%), N=11 | 6 (40%), N=15 | 0.92 |
| Fibrofatty replacement on endomyocardial biopsy, [n (%), N] | 5, N=8 | 0, N=3 | 6, N=22 | 0, N=1 | 4, N=5 | N/A |
| Genetic characteristics | ||||||
| 1 or more desmosome mutations [n (%)] | 13 (68%) | 7 (64%) | 28 (50%) | 4 (36%) | 8 (53%) | 0.23 |
| >1 desmosome mutation [n (%)] | 1 (5%) | 0 (0%) | 3 (5%) | 0 (0%) | 3 (20%) | 0.07 |
The Middle cohort consisted of 56 people with definite ARVD/C with a mean age of symptom onset or ARVD/C diagnosis at 34.3±8.1 years (min=22, max=48). Forty-eight had symptoms prompting their evaluation, and eight were screened due to a family history of ARVD/C prior to symptoms. The 11 individuals with probable ARVD/C in the Middle cohort had a mean age of 36.2±4.7 years (min=29, max=46). Nine had symptoms prompting their phenotypic screening, and two were evaluated for ARVD/C due to a family history of this condition prior to symptoms.
The Late cohort consisted of 15 people with definite ARVD/C with a mean age of symptom onset or a diagnosis of ARVD/C at 56.3±7.0 years (min=50, max=76). Thirteen underwent cardiac testing for symptoms, and two were evaluated due to a family history of ARVD/C prior to symptoms.
Phenotypic Characteristics
The results of phenotypic testing of all 112 participants in this study are shown in Table S1. Analysis was made with comparisons among the three groups, with separate categories for definite and probable ARVD/C. These characteristics varied only based on the presence of epsilon wave, which was most common in the Middle cohort (P=0.03), and abnormal SAECG, which was most common in the Late cohort (P=0.006) among the three groups. There is a decreased likelihood of an abnormal SAECG in the Young cohort (P=0.012), but no phenotypic characteristics were otherwise different in this cohort.
Desmosome Mutations
Thirteen individuals (67%) had one or more mutations identified in the Young cohort with definite ARVD/C (Tables 2 and S1). There were 13 different mutations: four altered a critically conserved nucleotide that forms the intron–exon splice site, one was a nonsense mutation that resulted in a premature termination codon, four were missense mutations disrupting highly conserved residues, and four were insertion–deletion mutations. The distribution of genes in which mutations occurred in each cohort is shown in Fig. 1.
Table 2.
Summary of mutations in the Young cohort
| Gene | Nucleotide change | Amino acid change | Previous report |
|---|---|---|---|
| With definite ARVD/C | |||
| PKP2 | 1368delA | N456fsX458 | Yes |
| 2146-1G>C | Mutant splice product | Yes | |
| 1171–2A>G | Mutant splice product | Yes | |
| 2489+1G>A | Mutant splice product | Yes | |
| 1613G>A | W538X | Yes | |
| 1271T>C | F424S | Yes | |
| 2011delC | R671fsX683 | Yes | |
| 145–148delCAGA | S50fsX110 | Yes | |
| DSG2 | 137G>A | R46Q | Yes |
| 523+2T>Ca | Mutant splice product | No | |
| 1038-1040delGAAa | K346del | No | |
| DSP | 1264G>A | E422K | No |
| JUP | 56C>T | T19I | Yes |
| With probable ARVD/C | |||
| PKP2 | 2509delA | V837fs930X | Yes |
| 2146-1G>C | Mutant splice product | Yes | |
| 1759G>A | V587I | Yes | |
| 2197–2202delCACACC ins G | A733fs X740 | Yes | |
| 1759delG | S587fs X 655 | No | |
| DSG2 | 3140C>G | T1047R | No |
| DSC2 | 658G>A | G220R | No |
Indicates that this mutation was found with another mutation in one or more individuals
Fig. 1.

Distribution of desmosome mutations in each cohort. The percentage of mutations in each gene is shown by cohort. Both definite and probable ARVD/C cases were included
A total of three novel desmosome gene mutations were identified in the Young cohort with definite ARVD/C (DSP p.E422K, DSG2 p.K346del, DSG2 c.523+2T>C; Fig. S1). The individual with the DSP p.E422K (1264G> A) mutation was evaluated for palpitations at age of 15 years. Echocardiogram showed a dilated right ventricle with severe right ventricular dysfunction. There was no family history of ARVD. One participant with recurrent syncopal events since the age of 17 years has two DSG2 mutations p.K346del and DSG2 c.523+2T>C (compound heterozygous). She has no family history of ARVD; she has two children, ages 7 and 8 years, each of whom carries one of the DSG2 mutations. Both are currently asymptomatic and have no phenotypic features of ARVD. DSG2 c.523+ 2T>C alters a critically conserved nucleotide leading to a mutant splice product. p.K346del (ca. 1038–1040 delGAA) causes in-frame deletion of a lysine residue in the extracellular domain EC3 of desmoglein-2. In the proband, it is possible that nonsense-mediated messenger ribonucleic acid (mRNA) decay of the mutant splice product (c.523+ 2T>C) contributed to the disease mechanism. However, RNA samples from the proband and her two children are not available for analysis.
Seven individuals (64%) had one or more mutations identified in the Young cohort with probable ARVD/C (Tables 2 and S1). There were seven different mutations: one altered a critically conserved nucleotide that forms the intron–exon splice site, three were missense mutations disrupting highly conserved residues, and three were insertion–deletion mutations. A total of three novel desmosome gene mutations were identified in the Young cohort with probable ARVD/C (DSG2 p.T1047R, DSC2 p.G220R, PKP2 p.V587delfsX655).
Twenty-eight individuals (50%) had one or more mutations identified in the Middle cohort with definite ARVD/C (Tables 3, and S1). There were 17 different mutations: two altered a critically conserved nucleotide that forms the intron–exon splice site, four were nonsense mutations that resulted in a premature termination codon, five were missense mutations disrupting highly conserved residues, five were insertion–deletion mutations, and one was a cryptic splice mutation.
Table 3.
Summary of mutations in the Middle cohort
| Gene | Nucleotide change | Amino acid change | Previous report |
|---|---|---|---|
| With definite ARVD/C | |||
| PKP2 | 145–148delCAGAa | S50fsX110 | Yes |
| 235C>T | R79X | Yes | |
| 1237C>Ta | R413X | Yes | |
| 1307–15del TAATGGGG1308insATTTAGTT | H436fsX447 | Yes | |
| 1613G>A | W538X | Yes | |
| 1642delG | V548fsX562 | Yes | |
| 2489+1G>A | Mutant splice product | Yes | |
| 2146-1G>C | Mutant splice product | Yes | |
| 2197–2202insGdel CACACC | A773fsX740 | Yes | |
| 2484C>T (a homozygous) | Cryptic splicing | Yes | |
| DSG2 | 1520G>A | C507Y | Yes |
| 2434G>T | G812C | Yes | |
| 1003A>Ga | T335A | Yes | |
| 146G>Aa | R49H | Yes | |
| 918G>Aa | W306X | Yes | |
| 829-1 delGCTTGAAGGGATa | G277fsX278 | Yes | |
| DSP | 1331A>G | I445V | Yes |
| With probable ARVD/C | |||
| PKP2 | 145–148delCAGA | S50fsX110 | Yes |
| 216insG | Q74fsX85 | Yes | |
| 2146-1G>C | Mutant splice product | Yes | |
| DSG2 | 146G>A | R49H | Yes |
Indicates that this mutation was found with another mutation in one or more individuals
Four individuals (36%) had one or more mutations identified in the Middle cohort with probable ARVD/C (Tables 3 and S1). There were four different mutations: one altered a critically conserved nucleotide that forms the intron–exon splice site, one was a missense mutation disrupting highly conserved residue, and two were insertion–deletion mutations.
Eight individuals (53%) had one or more mutations identified in the Late cohort with ARVD/C (Tables 4 and S1). There were ten different mutations: three altered a critically conserved nucleotide that forms the intron–exon splice site, six were missense mutations disrupting highly conserved residues, and one was an insertion–deletion mutation. A total of two novel desmosome mutations (Fig. S1) were identified in the Late cohort with definite ARVD/C (DSG2 p.D297N, PKP2 p. V725D). One individual had two desmosome gene variants that have previously been published as mutations (PKP2 p.S140F and DSG2 p.V56M), although they also occur rarely in controls and their association with disease has been questioned [18, 19]. However, he also has a clear splice site mutation (PKP2 ca. 2146-1G>C) and is included as positive for a desmosome gene mutation on that basis.
Table 4.
Summary of mutations in the Late cohort with ARVD/C
| Gene | Nucleotide change | Amino acid change | Previous report |
|---|---|---|---|
| PKP2 | 2489+1G>A | Mutant splice product | Yes |
| 2197–2202insGdelCACACC | A773fsX740 | Yes | |
| 2359C>T | L787F | Yes | |
| 2174T>A | V725D | No | |
| 1759G>A | V587I | Yes | |
| 2145+1G>C | Mutant splice product | Yes | |
| 419C>Ta | S140F | Yes | |
| 2146-1G>Ca | Mutant splice product | Yes | |
| DSG2 | 209G>Aa | V56M | Yes |
| 889G>A | D297N | No |
Indicates that this mutation was found with another mutation in one or more individuals
There were no differences in the prevalence of one or more desmosome mutations among the Young (67%), Middle (48%) and Late (53%; P=0.23) cohorts. PKP2 (Fig. 1) was the most common gene in which mutations were identified. There was no increase in frequency of compound heterozygous or digenic heterozygous mutations in the Young cohort compared to the Middle and Late cohorts (Table 1; P=0.072).
Low-Frequency Amino Acid Substitutions
Several low-frequency amino acid substitutions are more common in this cohort of 112 definite and probable ARVD/C participants than among 100 race-matched Coriell controls. A total of 15 missense variants were identified for further analysis (Table 5): six in PKP2, two in DSG2, one in JUP, three in DSP, and three in DSC2. These missense variants were chosen for further analysis because of their known low frequency in controls (< 5%) and their lack of known association with disease pathogenesis. We reasoned that these variants may be over-represented in ARVD/C compared to controls. Indeed, 21% of the total ARVD/C cohort compared to 13% of the controls had one or more of these variants (P=0.027; Fig. 2).
Table 5.
Summary of low-frequency amino acid substitutions
| Gene | cDNA change | Amino acid change | MUpro: prediction | PolyPhen: prediction | Frequency in total study cohort (N=115) | Frequency in Coriell controls (N=100) |
|---|---|---|---|---|---|---|
| PKP2 | 76G>A | D26N | Increased stability | Possibly damaging | 1/112 | 2/100 |
| 826C>T | P276S | Increased stability | Benign | 1/112 | 1/100 | |
| 1114G>C | A372P | Increased stability | Benign | 1/112 | 0/100 | |
| 1577C>T | T526M | Increased stability | Benign | 2/112 | 1/100 | |
| 2431C>A | R811S | Increased stability | Possibly damaging | 1/112 | 1/100 | |
| 2488G>C | A830P | Increased stability | Benign | 1/112 | 0/100 | |
| DSG2 | 473T>G | V158G | Increased stability | Probably damaging | 1/112 | 0/100 |
| 2648C>T | S883F | Increased stability | Benign | 1/112 | 0/100 | |
| JUP | 1942G>A | V648I | Increased stability | Benign | 4/112 | 2/100 |
| DSP | 4578C>A | N1526K | Increased stability | Benign | 1/112 | 0/100 |
| 4514C>T | A1505V | Increased stability | Benign | 1/112 | 0/100 | |
| 4609C>T | R1537C | Increased stability | Probably damaging | 4/112 | 3/100 | |
| DSC2 | 1787C>T | A596V | Increased stability | Possibly damaging | 1/112 | 1/100 |
| 1914G>C | Q638H | Increased stability | Possibly damaging | 1/112 | 0/100 | |
| 2687–2688insGA | E896fs X900 | N/A | N/A | 2/112 | 2/100 |
Fig. 2.
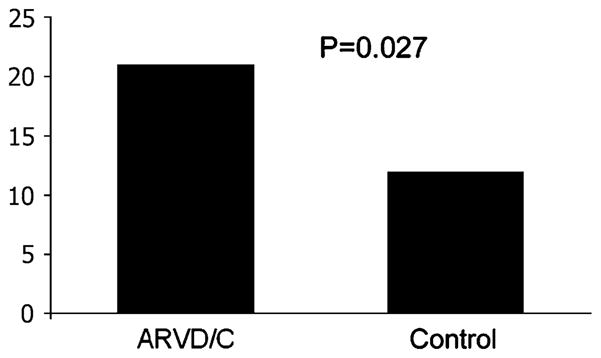
Percentage with low-frequency missense alleles. The percentage of participants with one or more of the low-frequency missense polymorphisms is shown. ARVD/C represents the percentage with both definite and probable disease; Control represents 100 race-matched Coriell controls
We sought to determine if these rare missense alleles influence the age of onset of ARVD/C by comparing their prevalence among the three cohorts. However, there is no difference in their frequency among the three cohorts (P=0.71). The presence of these variants also did not influence the age of onset of ventricular tachycardia (VT). Figure 3 shows the Kaplan–Meier curves documenting freedom from VT for individuals with (1) no desmosome mutations and none of the 15 missense variants, (2) one or more desmosome mutations and none of the 15 variants, and (3) one or more desmosome mutations and at least one of the 15 variants. The median cumulative arrhythmia-free survival was influenced by the presence of desmosome mutations, independent of any of these 15 low-frequency missense variants. The median cumulative arrhythmia-free survival with no desmosome mutation and none of the 15 variants is 45 years, and 32 years when one or more desmosome mutation is present without any of the 15 variants (P=0.02). When at least one desmosome mutation is present, the addition of any of the 15 variants did not influence the median cumulative arrhythmia-free survival.
Fig. 3.
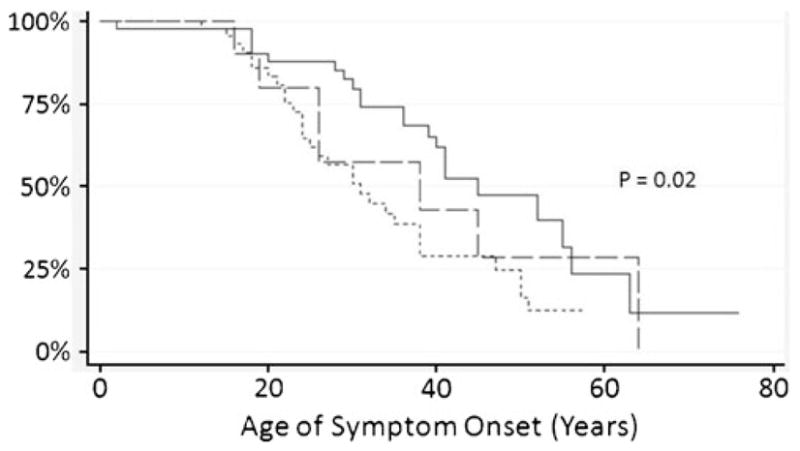
Proportion of individuals free from VT. Kaplan Meier curves demonstrate the proportion of individuals free from VT with one or more desmosome mutations and none of the 15 variants (- -), one or more desmosome mutations and at least one of the 15 variants (– –), and no desmosome mutations and none of the 15 missense variants (—). P=0.02 represents comparison between those with no desmosome mutation or missense variant and each of the other two groups; comparison between those with desmosome mutation with or without the other variants was not significant (P=0.85)
Discussion
Phenotypic heterogeneity in inherited cardiovascular disease is a common occurrence. Affected individuals within a family who harbor the same gene mutation often manifest wide clinical variability. This suggests that other modifying factors, including environmental exposures and other genetic influences, play a role in either exacerbating or attenuating disease manifestation. The exact impact of these factors is poorly understood.
For many inherited cardiovascular diseases, the presence of two or more mutations may result in earlier and more severe disease manifestation. For instance, in hypertrophic cardiomyopathy (HCM), people with homozygous, digenic heterozygous, or compound heterozygous mutations may have a higher incidence of SCD and may have more severe left ventricular hypertrophy [20, 21]. This led to a similar hypothesis in HCM that the presence of sarcomere gene mutations would be different among a cohort with childhood onset. However, Morita et al. [22] demonstrated sarcomere gene mutations in only 46 of 84 individuals (54%) with familial or sporadic HCM diagnosed before the age of 15 years. This did not differ from the overall prevalence of gene mutations (59%) in unrelated probands with HCM.
The genetics of early onset ARVD/C was previously less well characterized than early onset HCM. Recent studies have suggested that for the rare desmosome gene mutations (namely, DSG2 and DSC2), multiple mutations may be required to manifest the ARVD/C phenotype [23]. Multiple desmosome gene mutations have also been shown to have more frequent sudden death [24]. Even common polymorphisms (namely, PKP2 p.P366L, MAF:17%) may not be completely innocuous but, rather was suggested in one report to confer a more benign phenotype [25]. In our study, at least one desmosome mutation was found in 20 out 30 participants with definite ARVD/C or probable ARVD/C (67%) diagnosed before the age of 21 years. These results did not differ significantly from the Middle or Late cohorts (P=0.23). There was also no increase in prevalence of compound heterozygous or digenic heterozygous mutations in the Young cohort.
The age of onset of ARVD/C is not easily defined. The risk of SCD exists even in the early “concealed” phase of ARVD/C [3]. Progression of disease may not be uniform throughout life and likely involves sporadic periods of greater change as well as quiescent phases. After the age of 40 years, the incidence of SCD due to ARVD/C may decrease [26, 27]. The true incidence of later onset ARVD/C, however, may be severely underestimated due to the presence of other comorbidities such as coronary atherosclerosis and myocardial infarction. Phenotypic testing for ARVD/C after age of 50 years may also be misleading. SAECG for example, may be positive in up to 14% of the elderly with no clinical evidence of heart disease [28]. In this study, 53% of the Late cohort (mean age 56.3±7 years) had one or more desmosome mutation, not different from the Middle and Young cohorts. The reasons for later onset disease in this cohort may pertain to other genetic or environmental factors.
The autosomal dominant monogenic inheritance paradigm, where one gene mutation causes disease, is further challenged by the potential modifier effects of uncommon DNA variants that may influence disease. Crotti et al. [29] reported a family with Long QT syndrome (LQT2) in which the proband had a missense KCNH2 mutation p.A1116V and a common KCNH2 polymorphism p.K897T on the non-mutant allele. The proband had ventricular fibrillation and cardiac arrest at the age of 44 years. Family members with the same KCNH2 mutation p.A1116V without the p.K897T polymorphism, however, only had latent disease. Similar factors may be relevant in ARVD/C.
We attempted to explore the contribution of missense variants in ARVD/C by comparing age of onset as a marker of severity of disease. A total of 15 low-frequency (<5%) amino acid substitutions were analyzed among both cases and controls. The prevalence of these variants was higher (P=0.027) in the entire cohort of 112 probands with definite and probable ARVD/C. When we analyzed the Kaplan–Meier curves of VT-free survival, only the presence of desmosome mutations conferred a worse outcome. The addition of these low-frequency amino acid variants did not influence VT-free survival. The prevalence of these variants is also not significantly higher in the Young cohort. The increased prevalence of these variants in the disease population compared to controls does suggest a contributory role. However, the exact effect of these polymorphisms is difficult to determine without functional assays, and such assays are not currently available for desmosome protein variants.
Xu et al. [30] recently reported desmosome gene analysis in a cohort of 198 individuals with ARVD/C. In this cohort, 26% had a desmosome gene variant or mutation. Furthermore, among the 38 participants with a PKP2 mutation or variant, 16 also had a second mutation or variant present at low frequency among controls, representing 8% of the total cohort. This also suggests that additional desmosome gene variants may impact disease [30].
For this study, classification of all participants was based on the traditional 1994 Task Force Criteria (TFC) [17]. In a recent analysis of 105 individuals with proven ARVD/C based on the 1994 TFC, only three were reclassified as probable ARVD/C using the new TFC [31]. The impact of the new TFC criteria was predominantly felt in the probable ARVD/C group (64% were reclassified as ARVD/C) and in family members of probands (11% were reclassified as ARVD/C) [31, 32]. Because we included both definite and probable ARVD/C and applied these criteria equally to the three cohorts, the use of the newer criteria would not likely change our results.
Late potentials by SAECG are increasingly recognized as a marker for delay activation in ARVD/C. An abnormal SAECG (by the traditional TFC) could be a nonspecific finding in the elderly. An abnormal SAECG is also less commonly found in the Young cohort, suggesting a decrease in sensitivity in young individuals with ARVD/C. To satisfy a minor criterion, the new TFC only requires one of the three measured parameters in the SAECG to be abnormal [31]. While this increases sensitivity in Young cohort, it comes at a cost of decreasing specificity in the Late cohort.
The addition of a pathogenic mutation as a major criterion in the new TFC could encourage one to stop screening for mutations in other desmosome genes once a mutation is identified. As a result, a second mutation could be missed if comprehensive analysis is not performed. In addition, family members may be mistakenly counseled about their risk of inheritance if the proband is not thoroughly evaluated for desmosome gene mutations.
Study Limitations
Complete evaluation could not be performed in all first degree relatives, which limits our ability to associate familial segregation of many of these DNA variants with disease manifestations. Also, the age of diagnosis is dependent on the age at which a proband first sought evaluation for ARVD/C. Often, this follows the diagnosis of an affected person in the family. In the event that a proband is diagnosed as meeting TFC at the first evaluation, the true age of diagnosis would certainly precede it. On phenotypic evaluation, we relied on qualitative definitions of RV disease on imaging studies as recommended by the 1994 TFC rather than quantitative parameters for imaging studies that are proposed in the new TFC [17, 31]. The numbers of individuals included at early and late age are relatively small, and further multi-center analyses may help to confirm and extend these findings.
Conclusion
The phenotypic heterogeneity and variable penetrance of ARVD/C highlight the complex interplay of environmental and genetic factors contributing to this condition. In this analysis, the prevalence of desmosome mutations and of multiple mutations was similar in the Young, Middle, and Late cohorts. Certain rare missense variants were more common among ARVD/C cases of all ages compared to controls. This suggests that these rare desmosome gene variants may influence the development of ARVD/C but not the age of onset. Additional genetic and environmental modifiers likely impact age of onset of this condition.
Supplementary Material
Acknowledgments
This work was supported by funding from the National Institutes of Health (HL088072 to DPJ), the France-Merrick Foundation, JHU Friends in Red, and the Jeff Cooper CARE Foundation. The authors would also like to acknowledge the Johns Hopkins ARVD Program (http://www.arvd.com), which is supported by the Bogle Foundation, the Campanella family, the Wilmerding Endowment, the Dr. Francis P Chiaramonte Foundation, the Healing Hearts Foundation, Boston Scientific, Medtronic, and St Jude Medical. BY Tan was supported by funds from the National Medical Research Council Medical Research Fellowship and the Health Manpower Development Plan Fellowship by the Ministry of Health, Singapore.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s12265-010-9224-4) contains supplementary material, which is available to authorized users.
Conflicts of Interest Dr. Hugh Calkins receives honoraria for lectures from Boston Scientific, Medtronic, and St. Jude Medical and is a consultant for Medtronic. The authors have no other potential conflicts of interest to disclose.
Contributor Information
Boon Yew Tan, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA. Department of Cardiology, National Heart Centre, Singapore, Singapore.
Rahul Jain, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
A. Dénise den Haan, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
Yan Chen, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
Darshan Dalal, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
Harikrishna Tandri, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
Nuria Amat-Alarcon, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
Amy Daly, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
Crystal Tichnell, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
Cynthia James, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
Hugh Calkins, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
Daniel P. Judge, Email: djudge@jhmi.edu, Department of Medicine/Cardiology, Johns Hopkins University, School of Medicine, Baltimore, MD, USA. Division of Cardiology, Johns Hopkins University, Ross Building, #1049; Rutland Avenue, Baltimore, MD 21205, USA
References
- 1.Awad MM, Calkins H, Judge DP. Mechanisms of disease: Molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nature Clinical Practice Cardiovascular Medicine. 2008;5:258–267. doi: 10.1038/ncpcardio1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: Clinical impact of molecular genetic studies. Circulation. 2006;113:1634–1637. doi: 10.1161/CIRCULATIONAHA.105.616490. [DOI] [PubMed] [Google Scholar]
- 3.Dalal D, Molin LH, Piccini J, Tichnell C, James C, Bomma C, et al. Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation. 2006;113:1641–1649. doi: 10.1161/CIRCULATIONAHA.105.568642. [DOI] [PubMed] [Google Scholar]
- 4.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nature Genetics. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 5.Awad MM, Dalal D, Cho E, Amat-Alarcon N, James C, Tichnell C, et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. American Journal of Human Genetics. 2006;79:136–142. doi: 10.1086/504393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, SenChowdhry S, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. American Journal of Human Genetics. 2006;79:978–984. doi: 10.1086/509122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. American Journal of Human Genetics. 2007;81:964–973. doi: 10.1086/521633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 9.Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. Journal of Cardiovascular Electrophysiology. 2005;16:927–935. doi: 10.1111/j.1540-8167.2005.40842.x. [DOI] [PubMed] [Google Scholar]
- 10.Awad MM, Dalal D, Tichnell C, James C, Tucker A, Abraham T, et al. Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Human Mutation. 2006;27:1157. doi: 10.1002/humu.9461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.den Haan AD, Tan BY, Zikusoka MN, Llado LI, Jain R, Daly A, et al. Comprehensive desmosome mutation analysis in North Americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation: Cardiovascular Genetics. 2009;2:428–435. doi: 10.1161/CIRCGENETICS.109.858217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, et al. Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation. 2005;112:3823–3832. doi: 10.1161/CIRCULATIONAHA.105.542266. [DOI] [PubMed] [Google Scholar]
- 13.Taylor GP. The pathology of arrhythmogenic right ventricular cardiomyopathy. In: Redington AR, VanArsdell GS, Anderson RH, editors. Congenital diseases in the right heart. London: Springer; 2009. pp. 125–129. [Google Scholar]
- 14.Dalal D, James C, Devanagondi R, Tichnell C, Tucker A, Prakasa K, et al. Penetrance of mutations in plakophilin-2 among families with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Journal of the American College of Cardiology. 2006;48:1416–1424. doi: 10.1016/j.jacc.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 15.Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Journal of the American College of Cardiology. 2007;50:1813–1821. doi: 10.1016/j.jacc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Frigo G, Bauce B, Basso C, Nava A. Late-onset arrhythmogenic right ventricular cardiomyopathy. Journal of Cardiovascular Medicine (Hagerstown) 2006;7:74–76. doi: 10.2459/01.JCM.0000199781.77720.31. [DOI] [PubMed] [Google Scholar]
- 17.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. British Heart Journal. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christensen AH, Benn M, Tybjaerg-Hansen A, Haunso S, Svendsen JH. Missense variants in plakophilin-2 in arrhythmogenic right ventricular cardiomyopathy patients–disease-causing or innocent bystanders? Cardiology. 2010;115:148–154. doi: 10.1159/000263456. [DOI] [PubMed] [Google Scholar]
- 19.Posch MG, Posch MJ, Geier C, Erdmann B, Mueller W, Richter A, et al. A missense variant in desmoglein-2 predisposes to dilated cardiomyopathy. Molecular Genetics and Metabolism. 2008;95:74–80. doi: 10.1016/j.ymgme.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Nishi H, Kimura A, Harada H, Adachi K, Koga Y, Sasazuki T, et al. Possible gene dose effect of a mutant cardiac beta-myosin heavy chain gene on the clinical expression of familial hypertrophic cardiomyopathy. Biochemical and Biophysical Research Communications. 1994;200:549–556. doi: 10.1006/bbrc.1994.1483. [DOI] [PubMed] [Google Scholar]
- 21.Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. Journal of Medical Genetics. 2005;42:e59. doi: 10.1136/jmg.2005.033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, et al. Shared genetic causes of cardiac hypertrophy in children and adults. The New England Journal of Medicine. 2008;358:1899–1908. doi: 10.1056/NEJMoa075463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhuiyan ZA, Jongbloed JD, van der Smagt J, Lombardi PM, Wiesfeld AC, Nelen M, et al. Desmoglein-2 and desmocollin-2 mutations in dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients: Results from a multicenter study. Circulation: Cardiovascular Genetics. 2009;2:418–427. doi: 10.1161/CIRCGENETICS.108.839829. [DOI] [PubMed] [Google Scholar]
- 24.Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Spectrum of mutations and clinical impact in practice. Europace. 2010;12:861–868. doi: 10.1093/europace/euq104. [DOI] [PubMed] [Google Scholar]
- 25.Barahona-Dussault C, Benito B, Campuzano O, Iglesias A, Leung TL, Robb L, et al. Role of genetic testing in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Clinical Genetics. 2010;77:37–48. doi: 10.1111/j.1399-0004.2009.01282.x. [DOI] [PubMed] [Google Scholar]
- 26.Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–991. doi: 10.1161/01.cir.94.5.983. [DOI] [PubMed] [Google Scholar]
- 27.Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. The New England Journal of Medicine. 1988;318:129–133. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- 28.Aronow WS, Mercando AD, Epstein S. Usefulness of an abnormal signal-averaged electrocardiogram for predicting cardiac death in elderly persons without heart disease. The American Journal of Cardiology. 1995;75:1273–1274. doi: 10.1016/s0002-9149(99)80779-6. [DOI] [PubMed] [Google Scholar]
- 29.Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–1258. doi: 10.1161/CIRCULATIONAHA.105.549071. [DOI] [PubMed] [Google Scholar]
- 30.Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. Journal of the American College of Cardiology. 2010;55:587–597. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cox MGPJ, van der Smagt JJ, Noorman M, Wiesfeld AC, Volders PGA, van Langen IM, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy diagnostic task force criteria: Impact of new task force criteria. Circulation: Arrhythmia Electrophysiology. 2010;3:126–133. doi: 10.1161/CIRCEP.109.927202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.