

Summary
Although the critical role of T-cell receptor (TCR) microclusters in T-cell activation is now widely accepted, the mechanisms of regulation of these TCR-rich structures, which also contain enzymes, adapters, and effectors, remain poorly defined. Soon after microcluster formation, several signaling proteins rapidly dissociate from the TCR. Recent studies from our laboratory demonstrated that the movement of the adapters linker for activation of T cells (LAT) and Src homology 2 domain-containing leukocyte protein of 76 kDa (SLP-76) away from initial microcluster formation sites represents endocytic events. Ubiquity-lation, Cbl proteins, and multiple endocytic pathways are involved in the internalization events that disassemble signaling microclusters. Several recent studies have indicated that microcluster movement and centralization plays an important role in signal termination. We suggest that microcluster movement is directly linked to endocytic events, thus implicating endocytosis of microclusters as a means to regulate signaling output of the T cell.
Keywords: endocytosis, ubiquitylation, linker for activation of T cells, SLP-76, microclusters, T-cell signaling
Introduction
Effective T-cell activation is a vital part of the adaptive immune response and proper T-cell activation requires signaling from the T-cell receptor (TCR). T-cell signaling must be tightly controlled, as either too much or too little T-cell activation will result in a dysfunctional immune response. Ineffective TCR signaling results in severe immunodeficiency defects, while inappropriate signaling is associated with autoimmune diseases (1). The TCR is a multi-subunit structure consisting of antigen-binding α and β chains, signal transducing CD3 chains present as γε and δε dimers, and a TCRζ dimer. TCR signaling is initiated when recognition of a peptide bound to major histocompatibility complex (MHC) displayed on the surface of an antigen-presenting cell by the clonally defined α and β chains of the TCR, leads to activation of the signal transducing subunits (2). The striking diversity of TCRs and peptide-MHC ligands results in interactions with a broad range of affinities, and, consequently, an extensive array of signaling inputs can potentially be sent to the signaling machinery downstream of the TCR (3). The integrated response of the signaling molecules then distinguishes slight stimulatory variations and results in cell survival or differentiation versus death.
Two decades of extensive biochemical, pharmacological, and genetic studies have provided much information about the proximal TCR signaling machinery. Receptor engagement is rapidly followed by activation of protein tyrosine kinases (PTKs) that phosphorylate a number of downstream substrates, including the critical adapter protein linker for activation of T cells (LAT), which was cloned in our laboratory (4). Additional studies from our laboratory and others demonstrated that the LAT phosphotyrosines act as a scaffold to dock several Src homology 2 (SH2) domain-containing adapter proteins including Grb2, Grb2-related adapter downstream of Shc (Gads), and Grb2-related adapter protein (Grap) (5–8), which in turn are associated with other signaling proteins. SH2 domain-containing leukocyte protein of 76 kDa (SLP-76) is recruited to LAT through its interaction with the SH3 domain of Gads and acts as a platform for the recruitment of several signaling molecules including regulators of Ca2+ signaling and diacylglycerol production [phospholipase Cγ1 (PLCγ1) and interleukin-2 (IL-2)-inducible T-cell kinase (Itk)] as well as regulators of actin polymerization (NCK and Vav) and integrin activation [adhesion and degranulation-promoting adapter protein (ADAP)]. In addition, Grb2 recruits two known SH3 domain ligands, the Ras guanosine triphosphatase (GTPase) son of sevenless 1 (SOS1), and the E3 ligase and adapter Cbl, to the LAT complex. Together, integrated signals from these effector molecules regulate the cytoskeletal rearrangements, gene expression, and adhesion events that are associated with T-cell activation (9). Thus, TCR engagement induces the formation of LAT-based multiprotein complexes that initiate the intracellular signals required for T-cell activation (2).
In the past decade, imaging approaches have given us remarkable views of the dramatic molecular rearrangements that accompany T-cell activation. Since the initial visualization of the highly patterned immune synapse (IS) at the interface of a T cell and an antigen-presenting cell (APC) in fixed cells (10), many studies have examined the protein movements and morphological changes triggered by TCR activation in live cells. Images collected at the onset of T-cell activation showed that TCR-rich structures termed ‘microclusters’ are formed within seconds of TCR engagement at the periphery of the T cell, which are then centripetally transported (11–14). Studies from our laboratory (15–17) using immobilized stimulatory antibodies demonstrated that TCR-rich microclusters rapidly recruited TCR proximal PTKs, enzymes, and adapters. Over time, data from many experimental systems have shown that microclusters are sites of signal initiation in a T cell (18).
Proper termination of TCR signals is as important as proper initiation, since failure of control mechanisms could result in autoimmunity or a compromised immune response. It is clear that regulation occurs at various levels starting with the selection of coreceptors, which can either enhance or diminish the TCR response. CD28 and CD4 are well-known activating coreceptors, while the most prominent examples of inhibitory receptors are cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed death 1 (PD1) (19, 20). Activation of the TCR also triggers mechanisms within the TCR signaling machinery that can decrease TCR-mediated signaling (21). These include dephosphorylation by phosphatases as well as phosphorylation on inhibitory residues by kinases (22, 23). In addition, transmembrane adapter scaffolds involved in T-cell activation, such as LAT, assemble protein complexes that trigger negative feedback loops to extinguish T-cell activation (24). Finally, activation-induced internalization of membrane proteins followed by degradation may eliminate activated signaling molecules, thus attenuating signal transduction (25).
In this review, we concentrate on endocytosis as a regulatory mechanism in the context of other negative regulatory mechanisms for TCR signaling. Endocytosis refers to the internalization of molecules from the cell surface into membrane-limited vesicles that are transported to internal membrane compartments. There are innumerable examples of the removal of active complexes by the endocytosis of cell surface receptors, as regulation of signal transduction is a concern for all signaling systems. Often endocytosis terminates signaling, but receptors can also be transported to internal sites where the assembly of unique signaling complexes leads to the activation of specific signal transduction pathways. Thus, endocytosis can have both positive and negative effects, and the importance of crosstalk between endocytosis and signaling has been established in many different cell types (26–28).
Extensive studies examining the fate of the TCR have established that following stimulation, the number of cell surface TCR is decreased due to a combination of increased receptor internalization, decreased recycling, and increased degradation (29, 30). Much less is known about the cellular fate of adapters recruited to the TCR. Imaging studies have shown that the microclusters assembled by TCR activation initially contain TCR and signaling proteins, but soon thereafter, the signaling molecules and TCR dissociate (13–15). This raises the question of whether the TCR and signaling proteins have diverse cellular fates. In this review, we focus on the intracellular fate of signaling molecules that are recruited to the TCR upon TCR engagement, with special emphasis on the adapters LAT and SLP-76.
Signal regulation at the IS
About 10 years ago, the Kupfer laboratory (10) identified the IS as a specialized junction between T cells and antigen-presenting surfaces characterized by a central supramolecular activation cluster (cSMAC) enriched for TCR–CD3 and an integrin-rich peripheral supramolecular activation cluster (pSMAC). Subsequent studies identified the distal SMAC, an outer region which contains large molecules such as CD43 and CD45 (31, 32). The IS was initially proposed to be a site for effective T-cell activation, as the cSMAC is enriched for TCRs, antigenic peptide-MHC complexes, and signaling molecules such as the PTK Lck and phosphoinositide 3-kinase (PI3K) products (10, 33, 34). Furthermore, the observations that the IS is stable for several hours and that prolonged interaction between T cells and APCs was required for full T-cell activation (34) led to the hypothesis that the IS provided a mechanism for sustained TCR engagement and signaling. Another proposal was that the IS forms to enable T cells and APCs to communicate with each other and that the observed molecular rearrangements are involved in polarized secretion by T cells (35). Subsequently, several studies have shown that TCR signaling can be effectively induced prior to or in the absence of cSMAC formation (36–41), leaving the role of the IS controversial and unclear.
There is increasing evidence that the IS acts as a site for regulation of TCR signaling. Lee et al. (42) used a computational model and cellular data from T cells lacking the adapter CD2AP, which fail to form well segregated synapses, to propose that the IS acts as an adaptive controller. In this model, at low peptide-MHC concentrations the cSMAC boosts TCR triggering by concentrating antigen, TCR, and kinases, thus decreasing the time required for antigenic ligand to find TCR and trigger activation. However, at higher concentrations of peptide-MHC, the model predicts that the c-SMAC attenuates TCR activation due to increased TCR downregulation. In support of this model, Varma et al. (43) found that the cSMAC is enriched for the phosphatase CD45 and lysobisphosphatidic acid, an acidic phospholipid that is involved in sorting of membrane proteins into degradative pathways. Thus, it has been proposed that the c-SMAC can serve as a site of TCR endocytosis, followed by receptor degradation and signal termination.
TCR microclusters: sites of signal initiation
It soon became clear that the IS observed by static imaging of fixed cells (10) is not the first molecular rearrangement that occurs in response to TCR stimulation. Grakoui et al. (11) provided the first dynamic pictures of the events that occur as the TCR engages with lipid bilayers containing peptide-MHC. They observed that within the first 30 s after initial contact, TCR-rich small structures form at the periphery of the T-cell contact zone. Krummel et al. first reported receptor clusters in a cell–cell system, at the interface of T cells expressing GFP-receptors and APCs (44). These small, discrete structures enriched for TCR have subsequently been called microclusters, a term that has come into wide use.
Advances in high speed microscopy allowed real time visualization of the early events of T-cell activation with greater temporal and spatial accuracy. Studies from our laboratory (15–17) using anti-CD3 coated glass coverslips demonstrated that downstream components of the TCR signaling pathway, such as PTKs ZAP-70 and Lck, adapters LAT, SLP-76, Grb2, Gads, NCK, and Wiskott–Aldrich syndrome protein, and enzymes PLC-γ1, Vav, and c-Cbl, are rapidly recruited within seconds to TCR microclusters, while larger glycoproteins such as CD43 and the protein tyrosine phosphatase CD45 are excluded from them. Subsequent studies using lipid bilayers containing peptide-MHC and total internal reflection fluorescence (TIRF) microscopy verified and extended these results (13, 14). In this model system, microclusters are continuously generated at the periphery of the T cell peptide-MHC interface, play a predominant role in generation of sustained signals, and are ultimately transported and consolidated at the center of the contact to form a cSMAC (43). A study examining the relationship between TCR signaling and costimulation showed that the costimulatory receptor CD28 is recruited initially to TCR microclusters and is later sorted away from the TCR to a unique outer subregion of the cSMAC, where it assembles with kinases, and generates sustained signaling (45). Recently, using a photoactivatable agonist, Huse et al. (46) has refined the time-scale of cluster formation and demonstrated that adapters are recruited to microclusters within 4 s.
Cooperative protein–protein interactions appear to play a critical role in the nucleation and stabilization of the multi-molecular signaling clusters (47–51), and dynamic actin polymerization provides the force required for the movement of the microclusters (43, 52, 53). These rapidly assembled clusters of signaling complexes are the predominant sites of TCR-induced tyrosine phosphorylation and are coincident with cytosolic calcium elevations, thus establishing TCR microclusters as sites of signal initiation (15, 44). Furthermore, obstruction of cluster formation resulted in reduced levels of TCR signaling, indicating that microclusters play a crucial role in TCR-mediated signaling pathways (48, 51).
Dynamic movement of TCR microclusters
Real time imaging techniques have also demonstrated the dynamic nature and changing composition of TCR microclusters after they are generated. In our planar modeling system, signaling proteins initially cluster in close proximity to the TCR (15–17). More recently, fine spatial imaging studies indicate that signaling protein clusters may represent discrete interdigitating domains (49, 53). These structures could resemble the distinct protein and lipid domains observed by transmission electron microscopy of plasma membrane sheets from mast cells and T cells (54, 55). The electron micrographs show that Fc receptor I on mast cells and TCR/CD3 in T cells cluster independently of LAT. These previously segregated clusters then move close together in an activated cell.
Following initial recruitment, components of the complexes rapidly exit these structures with distinct dynamics (15–17). While the adapters Grb-2 and Gads were transiently recruited to TCR clusters, ZAP-70-containing clusters appeared rather stable. However, photobleaching studies revealed that at the population level there is a continuous flux of ZAP-70 and other signaling molecules into and out of the clusters (15, 49). In comparison, the adapter LAT appeared to depart the initial complexes in what appeared to be small vesicular intermediates. Strikingly, SLP-76 dynamically translocated in discrete structures to a perinuclear region after clustering. Studies tracking microclusters using peptide-MHC-bearing bilayers and in T cell–APC interfaces have revealed that signaling proteins including kinases and adapters dissociate from the microclusters as the TCRs translocate to the cSMAC as it forms (13, 14, 33). Thus in all model systems, TCR microclusters are dynamically regulated both temporally and spatially. While TCR engagement causes the transient assembly of clusters that contain all these components, the signaling molecules separate from the TCR soon thereafter. This observation raises the question of whether the TCR and signaling molecules have diverse cellular fates. The movement and dissipation of LAT and SLP-76 clusters may represent strategies such as endocytosis employed by the cell on a short time scale to regulate signaling. Alternatively, movement away from initial clusters containing TCR and ZAP-70 might destabilize LAT–SLP-76 complexes that require phosphorylation by ZAP-70 to maintain their integrity.
Endocytic regulation in T-cell signaling
Endocytosis of proteins at microclusters would be an efficient way to alter molecular proximity by rapidly targeting proteins to different subcellular locations, dramatically changing reaction rates and thus regulating signaling outcomes. In support of this hypothesis, transmission electron microscopy studies in activated mast cells and T cells revealed that molecules involved in endocytosis, such as clathrin and ubiquitin, localize in regions adjacent to membrane clusters, suggesting that the cellular endocytic machinery may be poised to quickly internalize signaling complexes in stimulated immune cells (54–56).
Mechanisms of endocytosis
It is generally accepted that there are numerous ways to internalize proteins from the cell surface and that signaling is influenced by the choice of endocytic pathway. While many studies of endocytic mechanisms have focused on receptor internalization, it is reasonable to assume that similar mechanisms are used to internalize complexes that lack receptors. A major pathway for uptake of signaling molecules relies on the coat protein clathrin, although it is now known that clathrin-independent pathways are also responsible for the internalization of some signaling molecules (57, 58). These diverse internalization routes play an important role in modulating receptor signaling.
Clathrin-mediated endocytosis has been studied for more than two decades and is the best characterized pathway. Internalization begins with the recognition of a peptide-based sorting signal within the cytoplasmic domain of a receptor or other protein by clathrin adapter proteins. Binding to the adapter provides a link between a protein being internalized and the clathrin coat. This binding results in the concentration of cargo proteins in clathrin-coated patches at the plasma membrane. These patches invaginate into clathrin-coated pits that are detached from the plasma membrane through the activity of the fission protein dynamin (57, 59, 60). The internalization of the TCR via clathrin-dependent endocytosis has been extensively studied and is one of the best understood endocytic routes in T cells (61, 62).
Clathrin-independent pathways are more diverse, differing in both cellular machinery and intracellular destinations for their cargo molecules. The sorting signals, methods of cargo selection, and concentration for these pathways are not well understood, and no adapter proteins have been identified for clathrin-independent routes (63). The first non-clathrin pathway described involved internalization through caveolae, unique structures coated with the protein caveolin (64–66). However, some cells, including lymphocytes which do not express caveolin, cannot form caveolae and still possess clathrin-independent pathways (67, 68). In particular, lymphocytes internalize the β chain of the IL-2 receptor via a clathrin-independent, non-caveolar pathway (69). It is now clear that there are many endocytic pathways generating uncoated vesicles with their own sets of preferred cargo. At first, many of these pathways were identified by determining the colocalization of cargo proteins with various marker proteins; however, attempts are now being made to define clathrin-independent endocytic pathways according to their molecular requirements. The criteria developed so far include dependence on caveolin, RhoA, cdc42, dynamin, and Arf6 (27, 63). Unfortunately, there are still many pathways that have not yet been described mechanistically and therefore remain identified only by the cargo carried by the endocytic vesicles.
The broad division into clathrin-dependent and -independent pathways also reflects a general difference in lipid requirements; usually the clathrin-independent pathways are more sensitive to cholesterol depletion than clathrin-mediated endocytosis (67, 70). This requirement for cholesterol combined with the preferential partitioning of many of the cargo molecules into lipid-ordered membrane domains (71, 72) led to the hypothesis that clathrin-independent uptake depends on the internalization of lipid rafts. For this reason, clathrin-independent endocytosis is also referred to as raft-dependent endocytosis, although this usage is becoming less common. Despite the uncertainty in the exact number and nature of clathrin-independent endocytic pathways, there is no doubt that they are of vital importance. Current estimates suggest that clathrin-independent pathways may account for 50% of the total cellular endocytic activity (73).
Endocytosis of the TCR
A large number of studies over the past 20 years have provided molecular and mechanistic details about the process of TCR endocytosis. Prior to encounter with antigen, TCRs are continuously endocytosed and re-expressed on the cell surface. Upon engagement by a peptide–MHC complex, the TCR is downregulated from the surface of the T cell (74). This downregulation appears to be due to a combination of increased receptor internalization, decreased recycling, and increased degradation (29, 30, 75, 76). In both stimulated and unstimulated cells, TCR is internalized into clathrin-coated vesicles via a dileucine endocytosis motif present in the CD3γ subunit (77–79). This motif consists of a DxxxLL sequence that binds the AP-2 clathrin adapter protein at the plasma membrane, which in turn links the TCR to the clathrin-dependent internalization machinery.
At least two distinct pathways exist for stimulation dependent TCR downregulation (80). The first pathway causes TCR recycling and is dependent on PKC-mediated activation. The second pathway is dependent on the PTK activity of Lck (81, 82). One mechanism of regulation by Lck involves ubiquitylation of CD3 and TCRζ chains by the ubiquitin ligase c-Cbl and leads to TCR degradation (the role of ubiquitylation in endocytosis is discussed further in a section below). Lck activity also induces phosphorylation of clathrin heavy chain, an event that positively correlates with ligand-induced TCR internalization (83). In addition, non-triggered TCR are endocytosed by the PKC-dependent pathway (30). TCR downregulation might attenuate signaling and/or might ensure an internal store of TCR that can be rerouted to the IS during the encounter with an APC.
Endocytosis of adapter proteins: SLP-76 is endocytosed in a novel, clathrin-independent pathway
Upon TCR activation of T cells, the adapter protein SLP-76 is recruited to signaling microclusters that form at the sites of receptor activation as described earlier (13, 15). Soon thereafter, SLP-76 moves away from these sites in small clusters. This medial movement has been observed in T cells activated with either plate-bound stimulation, by contact with peptide-containing lipid bilayers, or at the interface with superantigen-pulsed B cells (13, 15, 48). We undertook studies using SLP-76 fused to yellow fluorescent protein (SLP–YFP) to understand this movement. Multiple results indicated that it is an endocytic process (84). We began by demonstrating that perturbations that block endocytosis also inhibited the movement of SLP–YFP clusters. The first experiments examined SLP–YFP movement in T cells treated with tannic acid. Tannic acid crosslinks cell surface proteins thereby blocking both fusion and fission events at the plasma membrane (85, 86). SLP–YFP remained in the initial signaling complexes in the treated cells as a result of the block in endocytosis. In addition, SLP–YFP clusters did not move in T cells held at 16°C. This lowered temperature is commonly used to inhibit endocytosis (87, 88), and its efficacy in T cells was confirmed by reduced uptake of both transferrin and cholera toxin B (CTX). Microscopic techniques were then used to confirm that the SLP–YFP clusters had moved away from the plasma membrane. TIRF microscopy can be used to determine whether fluorescent molecules are near the plasma membrane, as the illumination field is confined to a region within 100–200 nm of the cell surface (89). In our studies, only immobile SLP–YFP could be observed by TIRF, and moving clusters located by epifluorescence disappeared when TIRF conditions were imposed. Direct visualization of vesicular structures containing SLP–YFP in fixed cells by immuno-electron microscopy showed clear association of gold particles marking the location of SLP–YFP on vesicles budding from the plasma membrane and associated with internal membranes (Fig. 1).
Fig. 1. SLP–YFP internalizes in uncoated structures.

Jurkat T cells expressing SLP–YFP were plated onto stimulatory coverslips, fixed after incubation at 37 °C for 5 min, stained with anti-GFP antibodies, and processed for electron microscopy. Enhanced gold staining is seen on uncoated pits, vesicles, and internal membranes. Arrowheads indicate pits and vesicles; Arrows indicate tubules, e-endosomes, G-Golgi, c-cap shaped structures similar to recycling endosomes. Insert shows higher magnification view of pits budding from the plasma membrane. Bar (main panel) = 280 nm, bar (insert) = 170 nm. Originally published in Traffic 2006;7:1143–1162, Publisher Wiley-Blackwell.
The endocytic pathway used to internalize SLP-76 is clathrin-independent. In electron micrographs, clathrin-coated structures never showed any SLP–YFP gold labeling, and in light level immunofluorescence studies, anti-clathrin antibodies failed to label the SLP–YFP clusters. In addition, there was no colocalization between SLP–YFP and internalized transferrin, which is a generally accepted marker for the clathrin-mediated endocytic pathway (57). Furthermore, the internalization of SLP–YFP was sensitive to cholesterol depletion under conditions that did not affect transferrin uptake. By these criteria, SLP-76 uptake is mediated by a clathrin-independent mechanism.
The SLP–YFP clusters lacked markers internalized by mechanistically defined clathrin-independent pathways. SLP–YFP was completely separate from internalized CTX and fluid phase markers, which are usually found in a cdc42-dependent, dynamin-independent pathway in cells that lack caveolin. SLP–YFP did not colocalize with MHC class I, which is found in the Arf6-regulated pathway (63). We were unable to determine the dynamin dependence of this pathway, as studies using dominant negative dynamin failed to have any effect on Jurkat T cells. The endocytic machinery required for SLP-76 internalization is not known.
SLP-76 is a peripheral passenger on the endocytosed membrane-limited vesicles, and it can be released and rebind during transport. Our electron micrographs consistently showed labeling on the cytosolic rather than the luminal side of vesicles, which is not surprising since SLP-76 is a cytosolic protein in unactivated T cells. Photobleaching experiments were used to probe the dynamics of SLP–YFP interactions with the vesicles. These studies showed that bleached molecules were replaced by unbleached ones with a half-life of 5 s. This rapid exchange is consistent with exchange between bound molecules and a cytosolic pool of SLP–YFP. In addition, all of SLP–YFP molecules in a cell could be photobleached by repetitive bleaching of a small number of internalized clusters. This finding indicates that there is a constant rapid reshuffling of SLP–YFP molecules such that all the SLP–YFP bound to one vesicle exchanges with molecules in the cytosol and then eventually with all of the SLP–YFP bound to other vesicles.
Since SLP-76 lacks any domains known to confer membrane association but does possess many protein–protein interaction domains (90), another protein probably links SLP-76 to the vesicle membranes. Of all the proteins tested, only two, the adapter Gads and the transmembrane adapter protein LAT, were found in the SLP-76 clusters. Moreover, the combination of these two additional proteins provides a plausible link to membranes. Gads is constitutively bound to SLP-76 via its SH3 domain, and Gads can bind to phosphorylated LAT (5, 91). Furthermore, immunofluorescence studies showed that the LAT found on SLP-76 clusters is phosphorylated and capable of binding Gads. Other studies have shown that without Gads, SLP-76 was not recruited to the initial microclusters, and no lateral movement of SLP-76 was seen. The phosphorylation sites in LAT are also needed for clustering of LAT and SLP-76 (48).
Endocytosed signaling complexes could regulate signaling by initiating new signals from intracellular sites or by targeting signaling molecules for degradation. The internalized SLP-76 clusters contain both phosphorylated LAT and phosphorylated SLP-76. Therefore these adapter proteins are in an activated state, capable of binding effector molecules, and signaling from these internalized complexes is possible. However, there is also evidence that SLP-76 is removed from the vesicles soon after internalization. Normally the SLP–YFP signal dissipated quickly at lower levels of SLP–YFP expression, and the fluorescent signal often disappeared before reaching the cell center. When endocytosis of the clusters was inhibited, the fluorescent signal persisted. These data suggest that following internalization, activated SLP-76 is removed from the clusters. The loss of this important adapter could lead to dissolution of the entire protein complex (51), thus attenuating signaling.
Endocytosis of adapter proteins: LAT is internalized in multiple pathways
Some molecules can be internalized by several different endocytic pathways in the same cell. When T cells are allowed to internalize transferrin, some transferrin-containing vesicles contain LAT as well as TCR/CD3 (92, 93). If CTX, which does not colocalize with transferrin in Jurkat T cells, is internalized during activation, some CTX containing vesicles also contain LAT. In addition, all the internalized SLP-76 clusters also contain LAT, although these vesicles contain neither transferrin nor CTX. Thus, LAT is internalized by clathrin-mediated endocytosis, a clathrin-independent pathway containing CTX, and a novel pathway defined by the presence of SLP-76. However, it appears that the amount of available SLP-76 controls the amount of LAT internalized via the latter pathway (84). At endogenous or near endogenous levels of SLP-76, about half of the LAT colocalizes with SLP-76. However, when SLP-76 is overexpressed eightfold, 85% of LAT is now found in the SLP-76 endocytic pathway. Other studies have suggested that inhibition of one endocytic pathway results in upregulation of other forms of endocytosis (94, 95). The presence of LAT in multiple pathways implies that LAT internalization will continue with high efficiency under most conditions. Alternatively, the presence of LAT in different pathways could indicate that LAT complexes with different compositions and thus different signaling capabilities are being formed with profound implications for T-cell signaling. In this context, the shift in LAT distribution caused by SLP-76 levels could modulate T-cell activation by affecting the composition, location, and signaling capabilities of endocytosed LAT complexes.
The dynamic movements of LAT and SLP-76 away from TCR-rich microclusters represent endocytic events (84, 93). In our studies using plate-bound stimulation, it appeared that endocytosis of these adapter-based complexes did not require internalization of the TCR. These data suggest that adapter proteins as well as receptors can serve as substrates for endocytic machinery and promote removal of specific components from signaling microclusters. Therefore, T-cell signaling may be modulated by the internalization of complexes composed of different proteins with the potential for regulation of diverse signaling cascades.
We demonstrated that both SLP-76 and LAT movement was inhibited after overexpression of a mutant form of the E3 ligase c-Cbl (93). Further evidence for the importance of ubiquitin in cluster internalization comes from the effects of the ubiquitin-interacting motif (UIM) of eps15 that is known to block clathrin-independent internalization of the EGF receptor (96). This construct did not affect the recruitment of SLP–YFP to TCR-based microclusters; however, in cells expressing the dominant negative UIM domain, the internalization of SLP–YFP clusters was severely inhibited (84). These observations prompted us to investigate the role of c-Cbl and ubiquitylation in endocytosis of cluster proteins.
Role of ubiquitylation
Ubiquitin machinery and the immune system
As described in the previous section, multiple endocytic sorting signals have been identified on the cytoplasmic domains of cell surface proteins, most of which are peptide sequences recognized by adapters involved in targeting molecules to clathrin-coated vesicles (97). Another sorting signal is provided by modification of target proteins by covalent attachment of ubiquitin. Ubiquitylation is a reversible post-translational modification that results in the conjugation of ubiquitin (Ub), a small 76 amino acid protein, to a target protein, usually on a lysine residue (98). Because ubiquitin itself harbors seven lysines that also can be used as targets for ubiquitylation, ubiquitin can polymerize into chains. The length and the topology of these chains are of importance for the cellular fate of the ubiquitylated protein. In general, monoubiquitylation plays a role in regulation of endocytosis and targeting for lysosomal degradation. Polyubiquitin conjugation through lysine residues at position 48 (Lys-48-linked) signals proteasomal destruction, whereas multiple monoubiquitylation and Lys-63-linked polyubiquitylation mediate non-proteolytic cellular functions (99). The ubiquitylation of a protein requires the successive actions of the ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2) and an ubiquitin ligase (E3), which catalyzes the transfer of activated ubiquitin to the lysine residue of the substrate protein and imparts specificity to the system (98). Ubiquitylation is reversible and may be removed by the action of de-ubiquitylating enzymes (100).
Post-translational modification by ubiquitylation modulates the immune system, potentially setting the balance between immune responses and tolerance (101). Several immune receptors and costimulatory molecules are modified by ubiquitin. In particular, ubiquitin ligases of both HECT (Itch, Nedd4) and RING (c-Cbl, Cbl-b, GRAIL) types play key roles in modulation of central and peripheral tolerance. The defective expression of several of these ubiquitin ligases has been related to the development of autoimmune disease in experimental murine models (102). Moreover, T-cell anergy requires upregulation of Cbl-b along with Itch and GRAIL to downregulate several signaling proteins through activation-dependent degradation (103). Interestingly, ubiquitin and Cbl-b appear to be enriched at the IS of antigen stimulated T cells (104). Ubiquitin-dependent endocytosis has also been utilized by viruses to downregulate key molecules of the host immune system as a mechanism of immune evasion (105). Viral RING-CH ligases and their cellular homologues, the membrane-associated RING-CH (MARCH) ligases, downregulate several critical immunoreceptors (106).
Although modification of plasma membrane proteins with ubiquitin was demonstrated several decades ago and receptor ubiquitylation has been implicated as a sorting signal that targets several activated receptors for internalization, proof that ubiquitin acts as an internalization signal came from relatively recent studies in yeast in which investigators found that lysine residues in the cytoplasmic domains of G-protein coupled receptors were crucial for rapid internalization of these proteins (101). In contrast, the evidence supporting the importance of receptor ubiquitylation in endocytosis in mammalian systems remains indirect. A recent study using a series of Epidermal growth factor receptor (EGFR) mutants that were defective in growth factor-stimulated ubiquitylation revealed that receptor internalization was uncoupled from receptor ubiquitylation (107). Similarly, although it has been suggested that ubiquitylation of MHC class II β chain regulates MHC class II internalization, ubiquitin-defective MHC class II is endocytosed at rates comparable to wildtype MHC class II (108). These findings suggest a more complex role for ubiquitylation than previously anticipated.
Cbl functions as an E3 ligase and an adapter to regulate TCR microclusters
The Cbl family of RING-finger domain-containing E3 ubiquitin ligases has important roles in the immune system. Mammalian cells contain three Cbl proteins: c-Cbl, Cbl-b, and Cbl-c. c-Cbl and Cbl-b are expressed in T cells. c-Cbl has well-established functions in regulating ubiquitylation, endocytosis, and downregulation of several receptor tyrosine kinases (RTKs) and non-RTKs in various systems (109, 110). In T cells, Cbl regulates ubiquitylation and downmodulation of the TCR. It has been reported that TCRζ and CD3δ chains are ubiquitylated upon TCR engagement (111). Ubiquitylation of TCRζ relies on ZAP-70, which links c-Cbl to phosphorylated TCRζ, thus selectively promoting ubiquitylation of activated TCR complexes (112). Another protein that links c-Cbl to the TCR is Src-like adapter protein, which seems to function as an adapter to target the ubiquitin ligase activity of c-Cbl to phosphorylated TCRζ chains, thus regulating surface TCR expression on thymocytes (113). In addition, Cbl ubiquitylates several molecules involved in TCR-mediated signaling including ZAP-70, LAT, PLC-γ1, phosphoinositide-3 kinase, Vav, and PKC-θ (93, 114).
The importance of Cbl family members in immune receptor signaling pathways has been clearly demonstrated by the phenotypes of gene knockout mice. Consistent with a role for Cbl proteins as negative regulators, both c-Cbl−/− and Cbl-b−/− mice display hyperactive signaling downstream of the TCR (115). Thymocytes from c-Cbl mutant mice exhibit enhanced phosphorylation of the PTK ZAP-70 and increased and sustained phosphorylation of its substrates, the adapters SLP-76 and LAT, probably because of deregulated ZAP-70 activity (116, 117).
How do the biochemical and genetic data on Cbl function in downmodulation of T-cell signaling components correlate to imaging models of T-cell activation? Fixed cell analysis showed that c-Cbl is recruited to microclusters within 2 min of activation (15). More recently, we examined the dynamics of c-Cbl movement in live cell imaging experiments (93). This study showed that c-Cbl was recruited to the clusters containing LAT and SLP-76 within seconds of TCR engagement. Soon after recruitment, Cbl clusters dissipated quickly, along with LAT and SLP-76. In striking contrast, expression of 70Z/3 Cbl, an oncogenic, dominant-negative version that is defective in linker/RING finger domain function, displayed persistent clusters that were severely inhibited in movement and dissipation. The same dominant-negative mutant of Cbl did not block LAT or SLP-76 recruitment into clusters but retained the adapters in clusters, thus preventing endocytosis and transport to endosomes. Similar inhibition of LAT movement was observed in primary CD4+ cells expressing 70Z/3 Cbl and in CD4+ cells from c-Cbl−/− mice, validating that the effects of 70Z/3 Cbl expression reflect c-Cbl function. These data indicate that c-Cbl is intimately involved in the sorting of LAT and SLP-76 into mobile endocytic structures. Thus, the RING domain of Cbl regulates LAT complex dynamics, and expression of E3 defective Cbl prolongs the association of LAT complexes with its attendant kinases. Consistent with this observation, persistent LAT clusters in 70Z/3 Cbl mutant expressing cells remain phosphorylated (Fig. 2). These effects could account for the ability of Cbl proteins to influence T-cell activation. It will be of interest to examine whether trafficking of other signaling proteins recruited to microclusters is regulated by Cbl proteins.
Fig. 2. Persistent LAT clusters caused by over expression of 70Z/3 Cbl contain phosphorylated LAT.
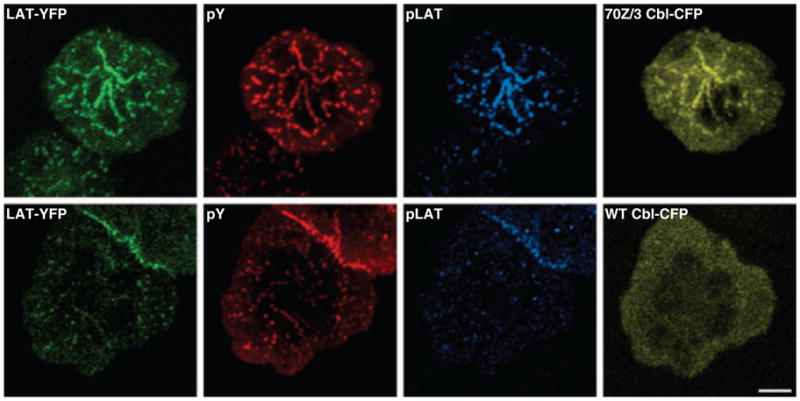
Jurkat T cells expressing LAT–YFP were transfected with either 70Z/3 Cbl–CFP (top row) or wildtype (WT) Cbl–CFP (bottom row). Twenty-four hours after transfection, the cells were plated onto stimulatory coverslips and fixed after incubation at 37 °C for 5 min. The cells were stained with mouse anti-phosphotyrosine (4G10) and rabbit anti-phospho-LAT191. LAT–YFP is shown in the green channel, anti-phosphotyrosine staining in the red channel, anti-phospho-LAT191 staining in cyan, and Cbl–CFP in yellow. In the cells expressing 70Z/3 Cbl-CFP, there are prominent clusters containing both LAT–YFP and 70Z/3 Cbl–CFP that show extensive staining for phosphotyrosine and phospho-LAT. In contrast, cells overexpressing WT Cbl–CFP have faint LAT clusters, no Cbl–CFP clusters, very little phosphotyrosine staining, and almost undetectable phospho-LAT staining. Bar = 5 μm.
Insight into the mechanisms by which c-Cbl regulates signaling cluster dynamics was obtained by examining domains of Cbl required for recruitment to and persistence at clusters. Cbl is a large multidomain protein and is composed of an N-terminal tyrosine kinase binding domain (TKB), a zinc-binding RING finger domain, several proline-rich sequences, multiple tyrosine residues that get phosphorylated upon TCR stimulation, and a C-terminal ubiquitin-associated domain (UBA) (115, 118). Mapping the requirement of Cbl domains revealed that recruitment to signaling clusters requires the proline-rich domain, while 70Z/3 Cbl persistence in clusters requires the TKB domain. The importance of c-Cbl proline-rich and TKB domains in c-Cbl recruitment and stabilization at signaling clusters is compatible with a role for LAT and ZAP-70. LAT interaction with the c-Cbl proline-rich domain can be mediated by Grb-2, PLC-γ, or NCK (2, 118), and ZAP-70 interacts with the TKB domain of c-Cbl (119). Interestingly, although neither LAT nor ZAP-70 binding were required individually for c-Cbl recruitment, simultaneous depletion of both molecules abolished c-Cbl recruitment to clusters. These data indicate a redundancy in the system and show that presence of either ZAP-70 or LAT is sufficient to stabilize c-Cbl recruitment to the same TCR-induced clusters, without participation of the other. Alternatively, it is possible that ZAP-70 and LAT are at distinct signaling clusters, and elimination of either signaling component allows for c-Cbl to be recruited to signaling clusters containing the other protein. Consistent with this scenario, in mast cell membranes, receptor-containing signaling domains and LAT-containing domains are discrete (55).
Once recruited to the microclusters, simultaneous association of c-Cbl with signaling molecules and the endocytic pathway would result in endocytosis of critical signaling molecules such as LAT and SLP-76. Ubiquitylation of LAT or other signaling proteins in the LAT complex by Cbl could facilitate their trafficking and subsequent degradation. Another possibility is that effects seen with the Cbl mutants on endocytosis results from defective ubiquitylation of endocytic adapter proteins. In fact, it has been proposed that ubiquitylation of a component of the endocytic apparatus is required for endocytosis of the α factor receptor in yeast (120) and growth hormone receptor in mammalian cells (121). Obviously, the precise sequence of molecular events linking c-Cbl to internalization of microcluster components may vary, and other models could account for the role of the c-Cbl RING domain. In any case, Cbl-dependent microcluster endocytosis opens up new avenues for understanding the fundamental process of signal regulation at these structures.
LAT ubiquitylation and endocytic sorting by Cbl
The essential role of LAT in T-cell development and activation has been demonstrated by studies performed in LAT-deficient cells and knockout mice showing impaired T-cell activation and T-cell development (122, 123). LAT is an integral membrane protein that possesses a membrane proximal palmitoylation motif and is constitutively membrane associated. It has a very short extracellular domain and a long cytoplasmic tail that contains several tyrosine phosphorylation motifs. Once phosphorylated, these motifs serve as docking sites for several enzymes and adapters. Thus, LAT serves as a scaffold that orchestrates T-cell signaling (2). In addition to containing tyrosines, the LAT cytoplasmic domain also contains several potential endocytic sorting motifs (97) including two lysines that could serve as sites for ubiquitylation. Indeed, LAT ubiquitylation has been seen in Jurkat T cells and in heterologous COS-7 cells expressing LAT and ubiquitin (93, 124). In contrast, no evidence was observed for SLP-76 ubiquitylation. Notably, in LAT immunoprecipitates, the predominant ubiquitylated band was detected at a size compatible with a monoubiquitylated species of LAT (93). Since monoubiquitylation has been implicated in the regulation of endocytosis and targeting for lysosomal degradation, it is enticing to speculate that this species could serve as a sorting signal for LAT internalization (125). In addition to the monoubiquitylated band, distinct higher molecular weight species were detected in LAT immunoprecipitates, consistent with the existence of multi-mono or polyubiquitylated LAT. However, immuno-blotting with antibodies to LAT did not detect the higher molecular weight species detected by antibodies directed to ubiquitin. Denaturing sequential immunoprecipitations confirmed that the detected species correspond to ubiquitylated LAT and ruled out the possibility that they correspond to proteins that coimmunoprecipitate with LAT (93). The inability to detect the high molecular weight bands with LAT antibody could indicate that these species are a small fraction of the total cellular LAT pool, thus making it difficult to detect. Consistent with this scenario, a decrease in LAT protein levels was not detected following T-cell activation in the defined time course over which dissipation of LAT clusters is seen, possibly indicating that upon TCR triggering only a small fraction of the cellular pool of LAT is recruited to signaling complexes, internalized, and degraded.
Analysis of sequences in LAT important for ubiquitylation identified a requirement of the LAT C-terminus for LAT ubiquitylation (124). However, in this analysis, a large segment of LAT, including phosphotyrosine docking sites through which c-Cbl may be recruited, were deleted. Thus, site directed mutagenesis and/or mass spectrometry studies of LAT to precisely determine which sequences in LAT are targeted for ubiquitylation are warranted. In an effort to assign a specific E3 to LAT ubiquitylation, LAT, c-Cbl, and ubiquitin were overexpressed in COS-7 cells (93). The expression of wildtype Cbl caused a modest increase in LAT ubiquitylation, predominantly the monoubiquitylated band, consistent with LAT being a target for Cbl-mediated ubiquitylation. In contrast, expression of 70Z/3 Cbl resulted in a significant decrease in all ubiquitylated LAT species. Thus, the RING activity of Cbl is required for LAT internalization and ubiquitylation. These data are consistent with a model in which Cbl-mediated ubiquitylation is required for the internalization of LAT and SLP-76 clusters. Upon expression of the Cbl RING mutant, the ubiquitylation of LAT and perhaps other signaling molecules is diminished, and as a result, microcluster endocytosis and dissipation are inhibited.
The linker for activation of B cells (LAB), the apparent counterpart of LAT in B cells, is also ubiquitylated (126) and internalized upon B-cell activation (127). Though the precise molecular mechanism by which this process occurs has not been investigated, LAB is tyrosine phosphorylated upon B-cell activation and associates with Cbl. These studies on LAT and LAB are the first reports of receptor-activated endocytosis of transmembrane adapter proteins.
Functional consequences of endocytosis
Despite the intense study of protein internalization routes in a variety of cells, the diverse biological implications of endocytosis are only beginning to be understood. Endocytosis has long been regarded as a mechanism to downregulate plasma membrane receptors. EGFR trafficking is one of the most well characterized models for studying endocytic pathways and how endocytosis affects signaling. Upon EGF binding, both EGF and EGFR are internalized and eventually degraded, which leads to a decreased number of receptors on the cell surface and negatively regulates receptor signaling. However, EGF–EGFR complexes in endosomes remain active and continue to signal after internalization (128, 129). In another example of the crosstalk between endocytosis and signaling, TGF-β receptors are endocytosed through clathrin-dependent and clathrin-independent routes that trigger a signaling response or degradation, respectively (130). Thus, endocytosis can have diverse effects on signal transduction.
In T cells, activation results in rapid internalization and degradation of triggered TCR (75). The importance of TCR downregulation in T-cell signaling pathways has been clearly demonstrated by the phenotypes of gene knockout mice with defective TCR internalization. For example, in mice lacking Cbl or CD2AP, T cells exhibit increased TCR levels on the cell surface and have exaggerated biological responses to antigenic stimulation (42, 115). In parallel with TCR degradation, several signaling components are known to be degraded (25, 103, 131, 132). Though the functional consequences of signaling component loss in stimulated cells is unclear, it is likely that controlled destruction of the signaling apparatus may serve to fine tune antigen receptor signaling.
Spatial translocation of signaling microclusters appears to have an important role in terminating TCR signals. Inhibition of cluster movement by expression of the 70Z/3 Cbl E3 ligase mutant, integrin ligation, or physical constraints appears to prolong and enhance TCR-mediated signals (53, 93, 133). In our plate-bound activation system, we have shown that LAT and SLP-based clusters are endocytosed rapidly upon TCR stimulation. We suggest that microcluster movement is directly linked to endocytic events and that the immobile clusters reflect a failure to internalize signaling complexes. These results implicate endocytosis in regulation of crucial features of TCR-induced signal transduction including signaling kinetics, amplitude, and spatial distribution. Consistent with this model, persistent LAT clusters that fail to get endocytosed remain phosphorylated (Fig. 2), indicating that the block in endocytosis prolongs signaling from LAT complexes at the plasma membrane. Furthermore, we observed an increase in expression levels of total LAT as well as in the levels of activated pools of LAT in cells expressing the 70Z/3 Cbl mutant. Cbl-mediated modulation of LAT endocytosis, levels, and ubiquitylation is consistent with the possibility that LAT ubiquitylation and endocytosis could lead to LAT degradation. At present, little is known about the physiological significance of modification of LAT by ubiquitin. But given our growing appreciation of the role of ubiquitylation in the control of endocytosis and signaling, it appears likely that it will have important functional consequences.
It must be noted that endosomes are gaining considerable attention as sites for assembly of signaling complexes (26, 134). The presence of signaling complexes on intracellular endosomal membranes observed in the EGF system suggests that the intracellular trafficking routes of signaling molecules have important implications for signaling. In T lymphocytes, a fraction of plasma membrane-associated Lck is internalized in endosomes along with ZAP-70 in CD2-triggered T cells (135). In our studies, endocytosed SLP-76 clusters contained phosphorylated LAT and SLP-76, indicating that they are signaling competent. Internalized LAT signaling complexes in endosomes might trigger qualitatively different signals than complexes located at the plasma membrane. Alternatively, functional signaling activity may be necessary for efficient degradation of the signaling complexes after internalization.
Conclusions and perspectives
In this review, we have examined how endocytosis affects signaling from TCR microclusters. From the moment that signaling proteins assemble in large multimeric complexes, disassembly mechanisms are triggered to limit the duration of signaling from these microclusters. Our studies indicate that endocytosis of adapter-based signaling complexes may serve to regulate signal transduction from TCR-based complexes. The idea that internalization of adapters, in addition to receptors, could regulate T-cell activation is relatively new, and the functional relevance of such mechanisms are just beginning to be understood. Given the essential scaffolding role of the adapter protein LAT, the regulated internalization of activated LAT signaling complexes might be an efficient strategy for rapidly controlling the amplitude, location, and duration of T-cell signaling, especially when compared with the task of dephosphorylating tyrosine residues in a microcluster or removing all bound effecter molecules. Our experiments indicated that Cbl proteins, ubiquitylation, and multiple internalization pathways are involved in the endocytic events that disassemble signaling microclusters. However, assessing the relevance of these factors in T cells will require much additional work. Despite years of study, the role of ubiquitylation in endocytosis remains controversial, even in well-characterized processes like the internalization of the EFGR. Understanding the complexities of T-cell signaling will be more difficult and will require experimentation across a broad range of model systems. Although there may be variations in how different models react, the underlying strategies leading to microcluster formation, function, and disassembly might be similar across systems, eventually allowing us to understand the events leading from receptor engagement to a final cellular response. More information on the role of endocytosis in these processes will open new avenues to understand the modulation of cell signaling pathways and might have important implications for the discovery of therapeutic agents.
Acknowledgments
We thank Dr. Roman Polishchuk for assistance with electron microscopy. This research was supported by the Intramural Research Program of the NIH, NCI, CCR.
References
- 1.Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435:590–597. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- 2.Samelson LE. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol. 2002;20:371–394. doi: 10.1146/annurev.immunol.20.092601.111357. [DOI] [PubMed] [Google Scholar]
- 3.Davis MM, Krogsgaard M, Huse M, Huppa J, Lillemeier BF, Li QJ. T cells as a self-referential, sensory organ. Annu Rev Immunol. 2007;25:681–695. doi: 10.1146/annurev.immunol.24.021605.090600. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell. 1998;92:83–92. doi: 10.1016/s0092-8674(00)80901-0. [DOI] [PubMed] [Google Scholar]
- 5.Zhang W, Trible RP, Zhu M, Liu SK, McGlade CJ, Samelson LE. Association of Grb2, Gads, and phospholipase C-γ1 with phosphorylated LAT tyrosine residues. Effect of LAT tyrosine mutations on T cell antigen receptor-mediated signaling. J Biol Chem. 2000;275:23355–23361. doi: 10.1074/jbc.M000404200. [DOI] [PubMed] [Google Scholar]
- 6.Lin J, Weiss A. Identification of the minimal tyrosine residues required for linker for activation of T Cell function. J Biol Chem. 2001;276:29588–29595. doi: 10.1074/jbc.M102221200. [DOI] [PubMed] [Google Scholar]
- 7.Paz PE, Wang S, Clarke H, Lu X, Stokoe D, Abo A. Mapping the Zap-70 phosphorylation sites on LAT (linker for activation of T cells) required for recruitment and activation of signalling proteins in T cells. Biochem J. 2001;356:461–471. doi: 10.1042/0264-6021:3560461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez-Villar JJ, et al. Phosphorylation of the linker for activation of T-cells by Itk promotes recruitment of Vav. Biochemistry. 2002;41:10732–10740. doi: 10.1021/bi025554o. [DOI] [PubMed] [Google Scholar]
- 9.Wu JN, Koretzky GA. The SLP-76 family of adapter proteins. Semin Immunol. 2004;16:379–393. doi: 10.1016/j.smim.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 10.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 11.Grakoui A, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- 12.Krummel MF, Davis MM. Dynamics of the immunological synapse: finding, establishing and solidifying a connection. Curr Opin Immunol. 2002;14:66–74. doi: 10.1016/s0952-7915(01)00299-0. [DOI] [PubMed] [Google Scholar]
- 13.Yokosuka T, et al. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- 14.Campi G, Varma R, Dustin ML. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med. 2005;202:1031–1036. doi: 10.1084/jem.20051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bunnell SC, et al. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J Cell Biol. 2002;158:1263–1275. doi: 10.1083/jcb.200203043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braiman A, Barda-Saad M, Sommers CL, Samelson LE. Recruitment and activation of PLCgamma1 in T cells: a new insight into old domains. EMBO J. 2006;25:774–784. doi: 10.1038/sj.emboj.7600978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barda-Saad M, Braiman A, Titerence R, Bunnell SC, Barr VA, Samelson LE. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat Immunol. 2005;6:80–89. doi: 10.1038/ni1143. [DOI] [PubMed] [Google Scholar]
- 18.Seminario MC, Bunnell SC. Signal initiation in T-cell receptor microclusters. Immunol Rev. 2008;221:90–106. doi: 10.1111/j.1600-065X.2008.00593.x. [DOI] [PubMed] [Google Scholar]
- 19.Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol. 2004;172:4661–4665. doi: 10.4049/jimmunol.172.8.4661. [DOI] [PubMed] [Google Scholar]
- 20.Leibson PJ. The regulation of lymphocyte activation by inhibitory receptors. Curr Opin Immunol. 2004;16:328–336. doi: 10.1016/j.coi.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 21.Acuto O, Di Bartolo V, Micheli F. Tailoring T-cell receptor signals by proximal negative feedback mechanisms. Nat Rev Immunol. 2008;8:699–712. doi: 10.1038/nri2397. [DOI] [PubMed] [Google Scholar]
- 22.Singer AL, Koretzky GA. Control of T cell function by positive and negative regulators. Science. 2002;296:1639–1640. doi: 10.1126/science.1071551. [DOI] [PubMed] [Google Scholar]
- 23.Veillette A, Latour S, Davidson D. Negative regulation of immunoreceptor signaling. Annu Rev Immunol. 2002;20:669–707. doi: 10.1146/annurev.immunol.20.081501.130710. [DOI] [PubMed] [Google Scholar]
- 24.Simeoni L, Lindquist JA, Smida M, Witte V, Arndt B, Schraven B. Control of lymphocyte development and activation by negative regulatory transmembrane adapter proteins. Immunol Rev. 2008;224:215–228. doi: 10.1111/j.1600-065X.2008.00656.x. [DOI] [PubMed] [Google Scholar]
- 25.Jang IK, Gu H. Negative regulation of TCR signaling and T-cell activation by selective protein degradation. Curr Opin Immunol. 2003;15:315–320. doi: 10.1016/s0952-7915(03)00048-7. [DOI] [PubMed] [Google Scholar]
- 26.Mills IG. The interplay between clathrin-coated vesicles and cell signalling. Semin Cell Dev Biol. 2007;18:459–470. doi: 10.1016/j.semcdb.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Lajoie P, Nabi IR. Regulation of raft-dependent endocytosis. J Cell Mol Med. 2007;11:644–653. doi: 10.1111/j.1582-4934.2007.00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donaldson JG, Porat-Shliom N, Cohen LA. Clathrin-independent endocytosis: a unique platform for cell signaling and PM remodeling. Cell Signal. 2009;21:1–6. doi: 10.1016/j.cellsig.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alcover A, Alarcon B. Internalization and intracellular fate of TCR-CD3 complexes. Crit Rev Immunol. 2000;20:325–346. [PubMed] [Google Scholar]
- 30.Geisler C. TCR trafficking in resting and stimulated T cells. Crit Rev Immunol. 2004;24:67–86. doi: 10.1615/critrevimmunol.v24.i1.30. [DOI] [PubMed] [Google Scholar]
- 31.Freiberg BA, et al. Staging and resetting T cell activation in SMACs. Nat Immunol. 2002;3:911–917. doi: 10.1038/ni836. [DOI] [PubMed] [Google Scholar]
- 32.Delon J, Kaibuchi K, Germain RN. Exclusion of CD43 from the immunological synapse is mediated by phosphorylation-regulated relocation of the cytoskeletal adaptor moesin. Immunity. 2001;15:691–701. doi: 10.1016/s1074-7613(01)00231-x. [DOI] [PubMed] [Google Scholar]
- 33.Ehrlich LI, Ebert PJ, Krummel MF, Weiss A, Davis MM. Dynamics of p56lck translocation to the T cell immunological synapse following agonist and antagonist stimulation. Immunity. 2002;17:809–822. doi: 10.1016/s1074-7613(02)00481-8. [DOI] [PubMed] [Google Scholar]
- 34.Huppa JB, Gleimer M, Sumen C, Davis MM. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat Immunol. 2003;4:749–755. doi: 10.1038/ni951. [DOI] [PubMed] [Google Scholar]
- 35.Bossi G, Trambas C, Booth S, Clark R, Stinchcombe J, Griffiths GM. The secretory synapse: the secrets of a serial killer. Immunol Rev. 2002;189:152–160. doi: 10.1034/j.1600-065x.2002.18913.x. [DOI] [PubMed] [Google Scholar]
- 36.Lee KH, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science. 2002;295:1539–1542. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 37.Richie LI, Ebert PJ, Wu LC, Krummel MF, Owen JJ, Davis MM. Imaging synapse formation during thymocyte selection: inability of CD3zeta to form a stable central accumulation during negative selection. Immunity. 2002;16:595–606. doi: 10.1016/s1074-7613(02)00299-6. [DOI] [PubMed] [Google Scholar]
- 38.Purtic B, Pitcher LA, van Oers NS, Wulfing C. T cell receptor (TCR) clustering in the immunological synapse integrates TCR and costimulatory signaling in selected T cells. Proc Natl Acad Sci USA. 2005;102:2904–2909. doi: 10.1073/pnas.0406867102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 2004;5:524–530. doi: 10.1038/ni1058. [DOI] [PubMed] [Google Scholar]
- 40.O’Keefe JP, Gajewski TF. Cutting edge: cytotoxic granule polarization and cytolysis can occur without central supramolecular activation cluster formation in CD8+ effector T cells. J Immunol. 2005;175:5581–5585. doi: 10.4049/jimmunol.175.9.5581. [DOI] [PubMed] [Google Scholar]
- 41.Brossard C, et al. Multifocal structure of the T cell - dendritic cell synapse. Eur J Immunol. 2005;35:1741–1753. doi: 10.1002/eji.200425857. [DOI] [PubMed] [Google Scholar]
- 42.Lee KH, et al. The immunological synapse balances T cell receptor signaling and degradation. Science. 2003;302:1218–1222. doi: 10.1126/science.1086507. [DOI] [PubMed] [Google Scholar]
- 43.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krummel MF, Sjaastad MD, Wulfing C, Davis MM. Differential clustering of CD4 and CD3zeta during T cell recognition. Science. 2000;289:1349–1352. doi: 10.1126/science.289.5483.1349. [DOI] [PubMed] [Google Scholar]
- 45.Yokosuka T, et al. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C theta translocation. Immunity. 2008;29:589–601. doi: 10.1016/j.immuni.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huse M, et al. Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity. 2007;27:76–88. doi: 10.1016/j.immuni.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 47.Hartgroves LC, Lin J, Langen H, Zech T, Weiss A, Harder T. Synergistic assembly of linker for activation of T cells signaling protein complexes in T cell plasma membrane domains. J Biol Chem. 2003;278:20389–20394. doi: 10.1074/jbc.M301212200. [DOI] [PubMed] [Google Scholar]
- 48.Bunnell SC, et al. Persistence of cooperatively stabilized signaling clusters drives T-cell activation. Mol Cell Biol. 2006;26:7155–7166. doi: 10.1128/MCB.00507-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Douglass AD, Vale RD. Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell. 2005;121:937–950. doi: 10.1016/j.cell.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Houtman JC, et al. Binding specificity of multiprotein signaling complexes is determined by both cooperative interactions and affinity preferences. Biochemistry. 2004;43:4170–4178. doi: 10.1021/bi0357311. [DOI] [PubMed] [Google Scholar]
- 51.Houtman JC, et al. Oligomerization of signaling complexes by the multipoint binding of GRB2 to both LAT and SOS1. Nat Struct Mol Biol. 2006;13:798–805. doi: 10.1038/nsmb1133. [DOI] [PubMed] [Google Scholar]
- 52.Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. Dynamic actin polymerization drives T cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity. 2001;14:315–329. doi: 10.1016/s1074-7613(01)00112-1. [DOI] [PubMed] [Google Scholar]
- 53.Nguyen K, Sylvain NR, Bunnell SC. T cell costimulation via the integrin VLA-4 inhibits the actin-dependent centralization of signaling microclusters containing the adaptor SLP-76. Immunity. 2008;28:810–821. doi: 10.1016/j.immuni.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 54.Lillemeier BF, Pfeiffer JR, Surviladze Z, Wilson BS, Davis MM. Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc Natl Acad Sci USA. 2006;103:18992–18997. doi: 10.1073/pnas.0609009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson BS, Pfeiffer JR, Surviladze Z, Gaudet EA, Oliver JM. High resolution mapping of mast cell membranes reveals primary and secondary domains of Fc(epsilon)RI and LAT. J Cell Biol. 2001;154:645–658. doi: 10.1083/jcb.200104049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilson BS, Pfeiffer JR, Oliver JM. Observing FcepsilonRI signaling from the inside of the mast cell membrane. J Cell Biol. 2000;149:1131–1142. doi: 10.1083/jcb.149.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benmerah A, Lamaze C. Clathrin-coated pits: vive la difference? Traffic. 2007;8:970–982. doi: 10.1111/j.1600-0854.2007.00585.x. [DOI] [PubMed] [Google Scholar]
- 58.Sandvig K, Torgersen ML, Raa HA, van Deurs B. Clathrin-independent endocytosis: from nonexisting to an extreme degree of complexity. Histochem Cell Biol. 2008;129:267–276. doi: 10.1007/s00418-007-0376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ungewickell EJ, Hinrichsen L. Endocytosis: clathrin-mediated membrane budding. Curr Opin Cell Biol. 2007;19:417–425. doi: 10.1016/j.ceb.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 60.Roth MG. Clathrin-mediated endocytosis before fluorescent proteins. Nat Rev Mol Cell Biol. 2006;7:63–68. doi: 10.1038/nrm1783. [DOI] [PubMed] [Google Scholar]
- 61.Boyer C, et al. T cell receptor/CD3 complex internalization following activation of a cytolytic T cell clone: evidence for a protein kinase C-independent staurosporine-sensitive step. Eur J Immunol. 1991;21:1623–1634. doi: 10.1002/eji.1830210707. [DOI] [PubMed] [Google Scholar]
- 62.Telerman A, Amson RB, Romasco F, Wybran J, Galand P, Mosselmans R. Internalization of human T lymphocyte receptors. Eur J Immunol. 1987;17:991–997. doi: 10.1002/eji.1830170715. [DOI] [PubMed] [Google Scholar]
- 63.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8:603–612. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pelkmans L, Helenius A. Endocytosis via caveolae. Traffic. 2002;3:311–320. doi: 10.1034/j.1600-0854.2002.30501.x. [DOI] [PubMed] [Google Scholar]
- 65.Parton RG, Joggerst B, Simons K. Regulated internalization of caveolae. J Cell Biol. 1994;127:1199–1215. doi: 10.1083/jcb.127.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anderson RG, Kamen BA, Rothberg KG, Lacey SW. Potocytosis: sequestration and transport of small molecules by caveolae. Science. 1992;255:410–411. doi: 10.1126/science.1310359. [DOI] [PubMed] [Google Scholar]
- 67.Orlandi PA, Fishman PH. Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J Cell Biol. 1998;141:905–915. doi: 10.1083/jcb.141.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deckert M, Ticchioni M, Bernard A. Endocytosis of GPI-anchored proteins in human lymphocytes: role of glycolipid-based domains, actin cytoskeleton, and protein kinases. J Cell Biol. 1996;133:791–799. doi: 10.1083/jcb.133.4.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Subtil A, Hemar A, Dautry-Varsat A. Rapid endocytosis of interleukin 2 receptors when clathrin-coated pit endocytosis is inhibited. J Cell Sci. 1994;107:3461–3468. doi: 10.1242/jcs.107.12.3461. [DOI] [PubMed] [Google Scholar]
- 70.Schnitzer JE, Oh P, Pinney E, Allard J. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J Cell Biol. 1994;127:1217–1232. doi: 10.1083/jcb.127.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kirkham M, Parton RG. Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta. 2005;1746:349–363. doi: 10.1016/j.bbamcr.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 72.Pelkmans L. Secrets of caveolae- and lipid raft-mediated endocytosis revealed by mammalian viruses. Biochim Biophys Acta. 2005;1746:295–304. doi: 10.1016/j.bbamcr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 73.Gong Q, Huntsman C, Ma D. Clathrin-independent internalization and recycling. J Cell Mol Med. 2008;12:126–144. doi: 10.1111/j.1582-4934.2007.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 75.Valitutti S, Muller S, Salio M, Lanzavecchia A. Degradation of T cell receptor (TCR)-CD3-zeta complexes after antigenic stimulation. J Exp Med. 1997;185:1859–1864. doi: 10.1084/jem.185.10.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu H, Rhodes M, Wiest DL, Vignali DA. On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity. 2000;13:665–675. doi: 10.1016/s1074-7613(00)00066-2. [DOI] [PubMed] [Google Scholar]
- 77.Minami Y, Samelson LE, Klausner RD. Internalization and cycling of the T cell antigen receptor. Role of protein kinase C. J Biol Chem. 1987;262:13342–13347. [PubMed] [Google Scholar]
- 78.Krangel MS. Endocytosis and recycling of the T3-T cell receptor complex. The role of T3 phosphorylation. J Exp Med. 1987;165:1141–1159. doi: 10.1084/jem.165.4.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dietrich J, et al. Ligand-induced TCR down-regulation is not dependent on constitutive TCR cycling. J Immunol. 2002;168:5434–5440. doi: 10.4049/jimmunol.168.11.5434. [DOI] [PubMed] [Google Scholar]
- 80.Lauritsen JP, Christensen MD, Dietrich J, Kastrup J, Odum N, Geisler C. Two distinct pathways exist for down-regulation of the TCR. J Immunol. 1998;161:260–267. [PubMed] [Google Scholar]
- 81.Luton F, Buferne M, Davoust J, Schmitt-Verhulst AM, Boyer C. Evidence for protein tyrosine kinase involvement in ligand-induced TCR/CD3 internalization and surface redistribution. J Immunol. 1994;153:63–72. [PubMed] [Google Scholar]
- 82.D’Oro U, Vacchio MS, Weissman AM, Ashwell JD. Activation of the Lck tyrosine kinase targets cell surface T cell antigen receptors for lysosomal degradation. Immunity. 1997;7:619–628. doi: 10.1016/s1074-7613(00)80383-0. [DOI] [PubMed] [Google Scholar]
- 83.Crotzer VL, Mabardy AS, Weiss A, Brodsky FM. T cell receptor engagement leads to phosphorylation of clathrin heavy chain during receptor internalization. J Exp Med. 2004;199:981–991. doi: 10.1084/jem.20031105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barr VA, et al. T-cell antigen receptor-induced signaling complexes: internalization via a cholesterol-dependent endocytic pathway. Traffic. 2006;7:1143–1162. doi: 10.1111/j.1600-0854.2006.00464.x. [DOI] [PubMed] [Google Scholar]
- 85.Newman TM, Severs NJ. Coated vesicles are implicated in the post-fusion retrieval of the membrane of rat atrial secretory granules. Cell Tissue Res. 1992;268:463–469. doi: 10.1007/BF00319153. [DOI] [PubMed] [Google Scholar]
- 86.Polishchuk R, Di Pentima A, Lippincott-Schwartz J. Delivery of raft-associated, GPI-anchored proteins to the apical surface of polarized MDCK cells by a transcytotic pathway. Nat Cell Biol. 2004;6:297–307. doi: 10.1038/ncb1109. [DOI] [PubMed] [Google Scholar]
- 87.Dunn WA, Hubbard AL, Aronson NN., Jr Low temperature selectively inhibits fusion between pinocytic vesicles and lysosomes during heterophagy of 125I-asialofetuin by the perfused rat liver. J Biol Chem. 1980;255:5971–5978. [PubMed] [Google Scholar]
- 88.Punnonen EL, Ryhanen K, Marjomaki VS. At reduced temperature, endocytic membrane traffic is blocked in multivesicular carrier endosomes in rat cardiac myocytes. Eur J Cell Biol. 1998;75:344–352. doi: 10.1016/s0171-9335(98)80067-8. [DOI] [PubMed] [Google Scholar]
- 89.Toomre D, Manstein DJ. Lighting up the cell surface with evanescent wave microscopy. Trends Cell Biol. 2001;11:298–303. doi: 10.1016/s0962-8924(01)02027-x. [DOI] [PubMed] [Google Scholar]
- 90.Clements JL. Known and potential functions for the SLP-76 adapter protein in regulating T-cell activation and development. Immunol Rev. 2003;191:211–219. doi: 10.1034/j.1600-065x.2003.00002.x. [DOI] [PubMed] [Google Scholar]
- 91.Liu SK, Fang N, Koretzky GA, McGlade CJ. The hematopoietic-specific adaptor protein gads functions in T-cell signaling via interactions with the SLP-76 and LAT adaptors. Curr Biol. 1999;9:67–75. doi: 10.1016/s0960-9822(99)80017-7. [DOI] [PubMed] [Google Scholar]
- 92.Bonello G, et al. Dynamic recruitment of the adaptor protein LAT: LAT exists in two distinct intracellular pools and controls its own recruitment. J Cell Sci. 2004;117:1009–1016. doi: 10.1242/jcs.00968. [DOI] [PubMed] [Google Scholar]
- 93.Balagopalan L, et al. c-Cbl-mediated regulation of LAT-nucleated signaling complexes. Mol Cell Biol. 2007;27:8622–8636. doi: 10.1128/MCB.00467-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kalia M, Kumari S, Chadda R, Hill MM, Parton RG, Mayor S. Arf6-independent GPI-anchored protein-enriched early endosomal compartments fuse with sorting endosomes via a Rab5/phosphatidylinositol-3′-kinase-dependent machinery. Mol Biol Cell. 2006;17:3689–3704. doi: 10.1091/mbc.E05-10-0980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Damke H, Baba T, van der Bliek AM, Schmid SL. Clathrin-independent pinocytosis is induced in cells overexpressing a temperature-sensitive mutant of dynamin. J Cell Biol. 1995;131:69–80. doi: 10.1083/jcb.131.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sigismund S, et al. Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci USA. 2005;102:2760–2765. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 98.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 99.Fang S, Weissman AM. A field guide to ubiquitylation. Cell Mol Life Sci. 2004;61:1546–1561. doi: 10.1007/s00018-004-4129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- 102.Gay DL, Ramon H, Oliver PM. Cbl- and Nedd4-family ubiquitin ligases: balancing tolerance and immunity. Immunol Res. 2008;42:51–64. doi: 10.1007/s12026-008-8034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Heissmeyer V, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–265. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- 104.Wiedemann A, et al. T-cell activation is accompanied by an ubiquitination process occurring at the immunological synapse. Immunol Lett. 2005;98:57–61. doi: 10.1016/j.imlet.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 105.Randow F, Lehner PJ. Viral avoidance and exploitation of the ubiquitin system. Nat Cell Biol. 2009;11:527–534. doi: 10.1038/ncb0509-527. [DOI] [PubMed] [Google Scholar]
- 106.Nathan JA, Lehner PJ. The trafficking and regulation of membrane receptors by the RING-CH ubiquitin E3 ligases. Exp Cell Res. 2009;315:1593–1600. doi: 10.1016/j.yexcr.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 107.Huang F, Goh LK, Sorkin A. EGF receptor ubiquitination is not necessary for its internalization. Proc Natl Acad Sci USA. 2007;104:16904–16909. doi: 10.1073/pnas.0707416104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Walseng E, Bakke O, Roche PA. Major histo-compatibility complex class II-peptide complexes internalize using a clathrin- and dynamin-independent endocytosis pathway. J Biol Chem. 2008;283:14717–14727. doi: 10.1074/jbc.M801070200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Swaminathan G, Tsygankov AY. The Cbl family proteins: ring leaders in regulation of cell signaling. J Cell Physiol. 2006;209:21–43. doi: 10.1002/jcp.20694. [DOI] [PubMed] [Google Scholar]
- 110.Thien CB, Langdon WY. Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol. 2001;2:294–307. doi: 10.1038/35067100. [DOI] [PubMed] [Google Scholar]
- 111.Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–178. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- 112.Wang HY, et al. Cbl promotes ubiquitination of the T cell receptor zeta through an adaptor function of Zap-70. J Biol Chem. 2001;276:26004–26011. doi: 10.1074/jbc.M010738200. [DOI] [PubMed] [Google Scholar]
- 113.Myers MD, et al. Src-like adaptor protein regulates TCR expression on thymocytes by linking the ubiquitin ligase c-Cbl to the TCR complex. Nat Immunol. 2006;7:57–66. doi: 10.1038/ni1291. [DOI] [PubMed] [Google Scholar]
- 114.Duan L, Reddi AL, Ghosh A, Dimri M, Band H. The Cbl family and other ubiquitin ligases: destructive forces in control of antigen receptor signaling. Immunity. 2004;21:7–17. doi: 10.1016/j.immuni.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 115.Rao N, Dodge I, Band H. The Cbl family of ubiquitin ligases: critical negative regulators of tyrosine kinase signaling in the immune system. J Leukoc Biol. 2002;71:753–763. [PubMed] [Google Scholar]
- 116.Thien CB, Bowtell DD, Langdon WY. Perturbed regulation of ZAP-70 and sustained tyrosine phosphorylation of LAT and SLP-76 in c-Cbl-deficient thymocytes. J Immunol. 1999;162:7133–7139. [PubMed] [Google Scholar]
- 117.Murphy MA, et al. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl− deficient mice. Mol Cell Biol. 1998;18:4872–4882. doi: 10.1128/mcb.18.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Thien CB, Langdon WY. c-Cbl and Cbl-b ubiquitin ligases: substrate diversity and the negative regulation of signalling responses. Biochem J. 2005;391:153–166. doi: 10.1042/BJ20050892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rao N, Lupher ML, Jr, Ota S, Reedquist KA, Druker BJ, Band H. The linker phosphorylation site Tyr292 mediates the negative regulatory effect of Cbl on ZAP-70 in T cells. J Immunol. 2000;164:4616–4626. doi: 10.4049/jimmunol.164.9.4616. [DOI] [PubMed] [Google Scholar]
- 120.Dunn R, Hicke L. Multiple roles for Rsp5p-dependent ubiquitination at the internalization step of endocytosis. J Biol Chem. 2001;276:25974–25981. doi: 10.1074/jbc.M104113200. [DOI] [PubMed] [Google Scholar]
- 121.Govers R, ten Broeke T, van Kerkhof P, Schwartz AL, Strous GJ. Identification of a novel ubiquitin conjugation motif, required for ligand-induced internalization of the growth hormone receptor. EMBO J. 1999;18:28–36. doi: 10.1093/emboj/18.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang W, Irvin BJ, Trible RP, Abraham RT, Samelson LE. Functional analysis of LAT in TCR-mediated signaling pathways using a LAT-deficient Jurkat cell line. Int Immunol. 1999;11:943–950. doi: 10.1093/intimm/11.6.943. [DOI] [PubMed] [Google Scholar]
- 123.Zhang W, et al. Essential role of LAT in T cell development. Immunity. 1999;10:323–332. doi: 10.1016/s1074-7613(00)80032-1. [DOI] [PubMed] [Google Scholar]
- 124.Brignatz C, Restouin A, Bonello G, Olive D, Collette Y. Evidences for ubiquitination and intracellular trafficking of LAT, the linker of activated T cells. Biochim Biophys Acta. 2005;1746:108–115. doi: 10.1016/j.bbamcr.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 125.Pickart CM. Ubiquitin in chains. Trends Biochem Sci. 2000;25:544–548. doi: 10.1016/s0968-0004(00)01681-9. [DOI] [PubMed] [Google Scholar]
- 126.Brdicka T, et al. Non-T cell activation linker (NTAL): a transmembrane adaptor protein involved in immunoreceptor signaling. J Exp Med. 2002;196:1617–1626. doi: 10.1084/jem.20021405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mutch CM, et al. Activation-induced endocytosis of the raft-associated transmembrane adaptor protein LAB/NTAL in B lymphocytes: evidence for a role in internalization of the B cell receptor. Int Immunol. 2007;19:19–30. doi: 10.1093/intimm/dxl118. [DOI] [PubMed] [Google Scholar]
- 128.Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009;315:683–696. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 129.Sorkin A. Internalization of the epidermal growth factor receptor: role in signalling. Biochem Soc Trans. 2001;29:480–484. doi: 10.1042/bst0290480. [DOI] [PubMed] [Google Scholar]
- 130.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 131.Penna D, Muller S, Martinon F, Demotz S, Iwashima M, Valitutti S. Degradation of ZAP-70 following antigenic stimulation in human T lymphocytes: role of calpain proteolytic pathway. J Immunol. 1999;163:50–56. [PubMed] [Google Scholar]
- 132.Rao N, et al. Negative regulation of Lck by Cbl ubiquitin ligase. Proc Natl Acad Sci USA. 2002;99:3794–3799. doi: 10.1073/pnas.062055999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310:1191–1193. doi: 10.1126/science.1119238. [DOI] [PubMed] [Google Scholar]
- 134.Sadowski L, Pilecka I, Miaczynska M. Signaling from endosomes: location makes a difference. Exp Cell Res. 2009;315:1601–1609. doi: 10.1016/j.yexcr.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 135.Marie-Cardine A, Fischer S, Gorvel JP, Maridonneau-Parini I. Recruitment of activated p56lck on endosomes of CD2-triggered T cells, colocalization with ZAP-70. J Biol Chem. 1996;271:20734–20739. doi: 10.1074/jbc.271.34.20734. [DOI] [PubMed] [Google Scholar]