

Abstract
Thrombotic thrombocytopenia purpura (TTP) caused by a deficiency in ADAMTS-13 activity is considered to involve a subset of thrombotic microangiopathy (TMA). Although concept of TTP is included under the umbrella of TMA, discrimination of TTP from TMA is occasionally difficult in an autoimmune disorder. Herein, we report a case with TTP associated with systemic lupus erythematosus (SLE). In this case, it was difficult to discriminate TTP from TMA and the measurement of ADAMTS-13 activity was useful for obtaining an accurate diagnosis. SLE patients having thrombocytopenia in complication with anemia should be considered a monitoring of ADAMTS-13 activity even though the patients lacked symptoms of TTP related to the microvascular coagulation.
1. Introduction
Thrombotic thrombocytopenic purpura (TTP) is a life-threatening syndrome first described by Moschocowitz in 1924 [1]. TTP is clinically characterized by five typical syndromes: thrombocytopenia with platelet consumption, hemolytic anemia characterized by schistocytes, renal impairment, neurological abnormalities, and fever. The classic pentad described above is observed in as few as 40% of TTP patients [2], and the clinical course of the syndrome is usually rapid, therefore, an accurate diagnosis and immediate treatment including plasma exchange is necessary [3]. Von Wiilebrand factor-(vWF)-specific metalloprotease, a disintegrin-like and metalloprotease with thrombospondin type 1 motif-13 (ADAMTS-13) is known to play an important role in the pathogenesis of TTP [4–6]. A severe deficiency of ADAMTS-13 activity prevents the physiological processing of large vWF multimers which can lead to a strong platelet aggregation, and leads to the formation of microthrombosis in terminal arterioles and capillaries. Besides TTP, microthrombosis without a deficiency in ADAMTS-13 activity can also be associated with other diseases such as hemolytic uremic syndrome (HUS), various autoimmune diseases, cytotoxic drugs, human immunodeficiency virus (HIV), malignancies, disseminating intravascular coagulopathy (DIC), and pre-eclampsia [7]. Recently, these pathological conditions characterized by microthrombosis including TTP, have been classified as thrombotic microangiopathies (TMA). In autoimmune disorders, TMA occasionally occurs in systemic lupus erythematosus (SLE), antiphospholipid antibody syndrome (APS) [8, 9], and in many rare autoimmune disorders [7]. Rigorous classification of TMA is important to decide on the appropriate therapy. In particular, the combination of TTP and SLE has been reported to show a worse prognosis than SLE or idiopathic TTP alone [10], therefore, an accurate diagnosis and an immediate treatment are required.
Taken together, we recently encountered a case of SLE associated with TTP. This case indicated the diagnostic difficulty of TTP associated with autoimmune diseases such as SLE and the diagnostic efficacy of immediate monitoring of ADAMTS-13 activity for distinguishing TTP from other TMA.
2. Case Report
A 15-year-old female was admitted to our hospital because of thrombocytopenia with hemolytic anemia. She had noticed small purpura on her arms and her legs during 2 weeks before the admission. At the same time, she started having headaches, polyarthritis, and shortness of breath on exertion. These symptoms progressively deteriorated, thus prompting her hospital visit. On her physical examination, body temperature was 38.2°C. Her consciousness was alert. Small purpura were scattered over her entire arms and legs. A malar rash was also recognized on her face. Broad spectrum antibiotic (doripenem hydrate, 1.5 g/day) was involved in the initial therapy because severe bacterial infection was suspected in the situation with undetermined diagnosis on the admission. However, bacterial cultures from her blood, urine, sputum, and throat swab revealed no signs of any bacterial infections and we subsequently excluded bacterial infection from the diagnosis. Titers of antibodies against various kinds of virus such as cytomegalovirus, Epstein-Barr virus, and parvovirus B19 were also negative. Laboratory findings revealed thrombocytopenia with hemolytic anemia. A summary of the laboratory tests on admission is shown in Table 1. The serum hemoglobin level was low at 6.4 g/dL (normal range; 12.0–16.0 g/dL), her platelet count was also low at 4000/μL (normal range; 15.0–35.0 × 104/μL). The number of white blood cells was within normal range. Although the serum levels of fibrinogen degradation products (FDP) and D-dimers were elevated to 17.4 μg/dL (normal range; less than 5.0 μg/dL) and 8.78 μg/dL (normal range; less than 2.0 μg/dL), respectively, suggesting DIC, the other parameters related to DIC, such as the serum level of fibrinogen, prothrombin time (PT), and activated partial thromboplastin time (aPTT) were normal. The serum level of lactate dehydrogenase (LDH) was elevated to 915 IU/L (normal range; 119–219 IU/L), and that of total bilirubin level was also elevated at 2.9 mg/dL (normal range; 0.2–1.3 mg/dL) with dominant elevation of indirect bilirubin. The blood urea nitrogen (BUN) and creatinine levels were within the normal range and urinalysis did not show any abnormal findings. Although her serum haptoglobin levels were decreased at less than 10 mg/dL (normal range; >43–180) thus suggesting hemolytic anemia, both the direct and indirect Coombs tests were negative. The patient's bone marrow cells obtained by aspiration revealed normal differentiation, however, a peripheral blood smear test clearly showed schistocytes in the RBC. Her antinuclear antibody (ANA) test was positive, and she also had anti-DNA antibodies, anti-SS-A antibodies, anti-RNP antibodies, and anticardiolipin antibodies. Her serum complement levels were decreased (CH50; 21U/mL, C3; 81 mg/dL, and C4; 8 mg/dL, normal range; CH50 32–49 U/mL, C3 65–135 mg/dL, C4 13–35 mg/dL, resp.) and increased immune complex formation was observed (ICc1q 3.3 μg/mL, normal range; 0–3.0 μg/mL). She thus fulfilled the criteria for systemic lupus erythematosus (SLE) based on these laboratory findings (positive for ANA, anti-DNA antibody, anti-cardiolipin antibody, and thrombocytopenia) and her symptoms, such as malar rash and polyarthritis. At this moment, her SLE disease activity score (SLEDAI) was 13, thus indicating high disease activity. On admission, TMA, seronegative autoimmune hemolytic anemia with thrombocytopenia (EVANS syndrome), CAPS, and DIC were considered as possible candidates for her thrombocytopenia with hemolytic anemia. We provisionally diagnosed her illness as TMA with complicated autoimmune disorder because of a negative Coombs test, hemolytic anemia with schistocytes and thrombocytopenia, and immunosuppressive therapies, which consisted of intravenous cyclophosphamide administration (500 mg/body) and intravenous methylprednisolone (1000 mg/body, 3 days) administration followed by oral administration of prednisolone (50 mg/day), were initially introduced. However, the immunosuppressive therapy did not ameliorate her illness, therefore, we suspected TTP. As expected, additional laboratory tests showed that her serum ADAMTS-13 activity was obviously suppressed to an undetectable level. Although she did not have typical symptoms of TTP such as renal dysfunction and abnormalities of the central nervous system, we diagnosed her illness as TTP. Plasma exchange therapy was immediately added to her treatment. The therapy was continued for 5 consecutive days, then, her symptoms and the abnormal findings of laboratory tests completely disappeared. The clinical course of the patient is summarized and illustrated in Figure 1. The serum level of ADAMTS-13 activity was restored from an undetectable level to 60% of normal on day 17, and 168% on day 47, concomitant with the disease amelioration. She was discharged without any symptoms.
Table 1.
Laboratory test results on admission.
| Blood test | |||
| WBC | 5100/mm3 | CH50 | 21 U/mL |
| Neutro | 45.5% | C3 | 81 mg/dL |
| Lymph | 45.5% | C4 | 8 mg/dL |
| Mono | 6.0% | IgG | 1525 mg/dL |
| Eo | 0% | IgA | 190 mg/dL |
| Baso | 0% | IgM | 40 mg/dL |
| RBC | 219/mm3 | ANA | ×320 |
| Reticuro | 7.2% | Speckled | ×320 |
| Hb | 6.4 g/dL | Anti-DNA antibody (RIA) | 27 IU/mL |
| Hct | 18.0% | Anti-SS-A antibody | 1160 U/mL |
| Plt | 0.4/mm3 | Anti-SS-B antibody | (−) |
| PT-INR | 1.11 | Anti-Topo-1 antibody | (−) |
| ATPP | 29.1 sec (contro; 30.3 sec) | Anti-RNP antibody | 184 U/mL |
| FDP | 17.4 μg/dL | Anti-Sm antibody | (−) |
| D-D | 8.78 μg/dL | MPO-ANCA | (−) |
| ESR | 66 mm/h | Anti cardiolipin antibody (IgG) | 13 U/mL |
| TP | 6.9 g/dL | Anti-CLβ2GP1 antibody | (−) |
| ALB | 3.7 g/dL | LAC | (−) |
| BUN | 10 mg/dL | Anti platlet antibody | (−) |
| Cr | 0.54 mg/dL | PA-IgG | 2150 ng/107 cell |
| UA | 3.0 mg/dL | IC-C1q | 3.3 μg/mL |
| Na | 136 mmol/L | Direct Coombs test | (−) |
| K | 3.9 mmol/L | Indirect Coobms test | (−) |
| CL | 102 mmol/L | CMV IgG | (+) |
| T-Bil | 2.9 mg/dL | CMV IgM | (−) |
| D-Bil | 0.9 mg/dL | CMV Ag | (−) |
| AST | 61 IU/L | EBV EBNA | (−) |
| ALT | 35 IU/L | EBV-VCAIgG | (+) |
| LDH | 915 IU/L | EBV Ag | (+) |
| CK | 77 IU/L | EBV-VCAIgM | (−) |
| CRP | <0.3 mg/dL | HBs Ag | (−) |
| Urianalysis | |||
| Glu | (−) | ||
| Protein | (−) | ||
| RBC | (−) | ||
| WBC | (−) | ||
| abnormal cyst | (−) | ||
Figure 1.
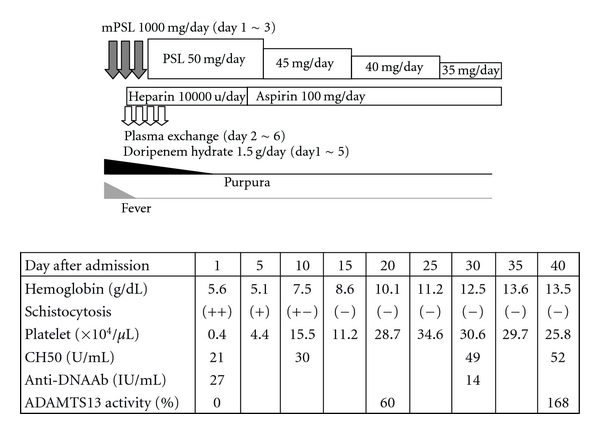
The clinical course in the present case. The patient was treated with intravenous administration with methylprednisolone (1000 mg/day for 3 days on the admission) followed by oral administration with prednisolone (50 mg/day). Daily plasma exchange and anticoagulant therapy was added on the second day. Plasma exchange was performed for 5 consecutive days and stopped at the sixth day of the admission. Subsequently, peripheral schistocytes diminished, her hemoglobin level and platelet count were improved, the complement level increased, the titer of anti-DNA antibody decreased, and the ADAMTS-13 activity increased. mPSL: methylprednisolone, and PSL: prednisolone.
3. Discussion
We herein reported a case of SLE associated with TTP. Because the patient lacked variable signs/symptoms of organ ischemia related to thrombosis in capillary arteries, it was difficult to determine the diagnosis of TTP at presentation. She was diagnosed with SLE based on her physical and laboratory findings (such as a malar rash, polyarthralgia, increased levels of several autoantibodies, and decreased serum complement concentrations). Decreased levels of ADAMTS-13 indicated that she had been suffering from TTP as well, and that her TTP contributed to the development of her thrombocytopenia and hemolytic anemia. TTP is classified as congenital and acquired type. Whereas congenital ADAMTS-13 deficiency is rare, loss of ADAMTS-13 activity is observed more frequently in acquired TTP. The ADAMTS-13 deficiency can be caused by an autoantibody and various other factors having an inhibitory effect on ADAMTS-13 activity [4].
Although the loss of ADAMTS-13 activity is the most important factor for diagnosis of TTP, it has been reported a few diseases condition resulted in ADAMTS-13 deficiency other than TTP. Nguyen and coworkers has reported that patients with severe sepsis revealed reduced ADAMTS-13 activity and the deficiency correlated with severity of thrombocytopenia and plasma level of interleukin-6 (IL-6) [11]. Indeed, mild- to-moderate ADAMTS-13 deficiency has been detected in systemic inflammation [12]. These reports suggest that inflammatory cytokines contribute to the deficiency of ADAMTS-13. Patients with SLE have been known to have an elevated serum level of cytokines due to aberrant activation of autoreactive lymphocytes. In the present case, some kind of cytokines may play an important role for deficiency of ADAMTS-13 in coordination with a production of inhibitory autoantibody against ADAMTS-13. Moreover, Lattuada et al. has reported that pregnant woman with HELLP syndrome (hemolytic anemia, elevated liver enzyme, and low platelets) had lower plasma levels of ADAMTS-13 activity [13]. Although some of clinical presentation TTP are shared by patients with severe sepsis and HELLP syndrome, the effective treatments differ in each diseases even though the deficiency of ADAMT-13 are observed in common with these diseases. No matter ADAMTS-13 deficiency observed, we should still take a possibility of other diseases condition into account in addition to TTP.
Although plasma exchange is the most effective treatment of TTP, it also can be used in other clinical manifestations related to SLE. Pugnoux et al. have reported that plasma exchange has beneficial effects for SLE-related diseases such as neurolupus, alveolar hemorrhage, and severe lupus nephritis [14]. Furthermore, Cerdas-Quesada have reported that refractory AIHA was successfully treated with plasma exchange [15]. The clinical manifestations of these diseases are shared with those of TTP. The rationale to use plasma exchange for the disease state is a removal of pathological autoantibodies, cytokines, and unknown harmful factors. In addition to TTP, the plasma exchange may be used for other complications of SLE as a supportive treatment until immunosuppressive therapy become effective. If immediate curative effects are required, plasma exchange may be an important strategy for certain SLE-rerated diseases even though TTP does not exist.
Similar to our case, autoimmune disorder occasionally makes it difficult to diagnose TTP, especially in the patients lacking typical clinical signs/symptoms of the disease. Moreover, it may be difficult to distinguish TTP from the other types of TMA. The possible differential diagnosis and the clinical differences among representative TMA including TTP are summarized in Table 2. HUS is characterized as renal diseases and usually associated with bacterial colitis (Escherichia coli O157 and Shigella-toxin-producing strain). HUS can be distinguished from TTP by diarrhea (D+HUS). Our patient had not shown any digestive symptoms; however, diarrhea negative (D-HUS) cases of HUS have been reported. EVANS syndrome, which is characterized by autoimmune hemolytic anemia (AIHA) with autoimmune thrombocytopenia, is also considered as one of important differential diagnosis from TTP. The detection of RBC bound immunoglobulin G and complement by the direct antiglobulin test (DAT; Coombs test) is useful in the diagnosis of AIHA [16]. The Coombs test in our patient was negative, although a small number of patients with AIHA have been reported to show a negative DAT [17], probably because of the lower affinity of IgG to RBC [18]. Therefore, the lack of these autoantibodies cannot conclusively exclude AIHA and/or EVANS syndrome. Catastrophic antiphospholipid syndrome (CAPS) which is an accelerated from APS [19] is a relatively new entity and thought to be a rare disease, although the exact thrombogenic mechanisms of CAPS remain unclear. The treatment of CAPS is not currently standardized and the prognosis is poor with death occurring in 50% of patients [20]. The diagnosis of TTP in patients with SLE could be difficult in SLE cases of positive for antiphospholipid antibody. In fact, laboratory findings in our patient revealed positivity for aCL, therefore, we also had to take CAPS into account as the possible diagnosis. However, the diagnosis of CAPS was not unlikely in the present case because of low titer of aCL. Regarding DIC, our patient was thought to have SLE with an elevation in the serum levels of FDP and D-dimer, suggesting DIC. Although the pathogenesis of DIC in SLE patients is still not fully understood, vasculitis has been considered to be involved in the pathogenesis of DIC in patients with SLE [21]. Immune complexes (ICs) activated by complements may injure the vascular endothelial cells, resulting in the release of tissue thromboplastin [21, 22]. Therefore, discrimination of TTP from a certain case with SLE may be difficult in such cases as the present case. In these situations, tests of the ADAMTS-13 activity seem to be the most useful tool for the diagnosis in TTP. Fujimura and Matsumoto categorized TTP using plasma levels of ADAMTS13 activity (<3%; severe, 3–25%; moderate, 25–50%; mild) for the purpose of distinguishing TTP from TMA. They also reported that the levels of ADAMTS-13 activity are lower in idiopathic TTP than in secondary TTP [23]. Generally, patients with autoimmune diseases tended to a have mild-to-moderate deficiency in ADAMTS-13 activity. In addition, Coppo et al. reported that patients with severe ADAMTS-13 deficiency in adult idiopathic TMA are characterized by various autoimmune manifestations, a lower platelet count, and mild renal involvement [24]. Although our patient was diagnosed as SLE, she had a severe deficiency in her ADAMTS-13 level (<0.3), and steroid treatment was not sufficiently effective for her symptoms. Her main symptoms were thought to have been caused by her TTP, and plasma exchange treatment were performed, and thereafter her clinical and laboratory data were dramatically improved.
Table 2.
Differential diagnosis of hemolytic anemia with thrombocytopenia. The diseases possibly resembling TTP are summarized in Table 1. TTP: thrombotic thrombocytopenic purpura, HUS: hemolytic-uremic syndrome, CAPS: catastrophic anti-phopholipid antibody syndrome, DIC: disseminating intravascular coagulopathy.
| TTP | HUS | EVANS syndrome | CAPS | DIC | |
|---|---|---|---|---|---|
| Tthrombocytopenia | (+) | (+) | (+) | (+) | (+) |
| Hemolytic anemia | (+) | (+) | (+) | (+−) | (+−) |
| Sschistocytes | (++) | (+) | (+−) | (+−) | (+−) |
| Clinical findings | Mental disorder | Diarrhea | Autoimmue reaction | Multiple organ infarction | Underlying disease |
| Renal dysfunction | Renal dysfunction | ||||
| Fever up | |||||
| Specific laboratory findings | Suppression of ADAMTS-13 activity | Positive for Shiga-toxin | Positive for Coombs test | Antiphospholipid antibodies | Elevation of FDP |
| Ttreatment | Plasma exchange | Hydration | Steroids | Anti-coagulant therapy | Anti-coagulant therapy |
| Steroids | Plasma exchange | Immunosuppression therapy (IVCY) | Steroids | Treatment for underlying disease | |
| 1mmunosuppression therapy(IVCY) | Iimmunosuppression therapy (IVCY) | ||||
| Anti-coagulant therapy | Plasma exchange |
Patient with congenital TTP, which results from a congenital deficiency in ADAMTS-13, may benefit from plasma infusion. Those with acquired TTP may also benefit from plasma exchange by the removal of the vWF-cleaving protease inhibitors, thereby restoring the ADAMTS-13 level. There is no proven benefit for plasma exchange in TMA (except TTP) associated with autoimmune-disorders. In such case, immunosuppressive therapy or B-cells depletion therapy by biologics may be a better choice than plasma exchange. Plasma exchange therapy has been widely used and is regarded as the for best treatment for TTP, however, the efficacy of plasma exchange has not been established in other type of TMA, and other therapeutic strategies may be a better choice, such as immunosuppressive agents, antibiotics (in cases of infection associated-TMA), and anticancer drugs (in cases of malignancy associated-TMA).
TTP has been sporadically reported in patients with SLE [25]. SLE and TTP share similar clinical symptom, therefore, complication of TTP in patients with SLE may be missed if careful and extensive studies are not performed. Plasma exchange is currently considered the most effective treatment for TTP, whereas immunosuppressive drugs (including steroids) are considered to be the most effective for SLE. TTP is a rare but life-threatening complication of SLE. Our present case suggested that prompt diagnosis of TTP by monitoring of the ADMATS-13 level if there is a suspicion of TTP, and the consequent intensive treatment including plasma exchange with adequate immunosuppressive therapy, are required to rescue patients from these life-threatening disorders.
Acknowledgments
This work was supported by the grants from The Institute for Environment and Gender-specific Medicine, Juntendo University Graduate School of Medicine. The authors are grateful to Dr. Yoshiaki Tokano for reviewing the paper.
References
- 1.Moschowitz E. Hyaline thrombosis of the terminal arterioles and capillaries: a hitherto undescribed disease. Proceedings of the New York Pathological Society. 1924;24:21–24. [Google Scholar]
- 2.Ridolfi RL, Bell WR. Thrombotic thrombocytopenic purpura. Report of 25 cases and review of the literature. Medicine. 1981;60(6):413–428. [PubMed] [Google Scholar]
- 3.Rock G, Shumak K, Buskard N, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. The New England Journal of Medicine. 1991;325(6):393–397. doi: 10.1056/NEJM199108083250604. [DOI] [PubMed] [Google Scholar]
- 4.Levy G, Nichols W, Lian E, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 5.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. Journal of Biological Chemistry. 2001;276(44):41059–41063. doi: 10.1074/jbc.C100515200. [DOI] [PubMed] [Google Scholar]
- 6.Soejima K, Mimura N, Hirashima M, et al. A novel human metalloprotease synthesized in the liver and secreted into the blood: possibly, the von Willebrand factor-cleaving protease? Journal of Biochemistry. 2001;130(4):475–480. doi: 10.1093/oxfordjournals.jbchem.a003009. [DOI] [PubMed] [Google Scholar]
- 7.Copelovitch L, Kaplan BS. The thrombotic microangiopathies. Pediatric Nephrology. 2008;23(10):1761–1767. doi: 10.1007/s00467-007-0616-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jorfen M, Callejas J, Formiga F, Cervera R, Font J, Ingelmo M. Fulminant thrombotic thrombocytopenic purpura in systemic lupus erythematosus. Scandinavian Journal of Rheumatology. 1998;27:76–77. doi: 10.1080/030097498441218. [DOI] [PubMed] [Google Scholar]
- 9.Musa M, Nounou R, Sahovic E, Seth P, Qadi A, Aljurf M. Fulminant thrombotic thrombocytopenic purpura in two patients with systemic lupus erythematosus and phospholipid autoantibodies. European Journal of Haematology. 2000;64(6):433–435. doi: 10.1034/j.1600-0609.2000.9l125.x. [DOI] [PubMed] [Google Scholar]
- 10.Zheng X, Kaufman R, Goodnough L, Sadler J. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103(11):4043–4049. doi: 10.1182/blood-2003-11-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen TC, Liu A, Liu L, et al. Acquired ADAMTS-13 deficiency in pediatric patients with severe sepsis. Haematologica. 2007;92(1):121–124. doi: 10.3324/haematol.10262. [DOI] [PubMed] [Google Scholar]
- 12.Mannucci PM, Vanoli M, Forza I, Canciani MT, Scorza R. Von Willebrand factor cleaving protease (ADAMTS-13) in 123 patients with connective tissue diseases (systemic lupus erythematosus and systemic sclerosis) Haematologica. 2003;88(8):914–918. [PubMed] [Google Scholar]
- 13.Lattuada A, Rossi E, Calzarossa C, Candolfi R, Mannucci PM. Mild to moderate reduction of a von Willebrand factor cleaving protease (ADAMTS-13) in pregnant women with HELLP microangiopathic syndrome. Haematologica. 2003;88(9):1029–1034. [PubMed] [Google Scholar]
- 14.Pagnoux C, Korach JM, Guillevin L. Indications for plasma exchange in systemic lupus erythematosus in 2005. Lupus. 2005;14(11):871–877. doi: 10.1191/0961203305lu2174rr. [DOI] [PubMed] [Google Scholar]
- 15.Cerdas-Quesada C. A life-threatening case of autoimmune hemolytic anemia successfully treated by plasma-exchange. Transfusion and Apheresis Science. 2010;42(3):235–237. doi: 10.1016/j.transci.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 16.Engelfriet C, Overbeeke M, von dem Borne A. Autoimmune hemolytic anemia. Seminars in Hematology. 1992;29(1):3–12. [PubMed] [Google Scholar]
- 17.Biagi E, Assali G, Rossi F, Jankovic M, Nicolini B, Balduzzi A. A persistent severe autoimmune hemolytic anemia despite apparent direct antiglobulin test negativization. Haematologica. 1999;84(11):1043–1045. [PubMed] [Google Scholar]
- 18.Kamesaki T, Oyamada T, Omine M, Ozawa K, Kajii E. Cut-off value of red-blood-cell-bound IgG for the diagnosis of Coombs-negative autoimmune hemolytic anemia. American Journal of Hematology. 2009;84(2):98–101. doi: 10.1002/ajh.21336. [DOI] [PubMed] [Google Scholar]
- 19.Asherson R. The catastrophic antiphospholipid syndrome. Journal of Rheumatology. 1992;19(4):508–512. [PubMed] [Google Scholar]
- 20.Bucciarelli S, Espinosa G, Cervera R. The CAPS Registry: morbidity and mortality of the catastrophic antiphspholipd syndrome. Lupus. 2009;18(10):905–912. doi: 10.1177/0961203309106833. [DOI] [PubMed] [Google Scholar]
- 21.Kerr L, Spiera H, Aledort L. Acute disseminated intravascular coagulation as a complication of systemic lupus erythematosus. New York State Journal of Medicine. 1987;87(3):181–183. [PubMed] [Google Scholar]
- 22.Hirohata S, Miyamoto T. Elevated levels of interleukin-6 in cerebrospinal fluid from patients with systemic lupus erythematosus and central nervous system involvement. Arthritis and Rheumatism. 1990;33(5):644–649. doi: 10.1002/art.1780330506. [DOI] [PubMed] [Google Scholar]
- 23.Fujimura Y, Matsumoto M. Registry of 919 patients with thrombotic microangiopathies across Japan: database of Nara Medical University during 1998–2008. Internal Medicine. 2010;49(1):7–15. doi: 10.2169/internalmedicine.49.2706. [DOI] [PubMed] [Google Scholar]
- 24.Coppo P, Schwarzinger M, Buffet M, et al. Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: the French TMA reference center experience. PLoS One. 2010;5(4) doi: 10.1371/journal.pone.0010208. Article ID e10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen MH, Chen MH, Chen WS, et al. Thrombotic microangiopathy in systemic lupus erythematosus: a cohort study in North Taiwan. Rheumatology. 2011;50:768–775. doi: 10.1093/rheumatology/keq311. [DOI] [PubMed] [Google Scholar]