

Abstract
Classical antifolates (4-7) with a tricyclic benzo[4,5]thieno[2,3-d]pyrimidine scaffold and a flexible and rigid benzoylglutamate were synthesized as dual thymidylate synthase (TS) and dihydrofolate reductase (DHFR) inhibitors. Oxidative aromatization of ethyl 2-amino-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (±)-9 to ethyl 2-amino-4-methyl-1-benzothiophene-3-carboxylate 10 with 10% Pd/C was a key synthetic step. Compounds with 2-CH3 substituents inhibited human (h) TS (IC50 = 0.26-0.8 μM), but not hDHFR. Substitution of the 2-CH3 with a 2-NH2 increases hTS inhibition by more than 10-fold and also affords excellent hDHFR inhibition (IC50 = 0.09-0.1 μM). This study shows that the tricyclic benzo[4,5]thieno[2,3-d]pyrimidine scaffold is highly conducive to single hTS or dual hTS-hDHFR inhibition depending on the 2-position substituents. The X-ray crystal structures of 6 and 7 with hDHFR reveal, for the first time, that tricyclics 6 and 7 bind with the benzo[4,5]thieno[2,3-d]pyrimidine ring in the folate binding mode with the thieno S mimicking the 4-amino of methotrexate.
Keywords: DHFR, TS, dual, inhibitor
1. Introduction
Because of its critical importance in the biosynthesis of purine and pyrimidine nucleic acids, folate metabolism is an attractive target for chemotherapy. Antifolates that target folate metabolism have found clinical utility as antitumor, antimicrobial, and antiprotozoal agents.1-5 Among the folate dependent enzymes, thymidylate synthase (TS) and dihydrofolate reductase (DHFR) have been of particular interest. TS is a key enzyme in the de novo synthesis of 2’-deoxythymidine-5’-monophosphate (dTMP) from 2’-deoxyuridine-5’-monophosphate (dUMP).6,7 The reaction requires 5,10-methylenetetrahydrofolate (5,10-CH2THF) as a cofactor for one carbon unit transfer and represents the only de novo pathway for intracellular dTMP synthesis. DHFR catalyzes the reduction of dihydrofolate to tetrahydrofolate, and is indirectly responsible for dTMP synthesis by maintaining the reduced folate pool.8
Raltitrexed (RTX)9, pemetrexed (PMX)10 and methotrexate (MTX)11 (Figure 1) are examples of TS and/or DHFR inhibitors used in the clinic. RTX, approved in several European countries, Australia, Canada, and Japan for the treatment of advanced colorectal cancer, is a TS inhibitor that undergoes rapid polyglutamylation by the enzyme folylpolyglutamate synthetase12,13 (FPGS). PMX, in combination with cisplatin, was approved for the treatment of malignant pleural mesothelioma and also for non-small cell lung cancer. With TS inhibition as the primary mechanism of action, PMX was reported to inhibit several other folate-dependent enzymes including DHFR, glycinamide ribonucleotide formyltransferase (GARFTase), and aminoimidazole carboxamideribonucleotide formyltransferase (AICARFTase).10 Similar to RTX, polyglutamyltion by FPGS is essential for the cytotoxicity of PMX.
Figure 1.
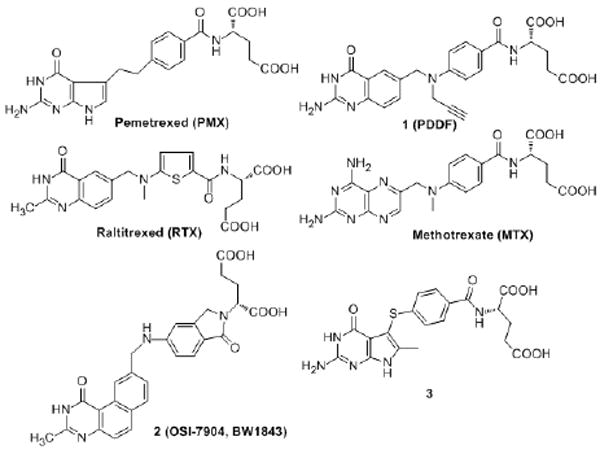
Antifolates
Classical antifolates, such as RTX and PMX, that have an N-benzoyl-L-glutamic acid side chain usually function as substrates for FPGS, which leads to high intracellular concentrations of these antitumor agents and increases TS inhibitory activity for some antifolates (RTX, 60-fold and PMX 130-fold) compared to their monoglutamate forms.9,10,14,15 Although polyglutamylation of certain antifolates (such as RTX and PMX) is necessary for their cytotoxic activity, it has also been implicated in toxicity to host cells, because of the longer cellular retention time of such polyanionic poly-glutamate metabolites.16 In addition, reduced expression of FPGS in tumor cells can lead to resistance to FPGS dependent classical antifolates such as PMX.17-19
To circumvent the potential tumor resistance problems associated with FPGS, classical antifolates should have high enzyme inhibitory potency as their monoglutamate forms and not require polyglutamylation by FPGS to exert their antitumor activity.20,21
It has been our long-standing goal not only to design potent TS and DHFR inhibitors but also to design and synthesize single agents that have potent dual TS and DHFR inhibitory activity simultaneously. Such dual inhibitors could act at two different sites (TS and DHFR) and might be capable of providing “combination chemotherapy” in single agents without the pharmacokinetic, overlapping toxicities and other disadvantages of two separate agents.20 This strategy may also lead to an improved toxicity profile.
Typically, antifolates that contain a 2-methyl-4-oxo substitution in the pyrimidine ring (such as RTX) are TS inhibitors, while 2-amino-4-oxo substitution in the pyrimidine ring (such as PMX) may provide affinity for both TS and DHFR. 2,4-Diamino substitutions on the pyrimidine ring of antifolates is usually associated with DHFR inhibitory activity.
In an attempt to prevent or minimize the potential problems associated with FPGS, including tumor resistance, and to develop dual TS and DHFR inhibitors, Gangjee et al.20 reported the synthesis of a classical antifolate N-{4-[(2-amino-6-methyl-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)thio]benzoyl}-L-glutamic acid, 3 (Figure 1), as a potent inhibitor of isolated hTS (IC50 = 42 nM) with a reasonable inhibition of human recombinant DHFR (IC50 = 2.2 μM) in its monoglutamate form thus providing dual inhibitory activity of TS and DHFR. Compound 3 was equipotent with 1 (Figure 1), a potent TS inhibitor, against hTS and was more potent than the clinically used RTX and PMX against isolated hTS in their monoglutamate forms. Molecular modeling (SYBYL 6.91)22 suggested that the 6-methyl group in compound 3 makes important hydrophobic contacts with Trp109 in hTS and also serves to lock the 5-position side chain into favorable, low energy conformations. Both these factors probably contribute to the high inhibitory activity of 3 in its monoglutamate form against hTS.
A potential advantage of compound 3 over RTX and PMX is that it is not a substrate for hFPGS, from CCRF-CEM cells, at concentrations up to 1045 μM.20 The lack of hFPGS substrate activity of 3 was attributed, in part, to the presence of the 6-methyl group on the pyrrolo[2,3-d]pyrimidine of 3. The 6-methyl group of 3 probably creates steric hindrance in its binding to the active site of hFPGS. Alternatively, the 6-methyl group of 3 may force the 5-position side chain into a conformation that is not conducive for binding to hFPGS. The fact that PMX, a pyrrolo[2,3-d]pyrimidine, much like 3, lacks a 6-methyl group and is a substrate for FPGS lends credence to the involvement of the 6-methyl moiety in preventing FPGS substrate activity in 3.
Tricyclic 2 (Figure 1) is a classical TS inhibitor with Ki = 0.09 nM.23,24 Although this compound is an excellent substrate for FPGS, it is subject to the addition of only one additional glutamic acid. Moreover, the high potency of 2 does not rely on polyglutamation as the monoglutamate form is equi-potent with the diglutamate form.25 Compound 2 is a noncompetitive TS inhibitor and its activity is not affected by the concentration of 5,10-CH2THF.23,24 In addition, it has been demonstrated that overexpression of the multidrug resistance proteins, MRP1 and MRP2, can confer tumor resistance to short term (4 h), but not long term (72 h), exposure of 2.26
In the course of our structure-based drug design program it was of interest to synthesize classical antifolates 4-7 with a tricyclic benzo[4,5]thieno[2,3-d]pyrimidine scaffold (Figure 2), as structural hybrides of 2 and 3. Compounds 4 and 5, similar to 2, have a 2-methyl-4-oxo pyrimidine ring which is generally associated with TS inhibition. In contrast, 6 and 7 have a 2-amino-4-oxo substituent, which could afford dual TS and DHFR inhibition, as observed for 3 and PMX. The 2-substitutions on 4-7 would access the importance of hydrogen-bonding at this position (6, 7) versus hydrophobic binding (4, 5) to biological activity. The size of a sulfur atom in 4-7 is larger than a nitrogen atom and smaller than two carbon atoms, thus the thiophene B-ring in benzo[4,5]thieno[2,3-d]pyrimidines 4-7 mimics both the smaller pyrrolo B-ring in 3 and the larger quinazoline B-ring in 2 (Figure 3).
Figure 2.
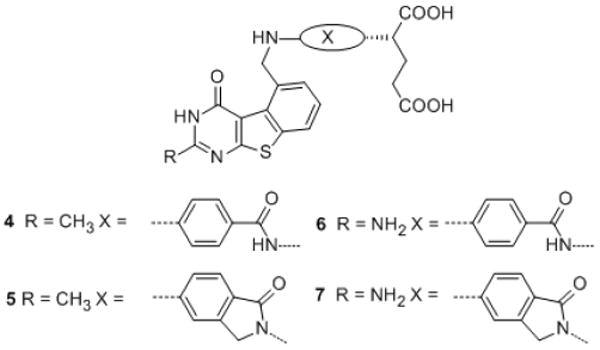
Target classical antifolates with the tricyclic benzo[4,5]thieno[2,3-d]pyrimidine scaffold
Figure 3.
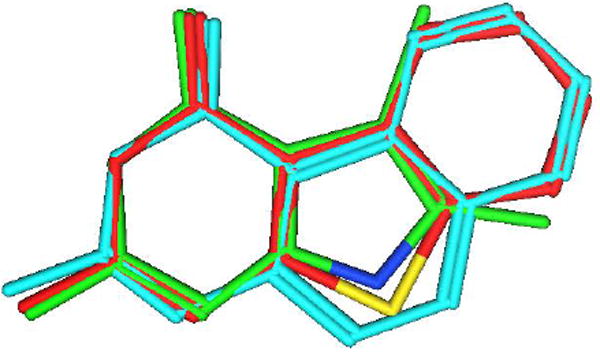
Superimposition of benzo[4,5]thieno[2,3-d]pyrimidine (red), pyrolo[2,3-d]pyrimidine (green) and benzo[f]quinazoline (cyan).
Similar to the C-ring in 2 and the 6-methyl group in 3, the benzo C-ring in 4-7 makes hydrophobic contacts with Trp109 in hTS and restricts the side chain to a conformation conducive for potent TS activity but perhaps not for FPGS substrate activity.
To explore the effects of side chain flexibility on biological activity, 4 and 6 have the same benzoylglutamate side chain as 3, while 5 and 7 have a more conformationally restricted 2-isoindolinylglutamate side chain like 2. Unlike other classical TS inhibitors, the glutamate side chain in 2 is part of an isoindolinone system, which restricts the side chain conformation. The crystal structure of ternary complex TS-dUMP-2 (PDB: 1SYN) 27 revealed that the binding of 2 and the nucleotide induced a closed conformation of the TS protein, similar to other antifolates. Surprisingly, however, the binding surface of 2 includes a hydrophobic patch from Val 77 that is normally buried.27,28,29,30
As shown in Figure 4, molecular modeling using MOE 2008.1031 revealed that when the central ring in 2 is truncated to a 5-member ring in the benzo[4,5]thieno[2,3-d]pyrimidine 5, and the substitution is moved from the 6-position to the 5-position, the resulting compound could bind to TS in a manner similar to 2.
Figure 4.
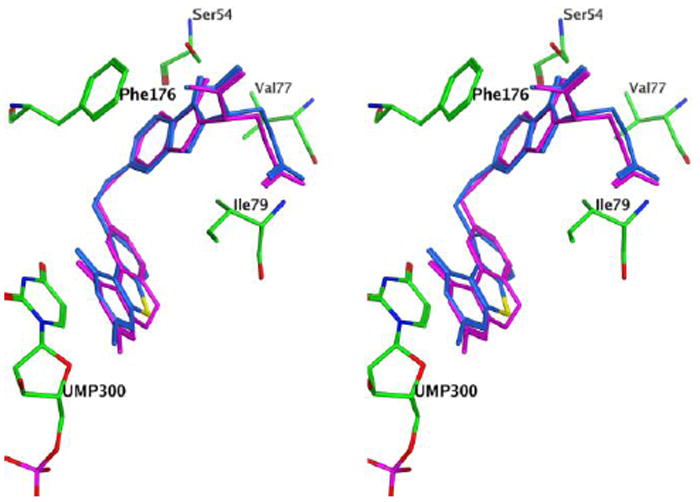
Stereoview of compound 5 (blue) superimposed on 2 (purple) in ecTS (green). Figure prepared with MOE 2008.10.31
2. Chemistry
It was envisioned that target compounds 4-7 would be synthesized via the coupling between the benzo[4,5]thieno[2,3-d]pyrimidine scaffold and the glutamate side chain.
The synthesis of benzo[4,5]thieno[2,3-d]pyrimidines started from commercially available α-methyl cyclohexanone (±)-8 to the thiophene intermediate (±)-9 (Scheme 1) via a Gewald reaction32 in 81% yield. The reaction was attempted in various solvents and bases with the optimized results obtained with ethanol and morpholine. Cyclization of 9 via the partially aromatized tricyclic intermediate should afford benzo[4,5]thieno[2,3-d]pyrimidines. To explore this strategy, (±)-10 (Scheme 1) was synthesized via the condensation of 9 and chloroformamidine hydrochloride.33 Aromatization of (±)-10 was expected to afford 12 (Scheme 1).
Scheme 1.
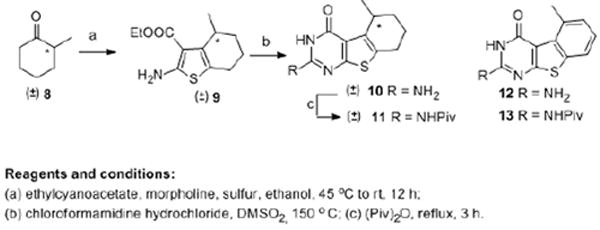
Synthesis of tricyclic thieno[2,3-d]pyrimidines 10 and 11
Rosowsky et al.34 reported aromatization of tetracyclic thieno-[2,3-d]pyrimidine via SeO2 in acetic acid at reflux. Attempts at this reaction for the conversion of 10 to 12 were unsuccessful. Gangjee et al.35 reported the oxidation of dihydropyrrolo[2,3-d]pyrimidines to their aromatic congeners via MnO2 oxidation. However, MnO2 oxidation for the aromatization of 10 was also unsuccessful. DDQ is reported36 to serve as a dehydrogenation agent to effect aromatization. Reaction of 10 with DDQ at reflux in dioxane for up to 24 h afforded no new product (TLC). Other solvents with different boiling points were also attempted at reflux and microwave conditions. Trace amounts of a new product was observed under certain conditions, however, the yields were poor and precluded characterization.
The poor solubility of (±)-10 in organic solvents could, in part, be responsible for the failure of aromatization. Thus, the 2-amino group in (±)-10 was protected with a pivaloyl group at reflux with the anhydride (Piv)2O (Scheme 1) to give 11, which was then subjected to DDQ oxidation under different reaction conditions. Unfortunately, no desired product was obtained.
The failure of the previous strategy prompted us to explore an alternate method, where the bicylic scaffold was aromatized first (Scheme 2). Bicyclic intermediate (±)-9 showed good solubility in most organic solvents. With toluene as the solvent and MnO2, SeO2 or DDQ as the oxidant, under bench-top conditions or microwave irradiation no desired product was obtained. A literature search revealed Pd/C oxidation.37-38 This allowed the conversion of (±)-9 to the fully aromatized 14. The solvent and time of the reaction were optimized for the aromatization with the optimal conditions being mesitylene as solvent at reflux for 48 h. Compared with (±)-9, the 1H NMR of 14 showed the disappearance of protons at δ 1.54-3.17 ppm and the appearance of three aromatic protons at δ 6.98-7.43 ppm, which confirmed aromatization. In addition, the appearance of benzylic protons at δ 2.38 as singlet also confirmed aromatization. With 14 in hand, cyclization was carried out to afford the tricyclic scaffold. The substitution at the 2-position of the benzo[4,5]thieno[2,3-d]pyrimidine are predicated by the reactant. Cyclization of 14 (Scheme 2) with chloroformamidine hydrochloride afforded the 2-amino-4-oxo benzo[4,5]thieno[2,3-d]pyrimidine 12 in 60% yield. Pivaloylation of 12 afforded 13. The reaction of 14 in acetonitrile with gaseous hydrogen chloride afforded the 2-methyl-4-oxo product 15 in 57% yield. The 2-methyl and 5-methyl groups of 15 occur in the 1H NMR at δ 2.35 and 2.95 respectively akin to similar dimethyl substituted quinazolines and benzoquinazolines.39-42 Free radical bromination of 13 and 15 with N-bromosuccinimide and a catalytic amount of benzoyl peroxide afforded intermediates 16 and 17 respectively.39-41 By limiting the amount of the NBS to just over one equivalent, the 5-methyl moiety of 15 was selectively brominated to afford 17, similar to that reported for quinazolines and benzoquinazolines.39-42 The 2-methyl moiety of 17 occurs in the 1H NMR at δ 2.69 and the 5-methylene protons occur at δ 5.84 similar to the corresponding quinazolines and benzoquinazolines.39-42
Scheme 2.
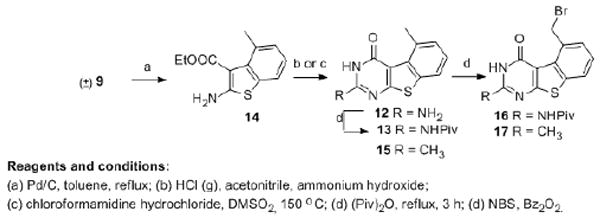
Synthesis of tricyclic thieno[2,3-d]pyrimidines 16 and 17
The benzoylglutamate side chain for 4 and 6 is commercially available, however the 2-isoindolinylglutamate side chain for 5 and 7 was synthesized via a literature method (Scheme 3).39 Esterification of 18 with SOCl2 in methanol afforded 19 in 91% yield. Radical bromination of 19 with N-bromosuccinimide and a catalytic amount of benzoyl peroxide afforded 20 in 48% yield. The 1H NMR of 20 showed the disappearance of protons at δ 2.69 ppm (CH3) and appearance of protons at δ 4.86 ppm (CH2Br). Treatment of 20 with excess diethyl L-glutamate hydrochloride and K2CO3 in DMF at room temperature afforded isoindoline 21 as an orange oil in 56% yield. The 1H NMR of 21 showed the appearance of protons at δ 4.51-4.83 ppm (isoindoline CH2) and δ 5.09-5.14 ppm (Gluα-CH). Reduction of the nitro group in 21 afforded the amine 22 in 92.5% yield.
Scheme 3.
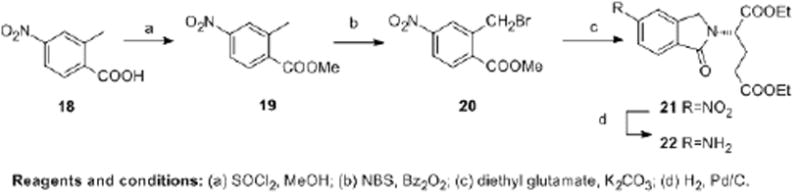
Synthesis of side-chain 22
As shown in Scheme 4, N-alkylation43 of 16 and 17 followed by hydrolysis of the ethyl ester (and the removal of pivaloyl protecting group in 6-7) with 1 N NaOH and subsequent acid workup afforded target compounds 4-7. The presence of glutamate in the side chain was confirmed from 1H NMR. The expected NH at δ 6.74-7.40 ppm, exchanged with D2O, and the benzylic protons occurred as a doublet at δ 5.13-5.24 ppm, which converts to a singlet upon D2O exchange, indicated the success of the coupling reaction. High resolution MS (HRMS) and the presence of the requisite protons of the side chain via NMR confirmed the structure of 4-7.
Scheme 4.
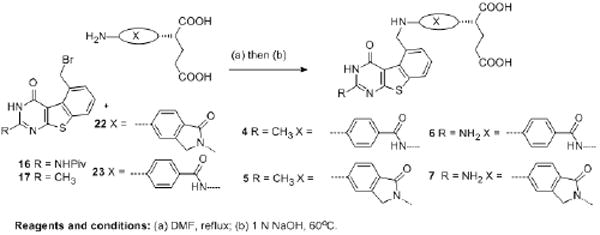
Synthesis of classical analogues 4-7
3. Biological Evaluation and Discussion
Compounds 4-7 were evaluated as inhibitors of TS and DHFR (Table 1) from three different sources; human, E. coli and T. gondii. Compound 1 (PDDF) was used as the standard compound for TS inhibition and MTX was used as the standard compound for DHFR inhibition. For comparison, the activities of PMX and lead compounds 2 and 3 are also provided. The classical benzo[4,5]thieno[2,3-d]pyrimidines 6-7 showed potent hTS inhibitory activity comparable to that of 1, 2 and 3 which supports the hypothesis that a 6-5-6 tricyclic system with a 5-substitution can inhibit TS in a similar fashion as the 6-6-6 tricyclic system with a 6-substitution. Compound 7 is the most potent hTS inhibitor in the series and is 2-fold more potent than compound 1 and equipotent with compound 2. Against isolated hTS, all four compounds 4-7 showed 50 to 100-fold greater activity than PMX, which relies on FPGS for its potent hTS inhibitory activity. Thus compounds 4-7, and in particular 6 and 7, have the potential to overcome the tumor resistance problems of PMX associated with lower levels of FPGS. With identical side chain substitution, the 2-amino analogs are more potent than the 2-methyl compounds. This indicates the importance of hydrogen bonding ability at the 2-position. Side chain flexibility could also play a role in enzyme inhibition. When the 2-substitution is methyl, the more rigid side chain analog 5 is 3-fold less potent than the more flexible 4 against hTS. However, this trend is reversed when the 2-substitution is an amino group, where the more rigid compound 7 is twice as potent as the flexible 6. These data suggest that the conformation of side chain glutamate along with the 2-substitution pattern dictates enzyme binding.
Table 1.
Inhibitory Concentrations (IC50 in μM) Against TS and DHFR.a
| compound | TS(μM)
|
DHFR(μM)
|
||||
|---|---|---|---|---|---|---|
| humanb | E. colib | T. gondiic | humand | E. colie | T. gondiic | |
| 1f | 0.064 | 0.048 | 0.072 | 11 | 11 | 0.26 |
| 2g | 0.032 | 0.023 | 0.027 | >20(16) | 20 | 0.71 |
| 3h | 0.053 | 0.21 | 0.09 | 2.1 | 22 | 0.23 |
| 4 | 0.26 | 0.82 | 1.7 | >20(35) | 15 | 1.4 |
| 5 | 0.8 | 0.85 | 3.7 | >20(24) | >20(38) | 1.8 |
| 6 | 0.068 | 0.017 | 0.14 | 0.09 | 0.4 | 0.02 |
| 7 | 0.034 | 0.05 | 0.17 | 0.1 | 0.4 | 0.01 |
| PMXh | 9.5 | 76 | 2.8 | 6.6 | 2300 | 0.43 |
| MTX | >18(29) | >18(14) | >18(36) | 0.022 | 0.0044 | 0.022 |
The percent inhibition was determined at a minimum of four inhibitor concentrations within 20% of the 50% point. The standard deviations for determination of 50% points were within ± 10% of the value given.
Kindly provided by Dr. Frank Maley, New York State Department of Health.
Kindly provided by Dr. Karen Anderson, Yale Univerisy, New Haven CT.
Kindly provided by Dr. J. H. Freisheim, Medical College of Ohio, Toledo, OH.
Kindly provided by Dr. R. L. Blakley, St. Jude Children’s hospital, Memphis TN.
Kindly provided by Dr. M. G. Nair, University of South Alabama.
Kindly provided by Dr. J. J. McGuire, Roswell Park Cancer Institute, Buffalo, NY.
Kindly provided by Dr. Chuan Shih, Eli Lilly and Co.
Compounds 4 and 5 are inactive against DHFR from different sources, while 6 and 7 are about equally potent against hDHFR and are about 5-times less potent than MTX. Compounds 6 and 7 also have different selectivity profile than MTX. MTX is 3-times more selective for E. coli (ec) DHFR than hDHFR, while 6 and 7 are more selective for hDHFR and the selectivity index is about 4. Inhibition of DHFR by 6 and 7 confirms our hypothesis that the replacement of the 2-methyl group by 2-amino group in benzo[4,5]thieno[2,3-d]pyrimidines can provide dual TS and DHFR inhibition. The inactivity of 4 and 5 against hDHFR is probably due to the absence of a salt bridge with Glu30 of hDHFR at the N1 and 2NH2. While compounds 6 and 7 can bind just like folate (PDB: 1U72) in which the 2-amino-4-oxo group binds to the enzyme with hydrogen bonding and the heterocycle and the benzoyl moieties bind to Phe31, Phe34 and Ile 60. The α-carboxylic acid of the glutamate makes ionic contact with Arg70. A second mode of binding would involve a 180° rotation about the C-2, NH2 bond (Figure 5), whereby the sulfur of the thiophene ring is now superimposed on the 4-oxo group of folate, with all other interactions being the same. It is important to note that binding of 6 and 7 in the flip mode (Fig. 5) also allows the sulfur of the thiophene ring to mimic the 4-amino of MTX. To determine which of these two modes of binding the molecule adopts to bind hDHFR, 6 and 7 were each cocrystalized with isolated hDHFR.
Figure 5.
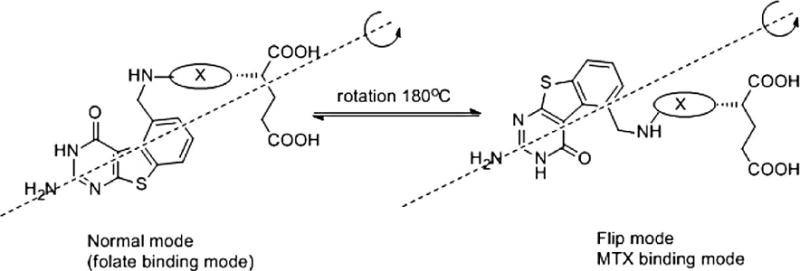
Proposed binding mode with DHFR
4. X--ray Crystal Structures
The X-ray crystal structures of the ternary complexes of tricyclic compounds 6 and 7 and NADPH with hDHFR were determined and refined to 1.35Ǻ resolution. These data show, for the first time, that the tricyclic benzo[4,5]thieno[2,3-d]pyrimidine antifolates 6 and 7 bind in a folate orientation such that the 2-NH2 and N3 interact with Glu30 and the thienosulfur occupies the N8 position observed in the binding of folic acid (Figures 6 and 7). These results are similar to those observed for the bicyclic structure of the N-{4-[2-amino-6-methyl-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-5-yl)thio]benzoyl}-L-glutamic acid44 in complex with active site mutants of hDHFR in which the thieno[2,3-d]pyrimidine ring sulfur in both complexes makes intermolecular contacts with the carbonyl of Ile7 (3.5 Ǻ), Val115 (3.7 Ǻ), the hydroxyl of Tyr121 (3.8 Ǻ) and the carbonyl of the nicotinamide ring (3.6Ǻ). Thus the steric addition of the benzo fused ring does not prevent the thieno S from making the same contacts with hDHFR as that of the 4-NH2 of MTX.45
Figure 6.
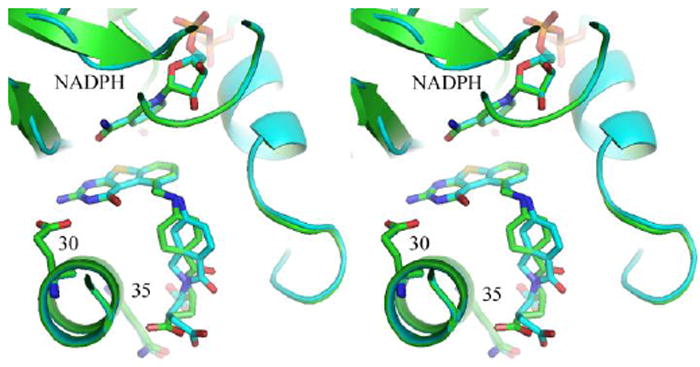
Stereoview of the X-ray crystal structures of 6 (green) and 7 (cyan) superimposed in the active site of hDHFR and NADPH ternary complex.
Figure 7.
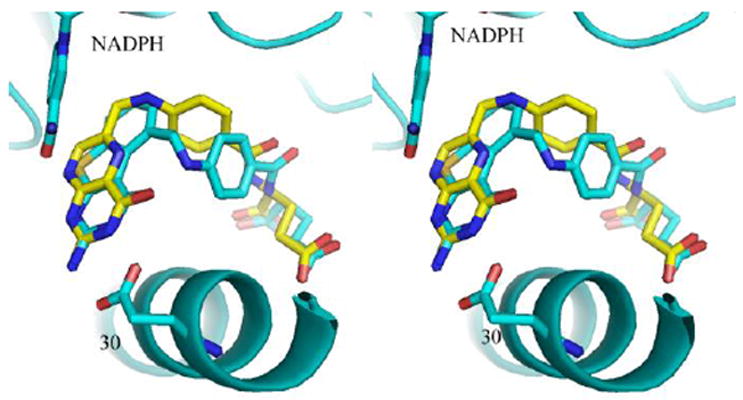
Stereo comparison of the structure of 6 (cyan) with that of folate (yellow) in hDHFR. Figure prepared with PyMol.46
Although molecular modeling studies suggest that both a flipped and folate orientation were possible for these structures (Figure 5), the observed crystal structures reveal, for the first time, only binding in the normal mode of folate orientation, as illustrated in Figure 7 that compares the binding of 6 with folate. The binding orientation of the 6-5-6-ring system is the same in 6 and 7 while the added ring in the isoindoline ring system of 7 shows, also for the first time, that the fused isoindolinyl ring system side chain of 7 is flipped compared to that of the normal p-aminobenzoylglutamate of 6 as illustrated in Figure 6. Despite this change, the functional groups of the L-glutamate side chain still occupy similar positions in the binding site. To our knowledge this is the first time that the fused conformationally restricted isoindolinyl ring system side chain has been compared side-by-side in a X-ray structure with the benzoyl ring in hDHFR.
Comparison of binding of 2 from the structure of ecTS 28 modeled into the binding site of 7 in hDHFR (from the X-ray crystal structure) shows that there are changes in the p-aminobenzoyl bridge conformation (Figure 8) that result from the displacement of the 6-6-6-ring system of 2 compared to the 6-5-6 ring system of 7. Similarly, a fit of 7 to that of 2 in ecTS (Figure 9) shows that contact with Val 77 is the same for both 7 and 2 (Figure 9).
Figure 8.
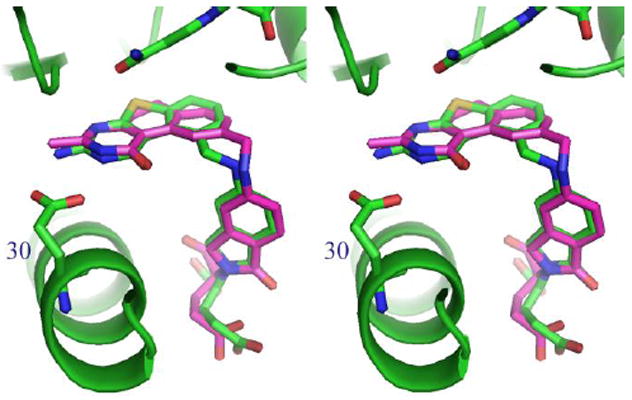
Model of 2 (violet) from ecTS 28 structure modeled to 7 in hDHFR (green).
Figure 9.
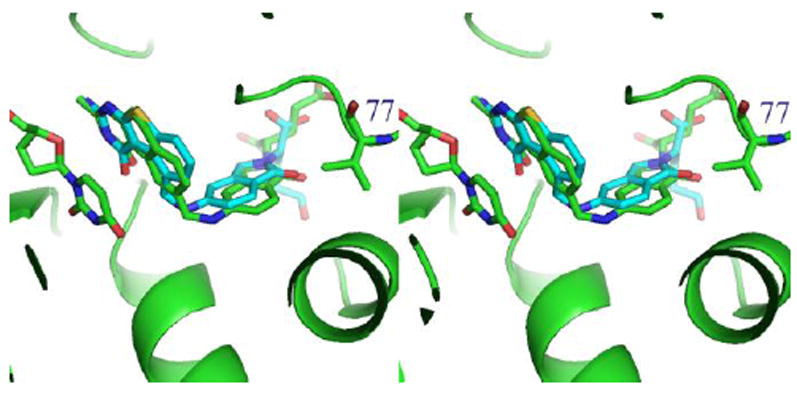
Stereoview of ecTS-2 28 (green) with compound 7 (cyan) from the crystal structure of hDHFR fit to the TS ligand. Also highlighted is residue Val77 in TS.
Conclusion
Compounds 4-7 were synthesized as classical antifolates with both rigid and flexible benzoylblutamates attached to the tricyclic benzo[4,5]thieno[2,3-d]pyrimidine scaffold. Compounds with a 2-CH3 moiety inhibited human TS but not human DHFR. Replacement of the 2-CH3 moiety with a 2-NH2 gave increased human TS inhibition and also human DHFR inhibition affording dual hTS/hDFHR inhibitors. The X-ray crystal structures of 6 and 7 show, for the first time, that tricyclics 6 and 7 bind in the folate rather than the MTX mode.
5. Experimental Section
All evaporations were carried out in vacuo with a rotary evaporator. Analytical samples were dried in vacuo (0.2 mmHg) in a CHEM-DRY drying apparatus over P2O5 at 80 °C. Melting points were determined on a MEL-TEMP II melting point apparatus with a FLUKE 51 K/J electronic thermometer and are uncorrected. Nuclear magnetic resonance spectra for proton (1H NMR) were recorded on either a Bruker WH-400 (400 MHz) spectrometer or a Bruker WH-300 (300 MHz) spectrometer. The chemical shift values are expressed in ppm (parts per million) relative to tetramethylsilane as an internal standard: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad singlet; exch, D2O exchangeable protons. Mass spectra were recorded on a VG-7070 double-focusingmass spectrometer or in a LKB-9000 instrument in the electron ionization (EI) mode. Chemical names follow IUPAC nomenclature. Thin-layer chromatography (TLC) was performed on Whatman Sil G/UV254 silica gel plates with a fluorescent indicator, and the spots were visualized under 254 and 365 nm illumination. All analytical samples were homogeneous on TLC in three different solvent systems. Proportions of solvents used for TLC are by volume. Column chromatography was performed on a 230-400 mesh silica gel (Fisher, Somerville, NJ) column. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. Element compositions are within 0.4% of the calculated values. Fractional moles of water frequently found in the analytical sample of antifolates could not be prevented in spite of 24-48 h of drying in vacuo and was confirmed where possible by the presence in the 1H NMR spectra. All solvents and chemicals were purchased from Aldrich Chemical Co. or Fisher Scientific and were used as received. For compounds 4-7, a single spot on TLC in three different solvent systems with three different Rf values confirmed >95% purity.
Ethyl 2-amino-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (9)
A mixture of sulfur (1.1 g, 36 mmol), 2-methylcyclohexanone (4.04 g, 36 mmol) , ethyl cyanoactetate (4.07 g, 36 mmol) and EtOH (150 mL) were placed in a round bottom flask and warmed to 45 °C and treated dropwise with morpholine (3.1 g, 36 mmol) over 15 min. The mixture was stirred for 5 h at 45 °C and 24 h at room temperature. Unreacted sulfur was removed by filtration, and the filtrate was concentrated under reduced pressure to afford an orange solid. The residue was loaded on a silica gel column packed with silica gel and eluted with 10% ethyl acetate in hexane. The fractions containing the desired product (TLC) were pooled and evaporated to afford 9 (6.97 g, 80.9 %) as an orange solid; mp 69.9-71 °C; Rf 0.44 (hexane/EtOAc 3:1); 1H NMR (DMSO-d6): δ 1.08-1.10 (d, 3 H, J = 6.6 Hz, 4-CH3), 1.25-1.28 (t, 3 H, J = 6.8 Hz, COOCH2CH3), 1.57-1.78 (m, 4 H), 2.39-2.43 (m, 2 H), 3.15-3.17 (m, 1 H), 4.10-4.24 (q, 2 H, J = 6.8 Hz, COOCH2CH3), 7.23 (s, 2 H, NH2 exch). Titled compound was used directly for next step without further characterization.
2-Amino-5-methyl-5,6,7,8-tetrahydro[1]benzothieno[2,3-d]pyrimidin-4(3H)-one (10)
A mixture of 9 (0.74 g, 3.28 mmol) and chloroformamidine hydrochloride (1.51 g, 13.12 mmol) in dimethyl sulfone (DMSO2 ) (4 g) was heated at 140° C for 4 h. The mixture was cooled to room temperature and water (15 mL) was added and ammonium hydroxide was used to neutralize the suspension. The brown solid, obtained by filtration, was washed with water and dried over P2O5 in vacuum. The solid was dissolved in methanol and silica gel was added. A dry silica gel plug was obtained after evaporation of the solvent. The plug was loaded on to a silica gel column and eluted with 5% methanol in chloroform. The fractions containing the desired product (TLC) were pooled and evaporated to afford 10 (0.46 g, 59.7 %) as a light yellow solid; mp > 300 °C; Rf 0.5 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6) δ 1.18-1.20 (d, 3 H, J = 6.8 Hz, CH3), 1.59-1.61 (m, 1 H), 1.71-1.83 (m, 3 H), 2.54-2.64 (m, 2 H), 3.15-3.18 (m, 1 H), 6.39 (s, 2 H, 2-NH2 exch), 10.73 (s, 1 H, 3-NH exch); Anal. (C11H13N3SO · 0.2 H2O): C, H, N, S.
2,2-Dimethyl-N-(5-methyl-4-oxo-3,4,5,6,7,8-hexahydro[1]benzothieno[2,3-d]pyrimidin-2-yl)propanamide (11)
To a 100 mL round-bottomed flask was added 10 (0.706 g, 3 mmol) and excess Piv2O (10 mL). The mixture was kept at reflux for 2 h. The excess Piv2O was removed under vacuum and the residue was made into silica gel plug. The silica gel plug obtained was loaded onto a silica gel column and eluted with 1:8 ethyl acetate/hexane. The fraction containing the desired product were pooled afford 11 (0.956 g, 70.3 %) as a white solid; mp 236.6-237.8 °C; Rf 0.31 (hexane/EtOAc 3:1); 1H NMR (DMSO-d6) δ 1.22 (s, 12 H, 4 CH3), 1.62-1.79 (m, 4 H), 2.58-2.73 (m, 2 H), 3.23 (br s, 1 H), 11.07 (s, 1 H, NH exch), 12.02 (s, 1 H, NH exch). Titled compound was used directly for next step without further characterization.
Ethyl 2-amino-4-methyl-1-benzothiophene-3-carboxylate (14)
A mixture of 9 (0.239 g, 1 mmol), 10 % Pd/C (0.239 g) and mesitylene (50 mL) were placed in a round bottom flask and kept at reflux for 1-2 days. The mixture was cooled to room temperature and Pd/C was removed by filtration, and the filtrate was concentrated under reduced pressure to afford a brown oil. The residue was loaded on a silica gel column and eluted with 10% ethyl acetate in hexane. The fractions containing the desired product (TLC) were pooled and evaporated to afford 14 (0.123 g, 52.1%) as an orange semi solid; Rf 0.38 (hexane/EtOAc 3:1); 1H NMR (DMSO-d6): δ 1.29-1.32 (t, 3 H, J = 6.8 Hz, COOCH2CH3), 2.38 (s, 3 H, CH3), 4.25-4.30 (q, 2 H, J = 6.8 Hz,COOCH2CH3), 6.98-7.04 (m, 2 H, C6H3), 7.44, 7.45 (d, 1 H, J = 4 Hz, C6H3), 7.43 (s, 2 H, NH2 exch). Titled compound was used directly for next step without further characterization.
2-Amino-5-methyl[1]benzothieno[2,3-d]pyrimidin-4(3H)-one (12)
A mixture of 14 (0.49 g, 2.07 mmol) and chloroformamidine hydrochloride (1.19 g, 10.37 mmol) in DMSO2 (2 g) was heated at 140° C for 2 h. The mixture was cooled to room temperature and water (15 mL) was added and ammonium hydroxide was used to neutralize the suspension. The brown solid, obtained by filtration, was washed with water and dried over P2O5 vacuum. The solid was dissolved in methanol and silica gel (1.0 g) was added. A dry silica gel plug was obtained after evaporation of the solvent. The plug was loaded on to a silica gel column and eluted with 5% methanol in chloroform. The fractions containing the desired product (TLC) were pooled and evaporated to afford 12 (0.29 g, 60.7 %) as a yellow solid; mp > 300 °C; Rf 0.41 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6) δ 2.87 (s, 3 H, CH3), 6.82 (s, 2 H, 2-NH2 exch), 7.13- 7.17 (m, 2 H, C6H3), 7.57, 7.59 (d, 1 H, J = 8.0 Hz, C6H3), 10.92 (s, 1 H, 3-NH exch). Titled compound was used directly for next step without further characterization.
2,2-Dimethyl-N-(5-methyl-4-oxo-3,4-dihydro[1]benzothieno[2,3-d]pyrimidin-2-yl)propanamide (13)
To a 100 mL round-bottomed flask was added 12 (1.34 g, 5.8 mmol) and excess Piv2O (4 eq). The mixture was kept at reflux for 2 h. The excess Piv2O was removed under vacuum and the residue was made into silica gel plug. The silica gel plug obtained was loaded onto a silica gel column and eluted with 1:8 ethyl acetate/hexane to afford 13 ( 1.299 g, 70.9 %) as a white solid, mp 178.3-179.5 °C; Rf 0.35 (hexane/EtOAc 3:1); 1H NMR (CDCl3) δ 1.35 (s, 9 H, 3 CH3 of Piv), 3.05 (s, 3 H, CH3), 7.28 (m, 2 H, C6H3), 7.56, 7.59 (d, 1 H, J = 8.0 Hz C6H3), 8.14 (s, 1 H, NH exch), 11.86 (s, 1 H, NH exch). Titled compound was used directly for next step without further characterization.
2,5-Dimethyl[1]benzothieno[2,3-d]pyrimidin-4(3H)-one (15)
To a 100 mL round flask were added 14 (2.35 g, 10 mmol) and CH3CN (50 mL). Vigorous stirring afforded, a clear solution. Anhydrous HCl gas was bubbled into the solution for 1 h to give a thick precipitate, which then redissolved into the acid solution. Anhydrous HCl gas was added for an additional 3 h after which the reaction mixture became clear. Evaporation of the solvent under reduced pressure afforded a residue that was dissolved in water. Concentrated aqueous NH4OH was added to afford a suspension at pH = 8. The precipitate was collected by filtration, washed with water and dried over P2O5 in a vacuum to afford 15 (1.0 g, 57%) as a yellow solid; mp > 300 °C; Rf 0.58 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6) δ 2.35 (s, 3 H, 2-CH3), 2.95 (s, 3 H, 5-CH3). 7.29 (m, 2 H, C6H3), 7.78 (s, 1 H, C6H3), 12.54 (s, 1H, 3-NH exch). HRMS calcd for C12H10N2OS 231.0592, found 231.0584.
5-(Bromomethyl)-2-methyl[1]benzothieno[2,3-d]pyrimidin-4(3H)-one (17)
To a 100 mL flask were added 15 (0.736 g, 3.2 mmol) and benzene (30 mL). The suspension was stirred at 60 °C for 30 min to afford a clear solution, followed by the addition of N-bromosuccinimide (0.620 g, 3.49 mmol) and benzoyl peroxide (50 mg). The mixture was maintained at reflux for 4h and then cooled to room temperature and washed with water, and evaporated to afford a yellow solid. The solid was dissolved in methanol and silica gel ( 1.5 g) was added. A dry silica gel plug was obtained after evaporation of the solvent. The plug was loaded on to a silica gel column and eluted with 6 % ethyl acetate in hexane to afford 17 (393 mg, 39%) as a white solid; mp 217.8-219.1 °C; Rf 0.3 (hexane/EtOAc 3:1); 1H NMR (CDCl3) δ 2.69 (s, 3 H, 2-CH3), 5.84 (s, 2 H, CH2Br), 7.44-7.47 (t, 1 H, J = 7.2, C6H3), 7.57-7.58 (d, 1 H, J = 7.2 Hz, C6H3), 7.80-7.82 (d, 1 H, J = 7.2 Hz, C6H3). HRMS calcd for C12H9N2OSBr 307.9619, found 307.9613.
2,2-Dimethyl-N-(5-methyl-4-oxo-3,4-dihydro[1]benzothieno[2,3-d]pyrimidin-2-yl)propanamide (16)
A solution of 13 (1.1 g, 3.49 mmol) in 1,2- dichloroethane (50 mL) was treated with N-bromosuccinimide (0.62 g, 3.49 mmol) and benzoyl peroxide (50 mg), and the mixture was maintained at reflux for 1 day. The mixture was cooled to room temperature and washed with water, and evaporated to an orange solid. The solid was dissolved in methanol and silica gel ( 1.5 g) was added. A dry silica gel plug was obtained after evaporation of the solvent. The plug was loaded on to a silica gel column and eluted with 6 % ethyl acetate in hexane to afford 16 (0.577 g, 41.9 %) as an orange solid; mp 217.8-219.1 °C; Rf 0.3 (hexane/EtOAc 3:1); 1H NMR (CDCl3) δ 1.27 (s, 9 H, 3 CH3 of Piv), 5.71 (s, 2 H, CH2Br), 7.30, 7.33 (d, 1 H, C6H3), 7.43, 7.46 (d, 1 H, J = 7.6 Hz, C6H3), 7.64, 7.66 (d, 1 H, J = 4 Hz, C6H3), 8.07 (s, 1 H, NH exch), 11.99 (s, 1 H, NH exch). Titled compound was used directly for next step without further characterization.
Methyl 2-methyl-4-nitrobenzoate (19)
Thionyl chloride (4.3 g, 36.45 mmol) was added dropwise to a stirred solution of 18 (3 g, 16.57 mmol) in MeOH (25 mL) while maintaining the internal temperature below 12 °C. When the addition was complete the mixture was left to stand at room temperature for 12h to result a white precipitation. The mixture was filtered and the filtrate was concentrated under reduced pressure to afford white solid. The solid was washed with hexane and ethyl ether to afford 19 (2.95 g, 91.3 %); mp 153.7-154.4 °C (lit.39 mp 153-154 °C); Rf 0.43 (hexane/EtOAc 3:1); 1H NMR (CDCl3) δ 2.69 (s, 3 H, CH3), 3.95 (s, 3 H, COOCH3), 8.02-8.11 (m, 3 H, C6H3).
Methyl 2-(bromomethyl)-4-nitrobenzoate (20)
A solution of 19 (2.57 g, 13.19 mmol) in 1,2-dichloroethane (100 mL) was treated with N-bromosuccinimide (2.3 g, 13.19 mmol) and benzoyl peroxide (0.26 g), and the mixture was kept at reflux for 2 days, then cooled, washed with water, and evaporated to a yellow oil (3.52 g). The oil was dissolved in acetone and silica gel ( 4.0 g) was added. A dry silica gel plug was obtained after evaporation of the solvent. The plug was loaded on to a silica gel column and eluted with 10 % ethyl acetate in hexane to afford 20 (1.73 g, 48.2 %) as a yellow oil; Rf 0.53 (hexane/EtOAc 3:1); 1H NMR (CDCl3) δ 3.89 (s, 3 H, COOCH3), 4.86 (s, 2 H, CH2Br), 7.98-8.23 (m, 3 H, C6H3).
Diethyl (2S)-2-(5-nitro-1-oxo-1,3-dihydro-2H-isoindol-2-yl)pentanedioate (21)
The oil 20 (0.85 g, 3.1 mmol) was stirred for 16 h with diethyl glutamate hydrochloride (1.54 g, 6.4 mmol) and powdered K2CO3 (1.7 g, 12 mmol) in DMA (3 mL) under argon. The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (3 × 20 mL). The combined ethyl acetate solutions were washed twice with brine, dried, and evaporated to an orange oil. The oil was dissolved in methanol and silica gel was added. A dry silica gel plug was obtained after evaporation of the solvent. The plug was loaded on to a silica gel column and eluted with 25 % ethyl acetate in hexane to afford 21 (0.63 g, 55.7 %) as an orange oil; Rf 0.44 (hexane/EtOAc 1:1); 1H NMR (CDCl3) δ 1.17-1.22 (t, 3 H, COOCH2CH3), 1.26-1.31 (t, 3 H, COOCH2CH3), 2.19-2.49 (m, 4 H, CHCH2CH2COOEt), 4.02-4.23 (2q, 4 H, COOCH2CH3), 4.51-4.83 (dd, 2 H, -CH2-), 5.09-5.14 (m, 1 H, CHCH2CH2COOEt), 8.00-8.03 (d, 1 H, C6H3), 8.36 (br s, 2 H, C6H3).
Diethyl (2S)-2-(5-amino-1-oxo-1,3-dihydro-2H-isoindol-2-yl)pentanedioate (22)
To a Parr hydrogenation bottle was added 21 (0.55 g, 1.51 mmol), 10% Pd/C (0.09 g) and acetyl acetate (30 mL). Hydrogenation was carried out at 55 psi for 12 h. After filtration, the organic phase was evaporated at vacuum to afford 22 (0.467 g, 92.5 %) as a orange oil; Rf 0.19 (hexane/EtOAc 1:1); 1H NMR (CDCl3) δ 1.17-1.19 (t, 3 H,COOCH2CH3), 1.20-1.25 (t, 3 H, COOCH2CH3), 2.20-2.51 (m, 4 H, CHCH2CH2COOEt), 4.02-4.28 (m, 6 H, 2 COOCH2CH3, NH2 exch), 4.22-4.51 (dd, 2 H, -CH2-), 5.03-5.07 (m, 1 H, CHCH2CH2COOEt), 6.69-6.73 (m, 2 H, C6H3), 7.60, 7.63 (d, 1 H, C6H3).
General Procedure for the Synthesis of Compounds 4-7
A stirred solution of the tricyclic bromide 16 or 17 (0.25 mmol) in dry DMF (5 mL) was treated with the appropriate amine 22 or 23 (1 mmol) and K2CO3 (95 mg, 0.69 mmol). The solution was stirred for 1 h at 80 °C under argon. The cooled reaction mixture was filtered and the filtrate was evaporated to obtain an orange solid. The solid was dissolved in methanol and silica gel was added. A dry silica gel plug was obtained after evaporation of the solvent. The plug was loaded on to a silica gel column and eluted with ethyl acetate: hexane (1: 1). The fractions containing the desired product (TLC) were pooled and evaporated to afford a solid, to which a combined solution of aqueous 1 N NaOH (3 mL) and methanol (12 mL) was added. The mixture was kept at reflux for 12 h. The methanol was evaporated under reduced pressure and the residue was dissolved in water (5 mL). The solution was cooled to 0 °C and carefully acidified to pH 3 with dropwise addition of 1 N HCl. The resulting suspension was left at 0 °C for 2 h and the precipitate was collected by filtration, washed with water (5 mL) and dried over P2O5/vacuum at 50 °C to afford target compounds 4-7.
N-(4-{[(2-methyl-4-oxo-3,4-dihydro[1]benzothieno[2,3-d]pyrimidin-5-yl)methyl]amino}benzoyl)-L-glutamic acid (4)
Using the general procedure described above with 17 and 23 afforded 4 (37 mg, 37 %) as a yellow solid; mp 219.6-221.3 °C; Rf 0.28 (MeOH/EtOAc, 1:6); Rf 0.32 (MeOH/ CHCl3, 1:6 + 1 drop of NEt3); Rf 0.34 (MeOH/CHCl3, 1:6 + 1 drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.22 (s, 1 H, 2-CH3), 1.88-2.01 (m, 2 H, Gluγ-CH2CH2), 2.29 (m, 2 H, Gluγ-CH2CH2), 4.31 (s, 1 H, Gluα-CH), 5.20 (s, 2 H, Benzylic CH2), 6.56-6.58 (d, 2 H, J = 9.0 Hz), 7.38-7.40 (d, 1 H, J = 7.5 Hz), 7.49 (d, 2 H, 2-NH2 exch), 7.58-7.59 (d, 2 H, J = 9.0 Hz), 7.88 (d, 1 H, J = 7.5 Hz) 8.02 (s, 1 H, CONH), 8.08 (d, 1H), 12.71 (s, 1 H, 3-NH exch); HRMS (ESI, pos mode) m/z [M + H+] calcd for C24H23N4O6S 495.1338, found 495.1345.
(2S)-2-(5-{[(2-methyl-4-oxo-3,4-dihydro[1]benzothieno[2,3-d]pyrimidin-5-yl)methyl]amino}-1-oxo-1,3-dihydro-2H-isoindol-2-yl)pentanedioic acid (5)
Using the general procedure described above with 17 and 22 afforded 5 (35 mg, 34 %) as a yellow solid; mp 236.4-237.7 °C; Rf 0.29 (MeOH/EtOAc, 1:6); Rf 0.32 (MeOH/ CHCl3, 1:6 + 1 drop of NEt3); Rf 0.36 (MeOH/CHCl3, 1:6 + 1 drop of gl. HOAc);1H NMR (DMSO-d6): δ 1.22 (s, 3 H, 2-CH3), 1.95 (m, 2 H, Gluβ-CH2), 2.18 (m, 2 H, Gluγ-CH2), 4.22 (s, 2 H, isoindolinyl CH2), 4.67-4.69 (m, 1 H, Gluα-CH), 5.21 (s, 2 H, Benzylic CH2), 6.65 (s, 1 H, CH), 6.81 (s, 1 H, NH exch), 7.30-7.32 (d, 1 H, J = 6.0 Hz), 7.37-7.41 (d, 1 H, J = 12 Hz), 7.49-7.51 (d, 1 H, J = 6.0 Hz), 7.88-7.90 (d, 1 H, J = 6.0 Hz), 8.03 (s, 1 H),12.76 (s, 1 H, 3-NH exch), HRMS (ESI, pos mode) m/z [M + H+] calcd for C25H23N4O6S 507.1333, found 507.1362.
N-(4-{[(2-amino-4-oxo-3,4-dihydro[1]benzothieno[2,3-d]pyrimidin-5-yl)methyl]amino}benzoyl)-L-glutamic acid (6)
Using the general procedure described above with 16 and 23 afforded 6 (39 g, 32 %) as an orange solid; mp > 300 °C; Rf 0.30 (MeOH/EtOAc, 1:6); Rf 0.34 (MeOH/ CHCl3, 1:6 + 1 drop of NEt3); Rf 0.34 (MeOH/CHCl3, 1:6 + 1 drop of gl. HOAc);1H NMR (DMSO-d6): δ 1.85-1.98 (m, 2 H, Gluγ-CH2CH2), 2.27-2.32 (m, 2 H, Gluγ-CH2CH2),4.26-4.34 (m, 1 H, Gluα-CH), 5.10-5.14 (d, 2 H, Benzylic CH2), 6.55 (s, H, CH2NH), 6.55-6.58 (d, 2 H, J = 9.0 Hz, 2 CH), 6.94 (br s, 2 H, 2-NH2 exch), 7.19-7.24 (t, 1 H, J = 7.5 Hz), 7.38-7.40 (d, 1 H, J = 7.5 Hz), 7.56-7.59 (d, 2 H, J = 9.0 Hz), 7.68-7.70 (d, 1 H, J = 7.5 Hz), 7.97-8.00 (d, 1 H, J = 6.9 Hz, NH exch), 11.19 (s, 1 H, 3-NH exch), 12.66 (s, 2 H, 2COOH exch); HRMS (ESI, pos mode) m/z [M + H+] calcd for C23H22N5O6S 496.1291, found 496.1316.
(2S)-2-(5-{[(2-Amino-4-oxo-3,4-dihydro[1]benzothieno[2,3-d]pyrimidin-5-yl)methyl]amino}-1-oxo-1,3-dihydro-2H-isoindol-2-yl)pentanedioic acid (7)
Using the general procedure described above with 16 and 22 afforded 7 (40 mg, 32 %) as a orange solid; mp > 300 °C; Rf 0.27 (MeOH/EtOAc, 1:6); Rf 0.30 (MeOH/ CHCl3, 1:6 + 1 drop of NEt3); Rf 0.33 (MeOH/CHCl3, 1:6 + 1 drop of gl. HOAc);1H NMR (DMSO-d6): δ 1.97-2.20 (m, 4 H, Gluβ-CH2, Gluγ-CH2), 4.21-4.25 (m, 2 H, isoindolinyl CH2), 4.68-4.71 (m, 1 H, Gluα-CH), 5.13, 5.14 (d, 2 H, benzylic CH2), 6.64 (br s, 2 H, C6H3), 6.74 (s, 1 H, NH exch), 6.89 (s, 2 H, 2-NH2 exch), 7.20-7.26 (t, 1 H, J = 7.5 Hz, C6H3), 7.30-7.33 (d, 1 H, J = 8.0 Hz, C6H3), 7.39-7.42 (dd, 1 H, C6H3), 7.69-7.72 (d, 1 H, J = 8.0 Hz, C6H3), 11.11 (s, 1 H, 3-NH exch), 12.78 (s, 2 H, 2COOH exch). HRMS (ESI, pos mode) m/z [M + H+] calcd for C24H21N5O6S, 508.1285; found, 508.1321.
Structure Determination and Refinement
Expression and purification of hDHFR were carried out as previously described.41 Recombinant hDHFR was washed in a Centricon-10 with 10 mM HEPES buffer, pH 7.4, and concentrated to 11.9 – 12.1 mg/mL for the two samples. The hDHFR protein was incubated with NADPH and an excess of compounds 6 and 7 for 1hr over ice prior to crystallization using the hanging drop vapor diffusion method. The reservoir solution for inhibitor 6 contained 100 mM KPO4, pH 6.9, 66% saturated NH4SO4, 3% v/v ethanol and 70% saturated NH4SO4 for compound 7. Crystals of hDHFR complex grew over serval days at 14°C and were trigonal, space group H3. Data were collected to 1.35A resolution for both complexes using the remote access robot47-49 at liquid N2 temperatures on beamline 9-2 at the Stanford Synchrotron Research Laboratory (SSRL) imaging plate system. The data were processed using Mosflm.50 The diffraction statistics are shown in Table 2 for the two complexes.
Table 2.
Data collection and refinement statistics for hDHFR-NADPH-6 and 7 ternary complexes.
| Data collection | hDHFR NADPH-6 | hDHFR NADPH-7 | ||
|---|---|---|---|---|
| PDB accession # | 3ntz | 3nu0 | ||
| Space group | H3 | H3 | ||
| Cell dimensions (Ǻ) | 84.29 | 77.79 | 84.39 | 78.19 |
| Beamline | SSRL 9-2 | SSRL 9-2 | ||
| Resolution (Ǻ) | 1.35 | 1.35 | ||
| Wavelength (Ǻ) | 0.975 | 0.975 | ||
| Rsym (%) a,b | 0.061 (0.72) | 0.066 (0.45) | ||
| Rmerge | 0.053 (0.62) | 0.063 (0.39) | ||
| Completeness (%) a | 100.0 (100.0) | 100.0 (100.0) | ||
| Observed reflections | 169,016 | 569,239 | ||
| Unique reflections | 45,260 | 45,587 | ||
| I/(I) | 15.3 (1.6) | 27.0 (8.6) | ||
| Multiplicity a | 3.7 (3.7) | 12.5 (12.1) | ||
| Refinement and model quality | ||||
| Resolution range (Ǻ) | 26.6 – 1.35 | 33.1 – 1.35 | ||
| No. of reflections | 45,259 | 45,587 | ||
| R-factor c | 18.6 | 18.0 | ||
| Rfree-factor d | 20.5 | 20.9 | ||
| Total protein atoms | 1817 | 1868 | ||
| Total water atoms | 164 | 187 | ||
| Average B-factor (Ǻ2) | 18.9 | 15.7 | ||
| Rms deviation from ideal | ||||
| Bond lengths (Ǻ) | 0.031 | 0.035 | ||
| Bond angles (°) | 2.84 | 2.99 | ||
| Luzzati | 0.159 | 0.145 | ||
| Ramachandran plot | ||||
| Most favored regions (%) | 97.8 | 97.8 | ||
| Additional allowed regions (%) | 2.2 | 2.2 | ||
| Generously allowed regions (%) | 0.0 | 0.0 | ||
| Disallowed regions (%) | 0.0 | 0.0 | ||
The values in parentheses refer to data in the highest resolution shell.
Rsym = ΣhΣi|Ih,i - <Ih>| / ΣhΣi|Ih,i|, where <Ih> is the mean intensity of a set of equivalent reflections.
R-factor = Σ|Fobs − Fcalc| / ΣFobs, where Fobs and Fcalc are observed and calculated structure factor amplitudes.
Rfree-factor was calculated for R-factor for a random 5% subset of all reflections.
The structures were solved by molecular replacement methods using the coordinates for hDHFR (u072) in the program Molref.50 Inspection of the resulting difference electron density maps made using the program COOT51 running on a MacG5 workstation revealed density for the ternary complex in both crystals. The final cycles of refinement were carried out using the program Refmac5 in the CCP4 suite of programs. The Ramachandran conformational parameters from the last cycle of refinement generated by PROCHECK52 showed that more than 98% of the residues have the most favored conformation and none are in the disallowed regions. Coordinated for these structures have been deposited with the Protein Data Bank (3ntz, 3nu0).
Dihydrofolate Reductase (DHFR) Assay 53
All enzymes were assayed spectrophotometrically in a solution containing 50 μM dihydrofolate, 80 μM NADPH, 50 mM Tris-HCl, 0.001 M 2-mercaptoethanol, and 0.001 M EDTA at pH 7.4 at 30° C. The reaction was initiated with an amount of enzymes yielding a change in optical density at 340 nm of 0.015 units/min.
Thymidylate Synthase (TS) Assay
TS was assayed spectrophotometrically at 30° C and pH 7.4 in a mixture containing 0.1 M 2-mercaptoethanol, 0.0003 M (6R,S)-tetrahydrofolate, 0.012 M formaldehyde, 0.02 M MgCl2, 0.001 M dUMP, 0.04 M Tris-HCl, and 0.00075 M NaEDTA. This was the assay described by Wahba and Friedkin54 except that the dUMP concentration was increased 25-fold according to the method of Davisson et al.55 The reaction was initiated by the addition of an amount of enzyme yielding a change in absorbance at 340 nm of 0.016 units/min in the absence of inhibitor.
Supplementary Material
Acknowledgments
This work is supported by National of Institute of Health, National Institute of Allergy and Infectious Diseases grant A1069966 (AG), National Cancer Institute grant CA 125153(AG) and General Medical Institute grant GM051670 (VC). The National Science Foundation, CHE0614785, is acknowledged for NMR facilities.
Footnotes
Supplementary Material Elemental Analysis and High-Resolution Mass Spectra (HRMS) (EI). This material is available free of charge via the Internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Jackson RC. In: Antifolate Drugs in Cancer Therapy. Jackman AL, editor. Humana Press; Totowa: 1999. pp. 1–12. [Google Scholar]
- 2.Gangjee A, Elzein E, Kothare M, Vasudevan Curr Pharm Design. 1996;2:263–280. [Google Scholar]
- 3.DeGraw JI, Colwell WT, Piper JR, Sirotnak FM, Smith RL. Curr Med Chem. 1995;2:630–653. [Google Scholar]
- 4.Huennekens FM, Duffy TH, Vitols KS. NCI monographs. 1987;5:1–8. [PubMed] [Google Scholar]
- 5.Berman EM, Werbel LM. J Med Chem. 1991;34:479–485. doi: 10.1021/jm00106a001. [DOI] [PubMed] [Google Scholar]
- 6.Chu E, Callender MA, Farrell MP, Schmitz JC. Cancer Chemother Pharmacol. 2003:80–897. doi: 10.1007/s00280-003-0625-9. [DOI] [PubMed] [Google Scholar]
- 7.Danenberg PV. Biochim Biophys Acta. 1977;47:73–92. doi: 10.1016/0304-419x(77)90001-4. [DOI] [PubMed] [Google Scholar]
- 8.Hitchings GH. Functions of Tetrahydrofolate and the Role of Dihydrofolate Reductase. In: Hitchings GH, editor. Cellular Metabolism in Inhibition of Folate Metabolism in Chemotherapy. Springer Verlag; New York: 1983. pp. 11–23. [Google Scholar]
- 9.Jackman AL, Taylor GA, Gibson W, Kimbell R, Brown M, Calvert AH, Judson IR, Hughes LR. Cancer Res. 1991;51:5579–5586. [PubMed] [Google Scholar]
- 10.Taylor EC, Kuhnt D, Shih C, Rinzel SM, Grindey GB, Barredo J, Jannatipour M, Moran RJ. Med Chem. 1992;35:4450–4454. doi: 10.1021/jm00101a023. [DOI] [PubMed] [Google Scholar]
- 11.Bertino JR, Kamen B, Romanini A. Folate Antagonists. In: Holland JF, Frei E, Bast RC, Kufe DW, Morton DL, Weichselbaum RR, editors. Cancer Medicine. Vol. 1. Williams and Wilkins; Baltimore, MD: 1997. pp. 907–921. [Google Scholar]
- 12.Gibson W, Bisset GMF, Marsham PR, Kelland LR, Judson IR, Jackman AL. Biochem Pharmacol. 1993;45:863–869. doi: 10.1016/0006-2952(93)90170-2. [DOI] [PubMed] [Google Scholar]
- 13.Sikora E, Jackman AL, Newell DF, Calvert AH. Biochem Pharmacol. 1988;37:4047–4054. doi: 10.1016/0006-2952(88)90094-9. [DOI] [PubMed] [Google Scholar]
- 14.Jackman AL, Newell DR, Gibson W, Jodrell DI, Taylor GA, Bishop JA, Hughes LR, Calvert AH. Biochem Pharmacol. 1991;42:1885–1895. doi: 10.1016/0006-2952(91)90586-t. [DOI] [PubMed] [Google Scholar]
- 15.Nair MG, Abraham A, McGuire JJ, Kisliuk RL, Galivan JH, Ferone R. Cell Pharmacol. 1994;1:245–249. [Google Scholar]
- 16.Bisset GMF, Bavetsias V, Thornton TJ, Pawelczak K, Calvert AH, Hughes LR, Jackman AL. J Med Chem. 1994;37:3294–3302. doi: 10.1021/jm00046a014. [DOI] [PubMed] [Google Scholar]
- 17.Barakat RR, Li WW, Lovelace C, Bertino JR. Gynecol Oncol. 1993;51:54–60. doi: 10.1006/gyno.1993.1246. [DOI] [PubMed] [Google Scholar]
- 18.McCloskey DE, McGuire JJ, Russell CA, Rowan BG, Bertino JR, Pizzorno G, Mini E. J Biol Chem. 1991;266:6181–6187. [PubMed] [Google Scholar]
- 19.Braakhuis BJM, Jansen G, Noordhius P, Kegel A, Peters G. J Biochem Pharmacol. 1993;46:2155–2161. doi: 10.1016/0006-2952(93)90604-u. [DOI] [PubMed] [Google Scholar]
- 20.Gangjee A, Devraj R, McGuire JJ, Kisliuk RL. J Med Chem. 1995;38:4495–4502. doi: 10.1021/jm00022a015. [DOI] [PubMed] [Google Scholar]
- 21.Marsham PR, Jackman AL, Barker AJ, Boyle FT, Pegg SJ, Wardleworth JM, Kimbell R, O’Connor BM, Calvert AH, Hughes LR. J Med Chem. 1995;38:994–1004. doi: 10.1021/jm00006a019. [DOI] [PubMed] [Google Scholar]
- 22.Tripos Inc.; 1699 South Hanley Road, St. Louis, MO 63144: [Google Scholar]
- 23.Pendergast W, Dickerson SH, Dev IK, Ferone R, Duch DS, Smith GK. J Med Chem. 1994;37:838–844. doi: 10.1021/jm00032a019. [DOI] [PubMed] [Google Scholar]
- 24.Beutel G, Glen H, Schoffski P, Chick J, Gill S, Cassidy J, Twelves C. Clin Cancer Res. 2005;11:5487–5495. doi: 10.1158/1078-0432.CCR-05-0104. [DOI] [PubMed] [Google Scholar]
- 25.Dev IK, Dallas WS, Ferone R, Hanlon M, McKee DD, Yates BB. J Biol Chem. 1994;269:1873–1882. [PubMed] [Google Scholar]
- 26.Hooijberg JH, Broxterman HJ, Kool M, Assaraf YG, Peters GJ, Noordhuis P, Scheper RJ, Borst P, Pinedo HM, Jansen G. Cancer Res. 1999;59:2532–2535. [PubMed] [Google Scholar]
- 27.Montfort WR, Weichsel A. Pharmacol Ther. 1997;76:29–43. doi: 10.1016/s0163-7258(97)00099-5. [DOI] [PubMed] [Google Scholar]
- 28.Stout TJ, Stroud RM. Structure. 1996;4:67–77. doi: 10.1016/s0969-2126(96)00010-x. [DOI] [PubMed] [Google Scholar]
- 29.Weichsel A, Montfort WR. Nature Struct Biol. 1995;2:1095–1101. doi: 10.1038/nsb1295-1095. [DOI] [PubMed] [Google Scholar]
- 30.Weichsel A, Montfort WR, Ciesla J, Maley F. Proc Natl Acad Sci. 1995;92:3493–3497. doi: 10.1073/pnas.92.8.3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vainio MJ, Johnson MS. J Chem Inf Model. 2007;47:2462–2474. doi: 10.1021/ci6005646. [DOI] [PubMed] [Google Scholar]
- 32.Gewald K, Schinke E, Böttcher H. Chem Ber. 1966;99:94–100. [Google Scholar]
- 33.Zhang M, Harper RW. Bioorg Med Chem Lett. 1997;7:1629–1634. [Google Scholar]
- 34.Rosowsky A, Chen KK, Lin M. J Med Chem. 1973;16:191–194. doi: 10.1021/jm00261a004. [DOI] [PubMed] [Google Scholar]
- 35.Gangjee A, Yu JM, Copper JE, Smith CD. J Med Chem. 2007;50:3290–3301. doi: 10.1021/jm070194u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yadav PP, Gupta P, Chaturvedi AK, Shukla PK, Maurya R. Bioorg Med Chem. 2005;13:1497–1505. doi: 10.1016/j.bmc.2004.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sengupta SK, Chatterjee S, Protopapa HK, Modeat EJ. J Org Chem. 1972;37:1323–1328. [Google Scholar]
- 38.Wartenberg FH, Koppe T, Wetzel W, Wydra M, Benz A. PCT Int Appl. 2001 CODEN: PIXXD2 WO 01/77099 A1 19980305. [Google Scholar]
- 39.Rosowsky A, Forsch RA, Null A, Moran RG. J Med Chem. 1999;42:3510–3519. doi: 10.1021/jm9807205. [DOI] [PubMed] [Google Scholar]
- 40.Pendergast W, Dickerson SH, Dev IK, Ferone R, Duch DS, Smith GK. J Med Chem. 1994;37:838–844. doi: 10.1021/jm00032a019. [DOI] [PubMed] [Google Scholar]
- 41.Patil SD, Jones C, Nair MG, Galivan J, Maley F, Kisliuk RL, Gaumont Y, Duch D, Ferone R. J Med Chem. 1989;32:1284–1289. doi: 10.1021/jm00126a023. [DOI] [PubMed] [Google Scholar]
- 42.Hughes LR, Jackman AL, Oldfield J, Smith RC, Burrows KD, Marsham PR, Bishop JAM, Jones TR, O’Connor BM, Calvert AH. J Med Chem. 1990;33:3060–3067. doi: 10.1021/jm00173a024. [DOI] [PubMed] [Google Scholar]
- 43.Marsham PR, Jackman AL, Hayter AJ, Daw MR, Snowden JL, O’Connor BM, Bishop JA, Calvert AH, Hughes LR. J Med Chem. 1991;34:2209–2218. doi: 10.1021/jm00111a042. [DOI] [PubMed] [Google Scholar]
- 44.Gangjee A, Li W, Kisliuk RL, Cody V, Pace J, Piraino J, Makin J. J Med Chem. 2009;52:4892–4902. doi: 10.1021/jm900490a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cody V, Luft JR, Pangborn W. Acta Crystallogr D Biol Crystallogr. 2005;61:147–155. doi: 10.1107/S0907444904030422. [DOI] [PubMed] [Google Scholar]
- 46.DELano Scientific LLC, PyMol [Google Scholar]
- 47.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 48.Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. J Appl Crystallogr. 2002;35:720–726. doi: 10.1107/s0021889802016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonzalez A, Moorhead P, McPhillips SE, Song J, Sharp K, Taylor JR, Adams PD, Sauter NK, Soltis SM. J Appl Crystallogr. 2008;41:176–184. [Google Scholar]
- 50.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P, Cowtan K. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 52.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 53.Kisliuk RL, Strumpf D, Gaumont Y, Leary RP, Plante L. J Med Chem. 1977;20:1531–1533. doi: 10.1021/jm00221a038. [DOI] [PubMed] [Google Scholar]
- 54.Wahba AJ, Friedkin M. J Biol Chem. 1962;237:3794–3801. [PubMed] [Google Scholar]
- 55.Davisson VJ, Sirawaraporn W, Santi DV. J Biol Chem. 1989;264:9145–9148. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.