

Abstract
Zn2+ is an important cofactor for insulin biosynthesis and storage in pancreatic β-cells. Correspondingly, polymorphisms in the SLC30A8 gene, encoding the secretory granule Zn2+ transporter ZnT8, are associated with type 2 diabetes risk. Using a genetically engineered (FRET)-based sensor (eCALWY-4), we show here that elevated glucose time-dependently increases free cytosolic Zn2+ ([Zn2+]cyt) in mouse pancreatic β-cells. These changes become highly significant (853 ± 96 pm versus 452 ± 42 pm, p < 0.001) after 24 h and are associated with increased expression of the Zn2+ importer family members Slc39a6, Slc39a7, and Slc39a8, and decreased expression of metallothionein 1 and 2. Arguing that altered expression of the above genes is not due to altered [Zn2+]cyt, elevation of extracellular (and intracellular) [Zn2+] failed to mimic the effects of high glucose. By contrast, increases in intracellular cAMP prompted by 3-isobutyl-1-methylxanthine and forskolin partially mimicked the effects of glucose on metallothionein, although not ZiP, gene expression. Modulation of intracellular Ca2+ and insulin secretion with pharmacological agents (tolbutamide and diazoxide) suggested a possible role for changes in these parameters in the regulation of Slc39a6 and Slc39a7 but not Slc39a8, nor metallothionein expression. In summary, 1) glucose induces increases in [Zn2+]cyt, which are then likely to facilitate the processing and/or the storage of insulin and its cocrystallization with Zn2+, and 2) these increases are associated with elevated expression of zinc importers. Conversely, a chronic increase in [Zn2+]cyt following sustained hyperglycemia may contribute to β-cell dysfunction and death in some forms of diabetes.
Keywords: Diabetes, Fluorescence Resonance Energy Transfer (FRET), Gene Expression, Pancreatic Islets, Zinc, Glucose Stimulation, Insulin Signaling, Zinc Homeostasis, Zinc Transporters, ZnT8
Introduction
The total Zn2+ content of the mammalian pancreas is high, and these ions are chiefly localized to the islet β-cell (1). Correspondingly, Zn2+ plays an important role in both insulin synthesis and storage (2–3). Indeed, total Zn2+ concentrations reach millimolar levels in the interior of the dense-core granule (4), where two Zn2+ ions coordinate six insulin monomers to form the hexameric structure on which insulin crystals are based (5). The physiological importance of Zn2+ homeostasis within the β-cell in man has been emphasized recently by the finding that a non-synonymous polymorphism (rs13266634 C/T) in the SLC30A8 gene, encoding the largely pancreatic, endocrine-restricted, granular zinc transporter ZnT8, is associated with an increased risk of type 2 diabetes (T2D)2 (6–9). Through the generation of mice bearing null alleles, we (10–11) and others (12) have shown previously that the absence of ZnT8 leads to the formation of amorphous β-cell secretory granules and mild glucose intolerance, consistent with a role for abnormal β-cell Zn2+ homeostasis in the pathogenesis of T2D (10–12).
A link between the zinc status of the body and diabetes has been known for many years (1). Several reports have shown that T2D patients display a marked decrease in total plasma Zn2+ and hyperzincuria compared with control subjects (13), suggesting the possibility that hyperglycemia may interfere with Zn2+ absorption in the kidney. Despite the consistent decrease in plasma Zn2+ observed across studies, the levels (total and free) of the ion within tissues (including muscle, kidney, and liver) are controversial (14–15). Reduced pancreatic Zn2+ has also been reported in a genetic model of T2D (16–18). However, no differences in the ultrastructural localization of Zn2+ were noted in β-cells from rats with monogenic (fa/fa Zucker) or more complex polygenic (Goto-Kakizaki) forms of diabetes (19).
Relatively little is known about how Zn2+ homeostasis is achieved in pancreatic β-cells and whether, and under what circumstances, changes in cytosolic Zn2+ ([Zn2+]cyt) may occur in these cells. When measured with a synthetic, intracellularly trappable probe (Zinquin), values for free [Zn2+] recorded from whole pancreatic islets were decreased after infusion of high glucose (20). However, the subcellular localization of this particular probe (and others of the same class) (21) is unclear, as it tends to compartmentalize to undefined granular structures (11). The above findings (20) are thus difficult to interpret in terms of the alterations that may occur at the molecular level to perturb intracellular Zn2+ homeostasis as glucose concentrations increase.
Using a newly developed, genetically encoded FRET-based probe (eCALWY-4), we have recently measured free [Zn2+]cyt in the β-cell line INS-1 (832/13) and found that it is buffered at 400 pm at low glucose concentrations (22). To tightly maintain this low free level of Zn2+, β and other cell types deploy a variety of mechanisms, including zinc transporters and binding proteins (23). There are two main classes of Zn2+ transporters. The ZnT family (coded by the Slc30a1–10 genes) is thought to move the ion from the cytosol to intracellular compartments or into the extracellular space (24). The ZiP (Zrt- and Irt-like proteins) family (coded by the Slc39a1–14 genes), on the other hand, is responsible for increases in cytosolic Zn2+ (25). Evidence for the expression of these transporters in the pancreas is quite limited, although most studies agree on the fact that Slc39a6 and Slc39a7, among the Zips, and Slc30a5 and Slc30a8, among the ZnTs, are the most abundant transporters in pancreatic islets (26–27), with other evidence suggesting that ZnT1 and ZnT4–9 are also additionally expressed (11, 28). Importantly, Zn2+ can also be transported into β-cells via voltage-gated L-type Ca2+ channels (29) opened upon glucose-induced depolarization of the plasma membrane (30).
Metallothioneins (Mt) are redox-active Zn2+ binding proteins with a dual function, playing a role in both Zn2+ homeostasis and in the regulation of the cellular redox state. In the latter context, these proteins release Zn2+ in response to oxidative damage (31), a condition often found in the tissues of T2D patients, probably reflecting chronic hyperglycemia (32).
The expression in the β-cell of the above array of proteins involved in Zn2+ homeostasis argues for the importance of tight control of cytosolic Zn2+ levels. Indeed, Zn2+ plays a role not only in the defense of β and other cells against oxidative damage (as part of the CuZn superoxide dismutase (33) and metallothioneins (34)), but may mediate apoptosis if the free concentrations of the ion decrease or increase above a safe set-point (35–36).
In the present study we show firstly, by imaging free [Zn2+]cyt in primary β-cells with eCALWY-4 (22), that glucose is able to induce a stable increase in the cytosolic concentration of these ions. Secondly, we show that high glucose leads to profound alterations in the expression of genes important for Zn2+ homeostasis in β-cells and explore the signaling mechanisms involved.
EXPERIMENTAL PROCEDURES
Animals
CD1 mice were housed with five mice per cage in a pathogen-free facility and were fed ad libitum with a standard mouse chow diet. Female mice were used at 10–12 week of age and were sacrificed by cervical dislocation as approved by the United Kingdom Home Office Animal Scientific Procedures Act, 1986.
Reagents
RPMI medium was obtained from Sigma. TRIzol reagent was from Invitrogen. ZnCl2, forskolin, 3-isobutyl-1-methylxanthine, tolbutamide, diazoxide, verapamil, N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine (TPEN), and 2-mercaptopyridine N-oxide (pyrithione) were from Sigma.
Islet Isolation and Dissociation into Single Cells
Pancreatic mouse islets were prepared as described elsewhere (37). The islets were allowed to recover overnight in culture medium (RPMI supplemented with 10% (v/v) heat-inactivated FCS, 2 mm glutamine, 100 units/ml penicillin, and 100 units/ml streptomycin). Islets were dissociated by 10-min incubation in Hanks'-based enzyme-free cell dissociation buffer (GIBCO, Invitrogen), before centrifugation and gentle pipetting (37). Dissociated cells were plated onto 24-mm sterile coverslips and allowed to recover overnight.
Generation of eCALWY-4-expressing Adenovirus and Infection of Primary Cells
eCALWY-4 and eCALWY-NB (non-binding) fragments (22) were PCR-amplified (forward, CTCGAGCGCCACCATGGGCCATAT; reverse, TCTAGAGGCCGCTTTACTTGTACAGCT) and cloned into the pCR2.1-TA vector (Invitrogen). The fragments were excised and cloned into plasmid pShuttle using Xho1/Xba1. Adenoviral constructs were generated as described elsewhere (38). Briefly, pShuttle-eCALWY constructs were digested with Pme1 and electroporated into competent BJ5183-AD-1 cells. Recombined pADeasy1 clones were screened, and positive clones were then digested with Pac1 and transfected into HEK293 cells for the generation of adenoviral particles. Primary islet cells were infected with eCALWY-4-expressing adenovirus (20–25 multiplicity of infection) for 4 h. The medium was then changed, and cells were allowed to express the protein for 48 h before imaging. Either 2 or 24 h before imaging, cells were incubated with different concentrations of glucose. These cultures typically contained 70–80% β-cells (39–40), and we confirmed the identity of individual β-cells by confocal microscopy (see Fig. 1B) in a limited number of experiments. Briefly, after infection with eCALWY-4-expressing adenovirus, primary cells were fixed in 3.7% paraformaldehyde and permeabilized in 0.1% Triton X-100 before immunostaining with a polyclonal anti-swine insulin antibody (1:200, DakoCytomation, Ely, UK) and an Alexa Fluor 568-coupled secondary antibody. Confocal imaging was performed as described elsewhere (40).
FIGURE 1.

Imaging the effect of elevated glucose on [Zn2+]cyt with eCALWY-4 in primary mouse β-cells. A, CD1 mouse islets were dissociated into single cells or clusters of cells, infected with eCALWY-4-expressing adenovirus, and plated on 24-mm coverslips. Cells were imaged with an Olympus IX microscope and excited at 433 nm, and the emission was recorded at 465 nm and 535 nm for cerulean and citrine, respectively (22). B, partially dissociated mouse islets seeded on converslips, were infected with eCALWY-4-expressing adenovirus, formaldehyde-fixed, permeabilized, and immunostained with anti-insulin antibody and Alexa Fluor 568-coupled secondary antibody, whereas DAPI was used for nuclear staining. Cell clusters containing insulin-positive cells were imaged by confocal microscopy. Scale bar = 10 μm. C, representative trace showing the changes in fluorescence ratio of a cell infected with eCALWY-4 adenovirus or eCALWY-nonbinding (D) and perifused with the indicated solutions. KRB, Krebs-Ringer buffer; TPEN, 50 μm; ZnPyr, 5 μm pyrithione plus 100 μm ZnCl2. E and F, analysis of free [Zn2+]cyt concentration after 2 and 24 h, respectively, of high glucose treatment. Bars represent mean ± S.E. Number of cell imaged (n) on three different days of experiments is given in brackets under each bar. ***, p < 0.001.
Imaging Free Cytosolic Zn2+
Cells were washed twice in Krebs Hepes-bicarbonate buffer (140 mm NaCl, 3.6 mm KCl, 0.5 mm NaH2PO4, 0.2 mm MgSO4, 1.5 mm CaCl2, 10 mm Hepes (pH 7.4), 2 mm NaHCO3), preequilibrated with 95:5 O2:CO2 and containing either 3 or 16.7 mm glucose. Zn2+ imaging was carried out as described by Vinkenborg et al. (22). Briefly, dissociated islet cells were maintained at 37 °C throughout with a heating stage (MC60, LINKAM, Scientific Instruments, Surrey, UK), and Krebs Hepes-bicarbonate buffer was perifused (1 ml/min) with additions as stated in Fig. 1C. 50 μm TPEN and 5 μm pyrithione were freshly prepared on each day of the experiments. Images were captured using an Olympus IX-70 widefield microscope with a 40× oil immersion objective and an Imago charge-coupled device camera (Till Photonics, Grafelfing, Germany) controlled by TILLvisION software (Till Photonics). For FRET measurements, a 455DRLP dichroic mirror (Chroma Technology) and two emission filters (Chroma Technology, D465/30 for cerulean and D535/30 for citrine), alternated by a filter changer (Lambda 10–2, Sutter Instruments), were used. Images were acquired at 1 Hz using a 100-ms exposure time and a 433-nm excitation wavelength. The fluorescence emission ratios were derived after subtracting background and calibrated for [Zn2+]cyt as described under “Results.”
RNA Extraction and Quantitative Real-time PCR
For RNA extraction, 100 islets were incubated for 1 h in RPMI (20 islets/ml) containing 3 mm glucose and subsequently transferred into a 60-mm Petri dish with culture medium containing either 3 or 16.7 mm glucose. Total RNA was obtained using TRIzol reagent (Invitrogen) and treated with a DNA-free kit (Applied Biosystems, Warrington, UK). Total RNA was then reverse-transcribed into cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems). cDNA (equivalent to 30 ng of RNA) was subject to quantitative real-time PCR using Power SYBR Green master mix (Applied Biosystems) in a 7500 fast real-time PCR system (ABI Biosystems) and analyzed by the comparative CT method. All real-time primers (supplemental Table 1) were generated using ABI Primer Express 3.0 software and were validated for specificity by dissociation curve (40).
Protein Extraction and Western Blotting (Immunoblotting) Analysis
For protein analysis, 250 (20 islets/ml of medium) islets were incubated for 1 h in RPMI containing 3 mm glucose before a further 24-h incubation with either 3 or 16.7 mm glucose. Islets were then washed twice in ice-cold PBS and lysed in ice-cold radioimmune precipitation assay buffer (50 mm Tris HCl (pH 8.0),150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) before sonication. Protein was assayed with a BCA kit (Pierce). Total protein extracts (30 μg) were resolved by SDS-PAGE (12% v/v acrylamide) and transferred to PVDF membranes, followed by immunoblotting with rabbit polyclonal anti mLIV-1 (ZiP6) and anti mKE4 (ZiP7, both used 1:200, Ref. 41), mouse monoclonal anti-metallothioneins (1:500, Abcam), and mouse monoclonal anti-tubulin (1:5000, Sigma clone B-5-1-2) antibodies. Secondary HRP-linked anti-rabbit antibodies (1:10000, GE Healthcare) were revealed by using ECL detection reagent (GE Healthcare). The intensity of the bands was quantified using ImageJ software, and the ratio between the intensity of target proteins and tubulin was determined.
Bioinformatic and Statistical Analysis
The promoter regions (-3000 to −1 bp upstream of the starting codon) of identified glucose-responsive genes were scanned and analyzed using Transcription Element Search System (TESS) software. Statistical significance was assessed by one or two-way analysis of variance or Student's two tailed t test with Bonferroni correction for multiple testing using GraphPad Prism and Excel software.
RESULTS
Elevated Glucose Promotes a Stable Increase in Cytosolic Free Zn2+ Concentrations in Primary β-Cells
Monitoring intracellular free Zn2+ in primary β-cells has so far only been achieved using synthetic chemical probes (11, 20). These sensors, however, suffer from limitations, such as a variable extent of compartmentalization into intracellular organelles, confounding both the measurements and their calibration. By generating an adenoviral expression construct based on our newly developed FRET-based Zn2+ probe, eCALWY-4 (22), we monitored cytosolic Zn2+ variations in primary β-cells and examined the time frames over which changes occurred. The ratio of citrine to cerulean emission (Fig. 1A) was plotted against time (C), and the minimum and maximum ratios were used to calculate the free [Zn2+]cyt using the following formula: [Zn2+] = Kd × (Rmax-R)/(R-Rmin), where the Kd for eCALWY-4 is 630 pm, Rmax represents the maximum fluorescence ratio (upon TPEN-mediated Zn2+ chelation), and Rmin is the minimum fluorescence ratio (obtained with 100 μm ZnCl2 in the presence of the Zn2+ ionophore, pyrithione, Ref. 22). Addition of TPEN or Zn2+-pyrithione did not affect the ratio of a non-binding sensor variant (Fig. 1D). Cells were incubated for 1 h at 3 mm glucose before application of medium containing either 3 or 16.7 mm glucose for 2 or 24 h. The free [Zn2+]cyt was calculated by averaging the concentration of the ion 1 min before application of TPEN. After 24 h of treatment, cells incubated at 16.7 mm glucose displayed a substantial and significant (p < 0.001) [Zn2+]cyt increase (853 ± 96 pm) compared with cells incubated at 3 mm glucose (452 ± 42 pm) (Fig. 1F). Although cytosolic Zn2+ concentrations were not significantly different after 2 h of incubation at high versus low glucose, a tendency toward an increase in the former case was observed (587 ± 101 pm at 16.7 mm glucose versus 496 ± 60 pm for cells incubated at 3 mm glucose, p = 0.431) (Fig. 1E), indicative of a time-dependent increase in [Zn2+]cyt in response to high glucose.
Regulation by Glucose of Genes Involved in Zn2+ Homeostasis
We hypothesized that the above variations in [Zn2+]cyt may, at least in part, reflect changes in the expression of genes known to regulate Zn2+ homeostasis in other cell types, namely ZiPs, ZnTs, and Mt. Thus, we measured the relative expression of the known mouse ZiPs (Slc39a), ZnT (Slc30a), and metallothionein (Mt) genes in pancreatic islets at varying glucose concentrations and at two time points. Firstly, after 1-h incubation at 3 mm glucose, mouse islets were cultured in medium supplemented with either 3 or 16.7 mm glucose for 24 h. Culture with 16.7 versus 3 mm glucose led to a significant increase in Slc39a6 (2.4-fold), Slc39a7 (2.4-fold), and Slc39a8 (6.8-fold) mRNA levels but a decrease in Mt-1 (3.2-fold) and Mt-2 (4-fold) gene expression (Fig. 2A). The change in ZiP6 expression was also significantly reflected at the protein level, as demonstrated by Western blotting (immunoblotting) analysis (Fig. 3). ZiP7 protein expression showed a tendency to increase in response to high glucose (Fig. 3A), although the difference was very small and did not reach significance (B). We were unable to perform immunoblot analysis on ZiP7 because of the lack of a specific antibody against the mouse protein. Immunoblot analysis of metallothioneins revealed no difference at high versus low glucose concentrations (data not shown). In contrast, no changes in mRNA levels were observed in the expression of any of the tested Slc30a (ZnT) family members in response to variations in glucose concentration (Fig. 2A).
FIGURE 2.

Effect of elevated glucose concentrations on the expression of the genes implicated in Zn2+ homeostasis in mouse pancreatic islets. Mouse islets were incubated for 1 h at 3 mm glucose before incubation for 24 h (A) or 2 h (B) with either 3 mm (white bars) or 16.7 (black bars) glucose. Total RNA was extracted, and qPCR analysis of Slc39a1–12, Slc30a1–10, Mt-1, and Mt-2 was performed. The mRNA levels were normalized to those of a housekeeping gene (cyclophilin A). Bars represent mean ± S.E. (n = 4). *, p < 0.05.
FIGURE 3.

Effect of elevated glucose concentrations on the expression of proteins implicated in Zn2+ homeostasis in mouse pancreatic islets. A, total cell lysates were loaded onto 12% SDS-PAGE, which was subsequently transferred onto a PVDF membrane (see “Experimental Procedures”). The membrane was blotted for ZiP6 (1:200), ZiP7 (1:200), and β-tubulin (1:1000) and with an HRP-lined anti-rabbit (1:5000) secondary antibody. B, quantification of three different immunoblot analyses for ZiP6 and ZiP7. The same area of interest was drawn around the specific bands for ZiP, ZiP7 as well as the corresponding tubulin band, and the intensity was measured using ImageJ software. The ratio of intensity between ZiP or ZiP7 signals and tubulin were plotted. *, p < 0.05.
To test whether the changes in cytosolic free Zn2+ observed (Fig. 1F) upon high glucose challenge were the consequence or the cause of the above changes in gene expression (Fig. 2A), further qPCR analyses were performed after 2 h of treatment (well before the [Zn2+]cyt increase reached statistically significant level (Fig. 1E)). This analysis was restricted to the genes that showed a difference when treated with high versus low glucose after 24 h. Significant changes of between 1.8- and 2.1-fold were observed at this earlier time point in Slc39a7 and Mt-2 expression, respectively, whereas both Slc39a6 and Slc39a8 showed a tendency to increase (and Mt-1 to decrease) at 16.7 versus 3 mm glucose (Fig. 2B).
The above findings suggested that the glucose-induced variations in gene expression were more likely to contribute to, rather than be the consequences of, changes in [Zn2+]cyt. To further explore this hypothesis, we exposed pancreatic islets to medium containing different glucose concentrations (as above), supplemented or not with 50 μm ZnCl2 for 24 h, to increase intracellular free Zn2+. To confirm that the concentration of extracellularly applied ZnCl2 was indeed able to induce an increase in intracellular free Zn2+, even in the absence of high glucose, we measured [Zn2+]cyt after incubation of dissociated islets with 3 mm glucose in the presence of 50 μm ZnCl2 for 24 h. As shown in Fig. 4A, extracellular ZnCl2 elevated free [Zn2+]cyt to a level comparable with that observed after incubation at high glucose concentrations (903 ± 89 pm versus 855 ± 138 pm). As anticipated (42), ZnCl2 supplementation strongly induced the metallothionein genes while decreasing the expression of the Slc39a genes tested (Fig. 4B). Thus, the increase in cytosolic free Zn2+ observed after 24 h of incubation with 16.7 mm glucose would appear likely to be a consequence of the changes in the expression of genes involved in controlling intracellular Zn2+ fluxes and binding.
FIGURE 4.

Effect of extracellular Zn2+ on [Zn2+]cyt and on the expression of Slc39a6, Slc39a7, Slc39a8, Mt-1, and Mt-2. A, dissociated mouse islets were infected with eCALWY-4-expressing adenovirus and subsequently incubated for 24 h in medium containing 16.7 mm glucose or 3 mm glucose with or without 50 μm ZnCl2 before imaging on an Olympus IX microscope. [Zn2+]cyt was calculated as described under “Results” and in the legend to Fig. 1. Bars represent mean ± S.E. The number of cells imaged (n) on three different days of experiments is given in brackets under each bar. **, p < 0.01; ***, p < 0.001. B, mouse islets were incubated for 1 h at 3 mm glucose before culture at either 3, 10, or 16.7 mm glucose for 24 h in the presence (black bars) or absence (white bars) of 50 μm ZnCl2. qPCR analysis of Slc39a6, Slc39a7, Slc39a8, Mt-1, and Mt-2 was performed. Values of expression were normalized to that of a housekeeping gene (cyclophillin A). The fold changes compared with the values obtained at 3 mm glucose are plotted. Bars represent mean ± S.E. (n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Effect of L-type Voltage-gated Ca2+ Channel (VGCC) Inhibition on Glucose-mediated [Zn2+]cyt Increase
It is known that influx of Zn2+ from the extracellular space can be mediated by voltage-gated Ca2+ channels, opened after glucose entry has caused KATP channels closure and membrane depolarization (29). To test whether the increase of [Zn2+]cyt reported here was via the glucose-induced opening of VGCCs, we measured intracellular Zn2+ levels after dissociated islets were exposed for 24 h to 16.7 mm glucose alone or with the addition of 20 μm verapamil, a concentration known to block the VGCCs in pancreatic β-cells (43). Despite the presence of the VGCC blocker, high glucose concentrations were still able to induce an increase in free [Zn2+]cyt to levels comparable with that found in the presence of 16.7 mm glucose alone (923 ± 137 pm for verapamil versus 855 ± 138 pm for the control condition, Fig. 5), suggesting that L-type Ca2+ channels do not play a pivotal role in the rise of cytosolic Zn2+ (Fig. 5).
FIGURE 5.

Effect of an L-type voltage-gated Ca2+ channel blocker on glucose-induced [Zn2+]cyt increase. Dissociated mouse islets were infected with eCALWY-4 expressing adenovirus before incubation with 3 or 16.7 mm glucose with or without 20 μm verapamil for 24 h before imaging on an Olympus IX microscope. [Zn2+]cyt was then calculated as explained under “Results” and in Fig. 1. Bars represent mean ± S.E. The number of cells imaged (n) on three different days of experiments is given in brackets under each bar. **, p < 0.01; ***, p < 0.001.
Regulation of Slc39a6, Slc39a7, and Slc39a8, and Mt-1 and Mt-22, by Intracellular cAMP
To examine the potential signaling mechanisms responsible for glucose-evoked changes in gene expression, we first investigated whether increased cAMP levels, normally elevated in β-cells exposed to high glucose concentrations (44–46), might mimic the effect of the sugar. Suggesting this as a possibility, in silico examination of the mouse Slc39a6, Slc39a7, Slc39a8, Mt-1, and Mt-2 genes revealed consensus binding sites for the cAMP responsive element binding protein (47) in the promoter regions of all the genes examined, with the exception of Mt-2 (supplemental Table 2). Accordingly, pancreatic islets were incubated in 3, 10, or 16.7 mm glucose in the presence or absence of the adenylate cyclase activator forskolin and the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (Fig. 6). Metallothionein expression was negatively modulated by elevation in cytosolic cAMP concentrations at 3 mm glucose, mimicking the effect of high glucose. Correspondingly, a cAMP responsive element binding protein consensus binding site was apparent in the Mt-1 promoter (supplemental Table 2). The Mt-2 promoter, however, contained no such site, indicating that cAMP elevation might act via other mechanisms than those involving the cAMP responsive element binding protein. By contrast, elevated glucose (16.7 mm), in combination with increased cAMP, tended to decrease the expression of the Slc39a genes examined, a trend that was significant in the case of Slc39a7. At low glucose concentrations, on the other hand, cAMP elevations failed to increase Slc39a6–8 gene expression (Fig. 6). Thus, increases in intracellular cAMP appear unlikely to explain the enhanced expression of Slc39a6, Slc39a7, and Slc39a8 in response to high glucose.
FIGURE 6.

Effect of elevated intracellular cAMP levels on Slc39a6, Slc39a7, Slc39a8, Mt-1, and Mt-2 expression. Mouse islets were incubated for 1 h at 3 mm glucose before incubation at either 3,10, or 16.7 mm glucose for 24 h in the presence (black bars) or absence (white bars) of 50 μm 3-isobutyl-1-methylxanthine and 10 μm forskolin. qPCR analysis of Slc39a6, Slc39a7, Slc39a, Mt-1, and Mt-2 was performed. Values of expression were normalized to that of cyclophillin A. The fold changes compared with the values at 3 mm glucose were plotted. Bars represent mean ± S.E. (n = 4). **, p < 0.01; ***, p < 0.001.
Effect of Sulfonylureas and Diazoxide on Gene Expression
Insulin release and rebinding to β-cell insulin receptors is believed to be responsible for the regulation by glucose of several genes in this cell type (48–49). To determine whether such a mechanism may be involved in the regulation of genes involved in Zn2+ homeostasis, we stimulated or blocked insulin secretion pharmacologically using the KATP channel modulators tolbutamide or diazoxide, respectively. The latter agent opens KATP channels and thus prevents cell depolarization, elevation in cytosolic Ca2+, and, therefore, insulin secretion (50). Pancreatic islets were incubated for 1 h at 3 mm glucose before further incubation with either 3 or 16.7 mm glucose for 24 h in the presence (or absence) of tolbutamide (51) or diazoxide (52). Despite showing no significant difference compared with the control case (supplemental Fig. 1), incubation with tolbutamide abolished the effects of glucose on the expression of Slc39a6-8. As shown in supplemental Fig. 1, tolbutamide, which promotes cytosolic Ca2+ increases and insulin secretion even in the absence of glucose metabolism, tended to evoke an increase in Slc39a6 and Slc39a7 expression even at low glucose concentrations, thereby at least partially mimicking the effects of incubation in high glucose (supplemental Fig. 1). Similarly, diazoxide eliminated the effect of the sugar to stimulate Slc39a6 and Slc39a7 gene expression (Fig. 7). Diazoxide also tended to decrease the expression of these genes, compared with the control case, at high glucose (Fig. 7). Surprisingly, perhaps, metallothionein gene expression was unchanged in the presence of either of these pharmacological agents. Slc39a8 expression was strongly up-regulated in the presence of diazoxide in combination with high glucose (Fig. 7).
FIGURE 7.

Effect of the KATP channel opener diazoxide on gene expression. CD1 mouse islets were incubated for 1 h at 3 mm glucose before a further 24 h in the presence of either 3 mm (white bars) or 16.7 mm (black bars) glucose in the presence or absence of 325 μm diazoxide. RNA was extracted, and Slc39a6, Slc39a7, Slc39a8, Mt-1, and Mt-2 gene expression was analyzed by qPCR. Values were normalized to the one of cyclophillin A, and the fold changes compared with control experiments at 3 mm glucose were plotted. Bars represent mean ± S.E. (n = 3). *, p < 0.05.
Effect of Diazoxide on Cellular Stress and Free [Zn2+]cyt
Because Slc39a8 was very strongly up-regulated in the presence of high glucose plus diazoxide, we questioned whether this was due to an increase in cellular oxidative stress. Thioredoxin-interactin protein TxNIP participates in the control of the redox state of the cell by inhibiting thioredoxin (53) and has been identified as one of the most glucose-inducible genes in human and rodent islets (54). We therefore measured the levels of TxNIP expression in response to glucose in the presence or absence of diazoxide. As expected (54) and shown in Fig. 8A, high glucose increased the level of TxNIP expression by about 3.3-fold. However, when islets were incubated with high glucose in the presence of diazoxide, the expression of TxNIP was 25 times higher than in control conditions, suggesting an increase in oxidative stress. We then measured free [Zn2+]cyt in the same conditions and found that the concentrations were significantly (p < 0.05) higher in the presence of high glucose and diazoxide than in the presence of high glucose alone (Fig. 8B). These data correlate with the increased expression of ZiP8 (Fig. 7) observed in the presence of high glucose and diazoxide.
FIGURE 8.

Effect of the KATP channel opener diazoxide on [Zn2+]cyt and the expression of the marker of cellular stress, TxNIP. A, mouse islets were incubated for 24 h in the presence of either 3 or 16.7 mm glucose supplemented (black bars) or not (white bars) with 325 μm diazoxide before RNA extraction and analysis of TxNIP gene expression by quantitative real-time PCR. Values were normalized to the one of cyclophillin A, and the fold changes compared with control experiments at 3 mm glucose were plotted. Bars represent mean ± S.E. (n = 3). *, p < 0.05. B, dissociated islets were infected with eCALWY-4-expressing adenovirus for 24 h and subsequently incubated for a further 24 h supplemented with 3 or 16.7 mm glucose in the presence or absence of 325 μm diazoxide before imaging on an Olympus IX microscope. [Zn2+]cyt was then calculated as explained under “Results” and in the legend to Fig. 1. The number of cells imaged (n) on three different days of experiments is given in brackets under each bar. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
DISCUSSION
The overall aim of this study was to test the hypothesis that glucose may stably regulate the free concentration of Zn2+ within the pancreatic β-cell and to explore the underlying mechanisms behind any observed changes. To address the first point, we generated an adenovirus to express our recently developed FRET probe for free cytosolic Zn2+ (22). This tool has allowed us, for the first time, to report, with high selectivity, changes in cytosolic free Zn2+ concentration within primary β-cells and to study its regulation by glucose and other agents. We show that the resting cytosolic Zn2+ concentrations (∼400 pm) are similar to those we have recently shown to pertain in clonal INS-1 (832/13) β-cells (22). This value is also in line with measurements made in other mammalian cells according to reports by many groups (55–57) but not others (58–59). Interestingly, we discovered that [Zn2+]cyt substantially and progressively increases in response to prolonged (24-h) exposure to high glucose levels in mouse pancreatic β-cells. We also show that these changes begin as early as 2 h after the increase in glucose concentration, in line with earlier results that demonstrate Zn2+ influx into mouse islet cells over shorter time frames (up to 15 min) using trappable chemical probes (29). Each of the above findings is in contrast, however, with earlier reports by Zalewski et al. (20), who observed a decrease in intracellular free Zn2+ in islet cells in response to the sugar. However, these earlier measurements were performed using whole islets and a synthetic probe (Zinquin) that localizes, at least partly, to dense core granules and other membrane-bound organelles, where the free concentration of the ion may be much higher than in the cytosol (11). Thus, the decrease in apparent free Zn2+ observed earlier may be due to confounding factors such as degranulation of the cells in response to glucose. Interestingly, when we blocked the L-type Ca2+ channel with verapamil, we found that the increase in free [Zn2+]cyt was comparable with that observed in the presence of high glucose alone, indicating that the flux of Zn2+ via the VGCC plays, in this case, a minor role.
We also found that the increase in cytosolic free Zn2+ concentration reported with eCALWY-4 was associated with increased expression of one of the key regulators of intracellular Zn2+ homeostasis, namely ZiP6, both at the mRNA and protein level. Furthermore, we demonstrated an increase in ZiP7 and ZiP8 mRNA expression, although the lack of a suitable specific antibody against the latter precluded confirmation of such change at the protein level. ZiP7 immunoreactivity displayed a small increase at high glucose concentrations, but upon quantification, these changes were not significant. It should be emphasized that the analysis of gene expression was performed on whole pancreatic islets. These structures contain a mixed cell population (α, β, Δ and pancreatic polypeptide producing cells (60)) so that changes in gene expression may conceivably reflect alterations in any of these cell types. However, given that β-cells represent 60–80% of all islet cells, it is nonetheless likely that changes in the latter cell type predominate.
In line with our findings here, ZiP8 overexpression elicited by tissue necrosis factor-α (TNF-α) induces an increase in intracellular Zn2+, measured with FluoZin-3 in lung epithelial cells (61). Moreover, down-regulation of ZiP6 is associated with diminished free cytosolic Zn2+, measured with the synthetic probe Newport green dichlorofluorescein, in dendritic cells (62). Both ZiP6 and ZiP8 localize to the plasma membrane of different cell types (61, 63, 64), although in T-cells (65), ZiP8 staining colocalized with lysosomal markers. ZiP7, on the other hand, is mainly localized to the Golgi apparatus (41), indicating that the observed increase in cytosolic free Zn2+ may result not only from enhanced influx of the ion from the extracellular space but also to release from (non secretory granule) intracellular compartments.
Surprisingly, perhaps, we did not observe any change in the expression of ZnT8/Slc30a8, the most abundant zinc transporter expressed in mouse pancreatic β-cells (10–11, 26), at high glucose concentrations, at least at the time points and under the culture conditions deployed here. Moreover, a recent report indicated that ZnT3 is expressed in pancreatic β-cells and that its expression increases in response to glucose (66). In contrast to these earlier findings but in line with Ref. 11, we failed here to observe detectable expression of the ZnT3-coding gene Slc30a3 or an increase in expression following glucose stimulation in primary β-cells. This might be explained by the fact that in the study by Smidt et al. (66), 4-week-old BALB/CA male mice were used, as opposed to the 12-week-old female CD1 mice used in the present study.
Despite the fact that we were unable to detect any changes at the protein level, the decrease in mRNA levels observed after 24 h at high glucose concentrations suggested a link between glucose metabolism and Mt expression.
A protective role for metallothioneins in diabetes has been suggested earlier from the observation that overexpression of these proteins in response to Zn2+ could attenuate streptozocytin-induced diabetes in mice (67) and may reflect the fact that they are able to increase the oxidative defense of pancreatic β-cells. The observation that an elevation in cAMP induces a decrease in metallothionein expression suggests that this might be the pathway through which glucose modulates Mt expression in pancreatic β-cells. However, such a mechanism would be in contrast to what is observed in adipocytes, where high levels of cAMP are correlated with increased metallothionein expression (68).
A previous study using oligonucleotide microarrays (69) has shown that Slc39a6 and Mt-1a expression are altered at the mRNA level in rat islets after 96 h of culture at varying glucose concentrations, but earlier time points were not examined. This study extends these earlier results and begins to dissect the intracellular signaling mechanisms that lie behind the glucose-induced changes in the expression of key regulators of intracellular Zn2+ homeostasis in the pancreatic β-cell. A model summarizing the changes that we report here is presented in Fig. 9. Whether altered zinc importer and metallothionein expression reflect alterations in the transcription of the corresponding genes or in mRNA (or protein) stability, or combinations of the above, were not addressed in this study and will require further detailed investigation in future.
FIGURE 9.
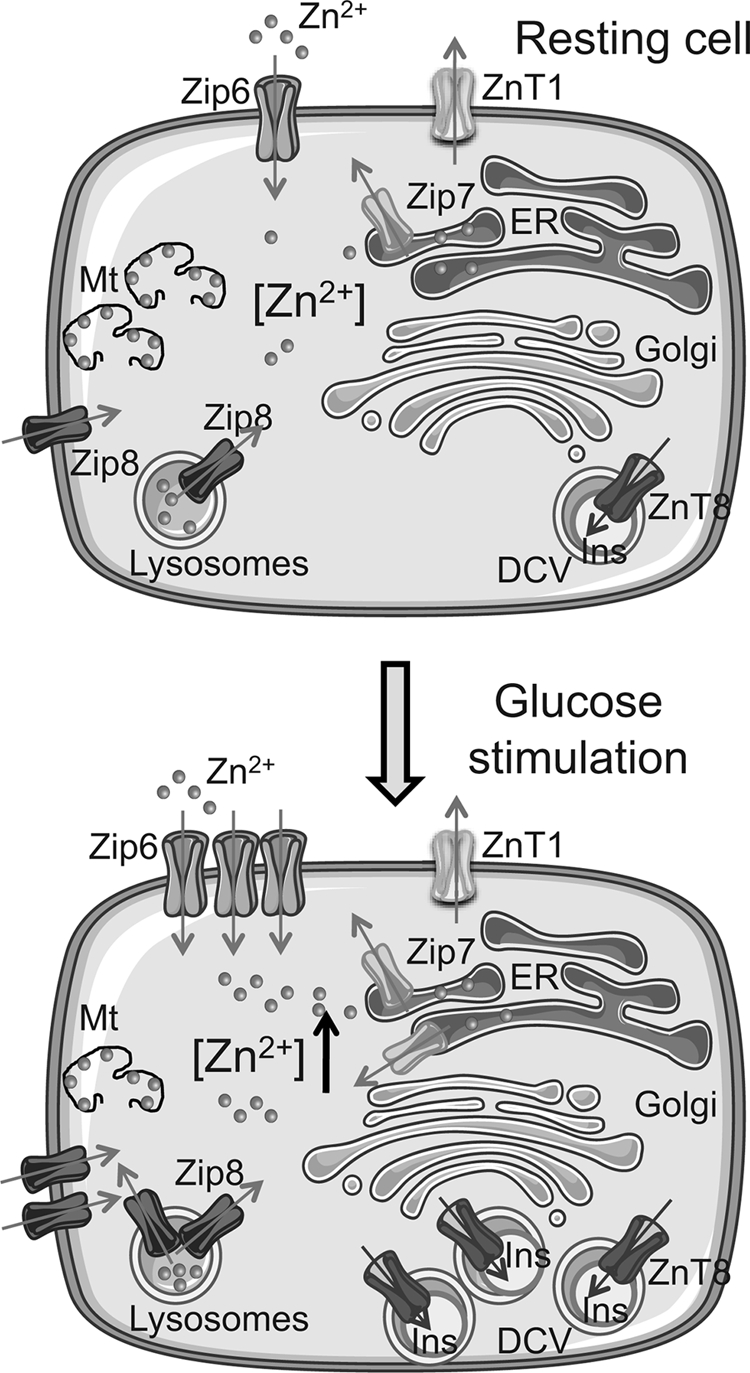
Proposed model of the effect of glucose on the regulation of Zn2+ homeostasis in pancreatic β-cells. Shown are glucose-induced increases in the levels of ZiP6 and ZiP7, likely to be both at the plasma membrane and intracellular membranes. ZiP8 levels may conceivably be increased both on lysosomes and plasma membrane. Metallothionein protein levels were unchanged at 24 h but seem likely to decrease at later time points following substantial decrease in Mt-1 and Mt-2 mRNA. Please see the Discussion for other details. The image was generated using Servier Medical Art.
Nonetheless, our findings implicate an unsuspected role for cAMP in the control by glucose of Mt-1 and Mt-2 mRNA expression, whereas an increase in cytosolic (and possibly nuclear) Zn2+ is unlikely to be involved in (and would rather be expected to antagonize) the regulation by glucose of these genes. On the other hand, a role for cAMP in the control of Slc39a6–8 expression by glucose seems unlikely, at least on the basis of the impact of a single large increase in the concentrations of intracellular cAMP, as achieved here by pharmacological means. Moreover, our results suggest that glucose might act via different mechanisms in each case. Either tolbutamide or diazoxide (which respectively mimic and reverse the effects of glucose on intracellular free Ca2+, thus affecting insulin secretion) block the effects of the sugar on Slc39a6 and Slc39a7, while exerting no effect on the action of glucose on the Mt-1 and Mt-2 mRNA level. The former result strongly implies a role for cytosolic Ca2+ increases in the action of the sugar on Slc39a6 and Slc39a7 expression. Although the effects downstream of Ca2+ may conceivably be mediated by enhanced insulin secretion and an autocrine (or paracrine) effect via β-cell insulin receptors (70), future studies will be required to explore this hypothesis.
Surprisingly, the expression of Slc39a8 increased enormously in the presence of high concentrations of glucose and diazoxide. In the same conditions, free [Zn2+]cyt was significantly higher than in the presence of high glucose alone. Although these findings seem likely to reflect an increase in oxidative stress, they also indicate that ZiP8 might play a pivotal role in modulating zinc homeostasis in pancreatic β-cells. We know that insulin secretion is correlated with zinc release (10), whose concentration will then rise locally close to the cell surface (71) and might therefore enter the cell via the L-type Ca2+ channel or ZiP proteins on the plasma membrane. However, in the presence of diazoxide, KATP channels are open, and therefore glucose is unable to stimulate insulin/Zn2+ release. Also, in the same conditions, we failed to observe an increase in the ZiP6 mRNA level. Despite these observations, we still noticed an increase in free [Zn2+]cyt, which may suggest an important role for ZiP8 in the glucose-induced increases in [Zn2+]cyt. ZiP8 has been found to be expressed both at the plasma membrane and on intracellular organelles, including lysosomes, in other cell types (65). Although similar analyses have yet to be performed in pancreatic β-cells, and we were unable to undertake an immunocytochemical analysis for ZiP8 because of the lack of a suitable antibody, we speculate that the increase in cytosolic Zn2+ observed might be due not only to the influx of the ion from the extracellular space but also to the release from intracellular compartments.
What may be the significance of the glucose-induced increase in cytosolic Zn2+ concentrations? We observed an initial increase in Slc39a6-8 and a decrease in Mt-1 and Mt-2 after just 2 h of exposure to elevated (16.7 mm) glucose, indicating that the up-regulation of these genes represents a relatively early event in response to the sugar and may be required for the enhanced synthesis or storage of insulin under these conditions (3). Moreover, it has been demonstrated that glucose can promote, via the polypirimide tract binding protein PTB1, the biogenesis of insulin-containing dense-core granules (72). However, although cells are able to synthesize insulin and other granule proteins de novo, this is not the case for Zn2+. Hence, the uptake (or reuptake) of Zn2+ ions is likely to be required to maintain adequate intracellular (and, as a consequence, intragranular) Zn2+ levels. Activation of the machinery required to achieve this (and, in the case of metallothioneins, a deactivation of that which would oppose the local bioavailability of the ions through its chelation in the cytosol) would appear to represent an important system for dealing with the need for enhanced insulin synthesis, storage, and secretion (Fig. 9). Emphasizing the importance of granular Zn2+, we have recently shown that mice lacking the granule-specific zinc transporter ZnT8 have reduced intragranular Zn2+, which is associated with defective insulin crystallization and processing in vivo (11, 73). These data confirmed previous observations made on a transgenic mouse bearing a mutant form of proinsulin (His-B10-Asp) unable to bind Zn2+, which showed similar defect in insulin storage (74). However, if hyperglycemia persists, as in the case of diabetes, then it is conceivable that elevated cytosolic (and possibly organellar) Zn2+ levels may contribute to failures in β-cell signaling and the acute regulation of insulin release and might lead to irreversible cell failure or death through apoptosis (35). Indeed, earlier studies (75) and our own unpublished observations3 indicate that elevations in intracellular free Zn2+ inhibit insulin secretion and lead to enhanced apoptosis.
Although high glucose induces insulin secretion, it also known to induce oxidative stress, as observed by others (76) and assessed by us with the measurement of expression of TxNIP. This change results in the suppression of the reducing activity of thioredoxin, which is expected in turn to prompt in an increase in reactive oxygen species generation and will therefore contribute further to oxidative stress. Enhanced oxidative stress might then prompt metallothioneins to release bound Zn2+, therefore increasing [Zn2+]cyt even more. This, together with the lowering of metallothionein expression, may further compromise β-cell survival at later time points. Although further studies are needed to assess the relative importance of changes in cytosolic [Zn2+] in the short and long term, it seems reasonable to speculate that tight control of free Zn2+ concentrations in β-cells may provide a means to prevent the decline in functional β-cell mass, which characterizes T2D (77).
Supplementary Material
Acknowledgment
We thank Prof. L. Huang for kindly providing the mKE4 (ZiP7) and mLIV1 (Zip6) antibodies.
This work was supported by Wellcome Trust Programme Grant 081958/Z/07/Z and Servier IdS. This work was also supported by a Royal Society Wolfson Research merit award (to G. A. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1 and Tables 1 and 2.
M. K. Loder and G. A. Rutter, unpublished data.
- T2D
- type 2 diabetes
- [Zn2+]cyt
- cytosolic Zn2+
- Mt
- metallothionein(s)
- TPEN
- N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine
- pyrithione
- 2-mercaptopyridine N-oxide
- KATP
- ATP-dependent K+ channel
- VGCC
- voltage-gated Ca2+ channel.
REFERENCES
- 1. Scott D. A., Fisher A. M. (1938) J. Clin. Invest. 17, 725–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Emdin S. O., Dodson G. G., Cutfield J. M., Cutfield S. M. (1980) Diabetologia 19, 174–182 [DOI] [PubMed] [Google Scholar]
- 3. Hutton J. C., Bailyes E. M., Rhodes C. J., Rutherford N. G., Arden S. D., Guest P. C. (1990) Biochem. Soc. Trans. 18, 122–124 [DOI] [PubMed] [Google Scholar]
- 4. Foster M. C., Leapman R. D., Li M. X., Atwater I. (1993) Biophys. J. 64, 525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hill C. P., Dauter Z., Dodson E. J., Dodson G. G., Dunn M. F. (1991) Biochemistry 30, 917–924 [DOI] [PubMed] [Google Scholar]
- 6. Saxena R., Voight B. F., Lyssenko V., Burtt N. P., de Bakker P. I., Chen H., Roix J. J., Kathiresan S., Hirschhorn J. N., Daly M. J., Hughes T. E., Groop L., Altshuler D., Almgren P., Florez J. C., Meyer J., Ardlie K., Bengtsson Boström K., Isomaa B., Lettre G., Lindblad U., Lyon H. N., Melander O., Newton-Cheh C., Nilsson P., Orho-Melander M., Råstam L., Speliotes E. K., Taskinen M. R., Tuomi T., Guiducci C., Berglund A., Carlson J., Gianniny L., Hackett R., Hall L., Holmkvist J., Laurila E., Sjögren M., Sterner M., Surti A., Svensson M., Tewhey R., Blumenstiel B., Parkin M., Defelice M., Barry R., Brodeur W., Camarata J., Chia N., Fava M., Gibbons J., Handsaker B., Healy C., Nguyen K., Gates C., Sougnez C., Gage D., Nizzari M., Gabriel S. B., Chirn G. W., Ma Q., Parikh H., Richardson D., Ricke D., Purcell S. (2007) Science 316, 1331–133617463246 [Google Scholar]
- 7. Scott L. J., Mohlke K. L., Bonnycastle L. L., Willer C. J., Li Y., Duren W. L., Erdos M. R., Stringham H. M., Chines P. S., Jackson A. U., Prokunina-Olsson L., Ding C. J., Swift A. J., Narisu N., Hu T., Pruim R., Xiao R., Li X. Y., Conneely K. N., Riebow N. L., Sprau A. G., Tong M., White P. P., Hetrick K. N., Barnhart M. W., Bark C. W., Goldstein J. L., Watkins L., Xiang F., Saramies J., Buchanan T. A., Watanabe R. M., Valle T. T., Kinnunen L., Abecasis G. R., Pugh E. W., Doheny K. F., Bergman R. N., Tuomilehto J., Collins F. S., Boehnke M. (2007) Science 316, 1341–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sladek R., Rocheleau G., Rung J., Dina C., Shen L., Serre D., Boutin P., Vincent D., Belisle A., Hadjadj S., Balkau B., Heude B., Charpentier G., Hudson T. J., Montpetit A., Pshezhetsky A. V., Prentki M., Posner B. I., Balding D. J., Meyre D., Polychronakos C., Froguel P. (2007) Nature 445, 881–885 [DOI] [PubMed] [Google Scholar]
- 9. Zeggini E., Weedon M. N., Lindgren C. M., Frayling T. M., Elliott K. S., Lango H., Timpson N. J., Perry J. R., Rayner N. W., Freathy R. M., Barrett J. C., Shields B., Morris A. P., Ellard S., Groves C. J., Harries L. W., Marchini J. L., Owen K. R., Knight B., Cardon L. R., Walker M., Hitman G. A., Morris A. D., Doney A. S., McCarthy M. I., Hattersley A. T. (2007) Science 316, 1336–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lemaire K., Ravier M. A., Schraenen A., Creemers J. W., Van de Plas R., Granvik M., Van Lommel L., Waelkens E., Chimienti F., Rutter G. A., Gilon P., in't Veld P. A., Schuit F. C. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 14872–14877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nicolson T. J., Bellomo E. A., Wijesekara N., Loder M. K., Baldwin J. M., Gyulkhandanyan A. V., Koshkin V., Tarasov A. I., Carzaniga R., Kronenberger K., Taneja T. K., da Silva Xavier G., Libert S., Froguel P., Scharfmann R., Stetsyuk V., Ravassard P., Parker H., Gribble F. M., Reimann F., Sladek R., Hughes S. J., Johnson P. R., Masseboeuf M., Burcelin R., Baldwin S. A., Liu M., Lara-Lemus R., Arvan P., Schuit F. C., Wheeler M. B., Chimienti F., Rutter G. A. (2009) Diabetes 58, 2070–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pound L. D., Sarkar S. A., Benninger R. K., Wang Y., Suwanichkul A., Shadoan M. K., Printz R. L., Oeser J. K., Lee C. E., Piston D. W., McGuinness O. P., Hutton J. C., Powell D. R., O'Brien R. M. (2009) Biochem. J. 421, 371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. el-Yazigi A., Hannan N., Raines D. A. (1993) Diabetes Res. 22, 67–75 [PubMed] [Google Scholar]
- 14. Cordova A. (1994) Biol. Trace Elem. Res. 42, 209–216 [DOI] [PubMed] [Google Scholar]
- 15. Raz I., Adler J. H., Havivi E. (1988) Diabetologia 31, 329–333 [DOI] [PubMed] [Google Scholar]
- 16. Kennedy M. L., Failla M. L. (1987) J. Nutr. 117, 886–893 [DOI] [PubMed] [Google Scholar]
- 17. Simon S. F., Taylor C. G. (2001) Exp. Biol. Med. 226, 43–51 [DOI] [PubMed] [Google Scholar]
- 18. Southon S., Kechrid Z., Wright A. J., Fairweather-Tait S. J. (1988) Br. J. Nutr. 60, 499–507 [DOI] [PubMed] [Google Scholar]
- 19. Søndergaard L. G., Stoltenberg M., Flyvbjerg A., Brock B., Schmitz O., Danscher G., Rungby J. (2003) APMIS 111, 1147–1154 [DOI] [PubMed] [Google Scholar]
- 20. Zalewski P. D., Millard S. H., Forbes I. J., Kapaniris O., Slavotinek A., Betts W. H., Ward A. D., Lincoln S. F., Mahadevan I. (1994) J. Histochem. Cytochem. 42, 877–884 [DOI] [PubMed] [Google Scholar]
- 21. Sensi S. L., Ton-That D., Weiss J. H., Rothe A., Gee K. R. (2003) Cell Calcium 34, 281–284 [DOI] [PubMed] [Google Scholar]
- 22. Vinkenborg J. L., Nicolson T. J., Bellomo E. A., Koay M. S., Rutter G. A., Merkx M. (2009) Nat. Meth. 6, 737–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liuzzi J. P., Cousins R. J. (2004) Annu. Rev. Nutr. 24, 151–172 [DOI] [PubMed] [Google Scholar]
- 24. Palmiter R. D., Huang L. (2004) Pfluegers Arch. 447, 744–751 [DOI] [PubMed] [Google Scholar]
- 25. Guerinot M. L. (2000) Biochim. Biophys. Acta 1465, 190–198 [DOI] [PubMed] [Google Scholar]
- 26. Chimienti F., Favier A., Seve M. (2005) Biometals 18, 313–317 [DOI] [PubMed] [Google Scholar]
- 27. Kambe T., Narita H., Yamaguchi-Iwai Y., Hirose J., Amano T., Sugiura N., Sasaki R., Mori K., Iwanaga T., Nagao M. (2002) J. Biol. Chem. 277, 19049–19055 [DOI] [PubMed] [Google Scholar]
- 28. Wijesekara N., Chimienti F., Wheeler M. B. (2009) Diabetes Obes. Metab. 11, 202–214 [DOI] [PubMed] [Google Scholar]
- 29. Gyulkhandanyan A. V., Lee S. C., Bikopoulos G., Dai F., Wheeler M. B. (2006) J. Biol. Chem. 281, 9361–9372 [DOI] [PubMed] [Google Scholar]
- 30. Ashcroft F. M., Proks P., Smith P. A., Ammälä C., Bokvist K., Rorsman P. (1994) J. Cell. Biochem. 55, 54–65 [DOI] [PubMed] [Google Scholar]
- 31. Maret W., Krezel A. (2007) Mol. Med. 13, 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lowell B. B., Shulman G. I. (2005) Science 307, 384–387 [DOI] [PubMed] [Google Scholar]
- 33. Crouch R. K., Gandy S., Patrick J., Reynolds S., Buse M. G., Simson J. A. (1984) Exp. Mol. Pathol. 41, 377–383 [DOI] [PubMed] [Google Scholar]
- 34. Chen H., Carlson E. C., Pellet L., Moritz J. T., Epstein P. N. (2001) Diabetes 50, 2040–2046 [DOI] [PubMed] [Google Scholar]
- 35. Bozym R. A., Chimienti F., Giblin L. J., Gross G. W., Korichneva I., Li Y., Libert S., Maret W., Parviz M., Frederickson C. J., Thompson R. B. (2010) Exp. Biol. Med. 235, 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim B. J., Kim Y. H., Kim S., Kim J. W., Koh J. Y., Oh S. H., Lee M. K., Kim K. W., Lee M. S. (2000) Diabetes 49, 367–372 [DOI] [PubMed] [Google Scholar]
- 37. Ravier M. A., Rutter G. A. (2005) Diabetes 54, 1789–1797 [DOI] [PubMed] [Google Scholar]
- 38. Luo J., Deng Z. L., Luo X., Tang N., Song W. X., Chen J., Sharff K. A., Luu H. H., Haydon R. C., Kinzler K. W., Vogelstein B., He T. C. (2007) Nat. Protoc. 2, 1236–1247 [DOI] [PubMed] [Google Scholar]
- 39. Elayat A. A., el-Naggar M. M., Tahir M. (1995) J. Anat. 186, 629–637 [PMC free article] [PubMed] [Google Scholar]
- 40. Meur G., Qian Q., da Silva Xavier G., Pullen T. J., Tsuboi T., McKinnon C., Fletcher L., Tavare J. M., Hughes S., Johnson P., Rutter G. A. (2011) J. Biol. Chem. 286, 13647–13656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang L., Kirschke C. P., Zhang Y., Yu Y. Y. (2005) J. Biol. Chem. 280, 15456–15463 [DOI] [PubMed] [Google Scholar]
- 42. Radtke F., Heuchel R., Georgiev O., Hergersberg M., Gariglio M., Dembic Z., Schaffner W. (1993) EMBO J. 12, 1355–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Szollosi A., Nenquin M., Henquin J. C. (2010) Br. J. Pharmacol. 159, 669–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Charles M. A., Fanska R., Schmid F. G., Forsham P. H., Grodsky G. M. (1973) Science 179, 569–571 [DOI] [PubMed] [Google Scholar]
- 45. Dyachok O., Idevall-Hagren O., Sågetorp J., Tian G., Wuttke A., Arrieumerlou C., Akusjärvi G., Gylfe E., Tengholm A. (2008) Cell Metab. 8, 26–37 [DOI] [PubMed] [Google Scholar]
- 46. Grill V., Cerasi E. (1973) FEBS Lett. 33, 311–314 [DOI] [PubMed] [Google Scholar]
- 47. Jansson D., Ng A. C., Fu A., Depatie C., Al Azzabi M., Screaton R. A. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10161–10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Da Silva Xavier G., Qian Q., Cullen P. J., Rutter G. A. (2004) Biochem. J. 377, 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leibiger B., Moede T., Schwarz T., Brown G. R., Köhler M., Leibiger I. B., Berggren P. O. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 9307–9312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bryan J., Aguilar-Bryan L. (1997) Curr. Opin. Cell Biol. 9, 553–559 [DOI] [PubMed] [Google Scholar]
- 51. Sánchez-Alvarez R., Paíno T., Herrero-González S., Medina J. M., Tabernero A. (2006) Glia 54, 125–134 [DOI] [PubMed] [Google Scholar]
- 52. Ma Z., Portwood N., Brodin D., Grill V., Björklund A. (2007) Diabetes 56, 1095–1106 [DOI] [PubMed] [Google Scholar]
- 53. Nishiyama A., Matsui M., Iwata S., Hirota K., Masutani H., Nakamura H., Takagi Y., Sono H., Gon Y., Yodoi J. (1999) J. Biol. Chem. 274, 21645–21650 [DOI] [PubMed] [Google Scholar]
- 54. Shalev A., Pise-Masison C. A., Radonovich M., Hoffmann S. C., Hirshberg B., Brady J. N., Harlan D. M. (2002) Endocrinology 143, 3695–3698 [DOI] [PubMed] [Google Scholar]
- 55. Krezel A., Maret W. (2006) J. Biol. Inorg. Chem. 11, 1049–1062 [DOI] [PubMed] [Google Scholar]
- 56. Li Y., Maret W. (2009) Exp. Cell Res. 315, 2463–2470 [DOI] [PubMed] [Google Scholar]
- 57. Taki M., Wolford J. L., O'Halloran T. V. (2004) J. Am. Chem. Soc. 126, 712–713 [DOI] [PubMed] [Google Scholar]
- 58. Bozym R. A., Thompson R. B., Stoddard A. K., Fierke C. A. (2006) Biol. Trace Elem. Res. 1, 103–111 [Google Scholar]
- 59. Dittmer P. J., Miranda J. G., Gorski J. A., Palmer A. E. (2009) J. Biol. Chem. 284, 16289–16297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Orci L., Malaisse-Lagae F., Ravazzola M., Rouiller D., Renold A. E., Perrelet A., Unger R. (1975) J. Clin. Invest. 56, 1066–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Besecker B., Bao S., Bohacova B., Papp A., Sadee W., Knoell D. L. (2008) Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L1127–1136 [DOI] [PubMed] [Google Scholar]
- 62. Kitamura H., Morikawa H., Kamon H., Iguchi M., Hojyo S., Fukada T., Yamashita S., Kaisho T., Akira S., Murakami M., Hirano T. (2006) Nat. Immunol. 7, 971–977 [DOI] [PubMed] [Google Scholar]
- 63. Chowanadisai W., Lönnerdal B., Kelleher S. L. (2008) Brain Res. 1199, 10–19 [DOI] [PubMed] [Google Scholar]
- 64. Taylor K. M., Morgan H. E., Johnson A., Hadley L. J., Nicholson R. I. (2003) Biochem. J. 375, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aydemir T. B., Liuzzi J. P., McClellan S., Cousins R. J. (2009) J. Leukoc Biol. 86, 337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Smidt K., Jessen N., Petersen A. B., Larsen A., Magnusson N., Jeppesen J. B., Stoltenberg M., Culvenor J. G., Tsatsanis A., Brock B., Schmitz O., Wogensen L., Bush A. I., Rungby J. (2009) PLoS ONE 4, e5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ohly P., Dohle C., Abel J., Seissler J., Gleichmann H. (2000) Diabetologia 43, 1020–1030 [DOI] [PubMed] [Google Scholar]
- 68. Trayhurn P., Duncan J. S., Wood A. M., Beattie J. H. (2000) Horm. Metab. Res. 32, 542–547 [DOI] [PubMed] [Google Scholar]
- 69. Bensellam M., Van Lommel L., Overbergh L., Schuit F. C., Jonas J. C. (2009) Diabetologia 52, 463–476 [DOI] [PubMed] [Google Scholar]
- 70. da Silva Xavier G., Varadi A., Ainscow E. K., Rutter G. A. (2000) J. Biol. Chem. 275, 36269–36277 [DOI] [PubMed] [Google Scholar]
- 71. Michael D. J., Ritzel R. A., Haataja L., Chow R. H. (2006) Diabetes 55, 600–607 [DOI] [PubMed] [Google Scholar]
- 72. Knoch K. P., Bergert H., Borgonovo B., Saeger H. D., Altkrüger A., Verkade P., Solimena M. (2004) Nat. Cell Biol. 6, 207–214 [DOI] [PubMed] [Google Scholar]
- 73. Wijesekara N., Dai F. F., Hardy A. B., Giglou P. R., Bhattacharjee A., Koshkin V., Chimienti F., Gaisano H. Y., Rutter G. A., Wheeler M. B. (2010) Diabetologia 53, 1656–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Carroll R. J., Hammer R. E., Chan S. J., Swift H. H., Rubenstein A. H., Steiner D. F. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 8943–8947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ghafghazi T., McDaniel M. L., Lacy P. E. (1981) Diabetes 30, 341–345 [DOI] [PubMed] [Google Scholar]
- 76. Bindokas V. P., Kuznetsov A., Sreenan S., Polonsky K. S., Roe M. W., Philipson L. H. (2003) J. Biol. Chem. 278, 9796–9801 [DOI] [PubMed] [Google Scholar]
- 77. Kahn S. E. (2001) Int. J. Clin. Pract. Suppl. 123, 13–18 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.