

Abstract
ZAP-70 and Syk are essential tyrosine kinases in intracellular immunological signaling. Both contain an inhibitory SH2 domain tandem, which assembles onto the catalytic domain. Upon binding to doubly phosphorylated ITAM motifs on activated antigen receptors, the arrangement of the SH2 domains changes. From available structures, this event is not obviously conducive to dissociation of the autoinhibited complex, yet it ultimately translates into kinase activation through a mechanism not yet understood. We present a comprehensive theoretical study of this molecular mechanism, using atomic resolution simulations and free-energy calculations, totaling >10 μs of simulation time. Through these, we dissect the microscopic mechanism coupling stepwise ITAM engagement and SH2 tandem structural change and reveal key differences between ZAP-70 and Syk. Importantly, we show that a subtle conformational bias in the inter-SH2 connector causes ITAM to bind preferentially to kinase-dissociated tandems. We thus propose that phosphorylated antigen receptors selectively recruit kinases that are uninhibited and that the resulting population shift in the membrane vicinity sustains signal transduction.
Keywords: Allosteric Regulation, Phosphotyrosine Signaling, Protein Self-assembly, SH2 Domains, Tyrosine Protein Kinase (Tyrosine Kinase), Free-energy Landscape, Modular Protein Domains, Molecular Dynamics Simulation
Introduction
Non-receptor Syk tyrosine kinases are an important class among eukaryotic signaling enzymes (1). The family consists of ZAP-70 and Syk, which are mostly found in T- and B-cells, respectively. These kinases help to propagate antigen-recognition signals initiated at the cell membrane. To do so, they first associate with the antigen-receptor complex and then phosphorylate downstream signaling and scaffolding proteins (2). Syk kinases are autoinhibited when isolated; activation follows their engagement of so-called ITAM motifs on the intracellular side of the antigen-receptor complex, through a molecular mechanism that is only partially understood. Consistent with their important cellular role, malfunction of Syk kinases underlies a range of immune system disorders (2, 3).
Both ZAP-70 and Syk contain a tandem of SH2 modules, which inhibit the catalytic activity of the enzyme by assembling onto the kinase domain. The SH2 tandem (tSH2)3 also serves as the docking platform for ITAM motifs. These motifs consist of two repeats of sequence YXX(L/I) connected by a spacer of variable length; doubly phosphorylated ITAM motifs are thus suitable targets for SH2 tandems. Moreover, this dual interaction results in greater affinity, typically in the nanomolar range; that of individual SH2 domains is 2 orders of magnitude weaker (4).
Despite the high sequence identity of Syk and ZAP-70 (∼60%), they differ noticeably in the thermodynamics of ITAM recognition (5–8). From a functional standpoint, this differentiation is likely related to the fact that Syk is more widely expressed, including in non-immune cells; the diversity of its ligands is therefore necessarily broader (3). However, in view of the similarity of the available tSH2 structures (9, 10), the molecular basis for this differentiation is not immediately clear.
The molecular mechanism by which ITAM recognition induces kinase activation also remains to be clarified. The atomic structure of the ZAP-70 tSH2 assembled onto the kinase domain (Fig. 1A) reveals few structural differences when compared with the tSH2 alone (6, 11). By contrast, ITAM engagement entails a noticeable reorientation of the SH2 modules in isolated tandems (Fig. 1B) (9). However, this rearrangement is not evidently incompatible with the tSH2-kinase interface as seen in the autoinhibited complex. Nevertheless, it has been hypothesized that catalytic activation is initiated by the disassembly of the tSH2 unit, in analogy with Src and Abl kinases (11). Full activation is believed to entail subsequent phosphorylation (e.g. by Lck or ZAP-70 itself) of specific sites in the tSH2-kinase linker and the catalytic domain (2).
FIGURE 1.

Available atomic-resolution structures of ZAP-70. A, structure of the autoinhibited ZAP-70 kinase complex (11). A bound ATP molecule and key regulatory tyrosines (Tyr315, Tyr319, Tyr597, and Tyr598) are highlighted. B, the SH2 tandem (represented as in A in the ITAM-bound (9) (opaque) and apo (6) (transparent) conformations. The ITAM peptide backbone is shown in red, with Tyr(P) and the residue at Tyr(P) + 3 detailed.
Here, we present a detailed theoretical study of the mechanism of regulation of Syk kinases by tSH2 and ITAM motifs. Atomic resolution molecular dynamics simulations and large-scale free-energy calculations of a comprehensive set of molecular constructs (Fig. 2) have been employed to dissect the microscopic mechanism of ITAM recognition by the tSH2 and to discern the impact of this event on the stability of the autoinhibited complex. We begin with a comparative analysis of the SH2 domains in Syk and ZAP-70, individually and in tandem, and rationalize the greater specificity of the latter in terms of their distinct structural dynamics, resulting from key differences in their sequence. Building on these insights, and focusing on ZAP-70, we assess how the conformational free-energy landscape of the tSH2 is reshaped either by the stepwise engagement of ITAM or by docking onto the kinase domain. From this analysis, we conclude that a subtle competition exists between these two events, in reflection of a differential conformational flexibility of the inter-SH2 domain region. Thus, we provide direct evidence in support of the notion that ITAM motifs contribute to the propagation of ZAP-70-mediated signals by selectively recruiting disassembled, and hence uninhibited, tandem-kinase complexes.
FIGURE 2.

Schematic overview of the constructs analyzed in this study and the combined simulation time in each case. Protein domains are colored as in Fig. 1. The tandem systems include ∼70,000–80,000 atoms (see ”Computational Procedures“). The total simulation time exceeds 11 μs. MD, molecular dynamics; TI, thermodynamic integration; US, umbrella sampling. PMF, potential of mean force.
COMPUTATIONAL PROCEDURES
Preparation of Molecular Systems and Simulation Details
All simulation models (Fig. 2) were constructed on the basis of available crystal structures of Syk (1A81) or ZAP-70 (2OQ1, 1M61, and 2OZO). All ionizable groups were assigned their most probable protonation state at neutral pH. Crystallographic water molecules within 5 Å of protein atoms were preserved in each simulation system. Missing atoms and N/C-terminal residues were modeled in, resulting in the full 256-residue tSH2 structure of ZAP-70 or Syk. For the tSH2-kinase system, Phe315 and Phe319 (mutated to obtain stable crystals of the autoinhibited state (11)) were replaced by wild-type Tyr315 and Tyr319, whereas missing regions of the core kinase domain were modeled on the basis of a complete crystal structure of the isolated domain. The tSH2ZAPITAM construct includes the co-crystallized 19-residue ITAM peptide; this corresponds to residues 69–87 of the T-cell receptor CD3-ζ chain. The analogous tSH2SykITAM construct includes the co-crystallized 18-residue ITAM peptide, which corresponds to residues 168–185 of the T-cell receptor CD3-ϵ chain. Both ITAM peptides are doubly phosphorylated. In the tSH2ZAPΔITAM and tSH2SykΔITAM simulations, the ITAM peptide was removed from the complex and replaced by solvent. These simulations, however, start in the bound conformation of the tandem, in contrast to tSH2ZAPApo, which starts in the unbound state. In the tSH2ZAPcITAM and tSH2ZAPnITAM constructs, either the C-terminal or the N-terminal half of the ITAM peptide was retained, respectively, deleting the other half. In the tSH2ZAPApo+cITAM construct, the C-terminal half of the ITAM peptide was modeled into the N-terminal SH2 (nSH2) domain of the apo state crystal structure, through least-squares fitting the nSH2 domains in the ITAM-bound and apo structures. For the cSH2ZAP and cSH2Syk simulations, the ITAM peptide along with the first ∼150 residues of the tSH2 structure (i.e. the nSH2 and I-linker domains) were removed, resulting in a C-terminal SH2 construct including residues 158–256.
Each of the tSH2 constructs was placed in a box of dimensions 9 × 9 × 9 nm3 and solvated with ∼22,000 water molecules (∼70,000 atoms in total). The tSH2-kinase system was placed in a box of dimensions 12 × 10 × 7 nm3 and solvated with ∼22,000 water molecules (∼80,000 atoms in total). The linker-helix constructs were placed in a box of dimensions 3 × 3 × 3 nm3 and solvated with ∼1,000 water molecules (∼3,000 atoms in total). The isolated cSH2 domains were placed in a truncated octahedral box of length ∼6.7 nm and solvated with ∼7,000 water molecules (∼22,000 atoms in total). Counter ions were added to neutralize each simulation box and to achieve a final salt concentration of 0.1 m NaCl. Each system was equilibrated over 2 ns, during which position restraints, applied to all non-hydrogen protein atoms, were gradually removed to relax the protein structure and solvent. The N and C termini of the linker, helical in both apo and ITAM-bound crystal structures, were subjected to weak helical restraints throughout the linker simulations to prevent unfolding. For the cSH2 constructs, additional 20-ns, restraint-free equilibrations were carried out. For every tSH2 and tSH2-kinase system, the solvation and equilibration steps were independently repeated 10 times to improve sampling. The final production simulations (25–35 ns) were also repeated ten times (Fig. 2).
All simulations were carried out with NAMD 2.6 (12), using the all-atom CHARMM22/CMAP force field (13). The simulations were performed at constant temperature (298 K) and pressure (1 atm), using a Langevin thermostat and a Nosé-Hoover Langevin barostat, under periodic boundary conditions. Electrostatic interactions were computed using the particle-mesh-Ewald algorithm; the real-space electrostatic and van der Waals interactions were cut off at 14 Å.
Energetics of EF-loop Gating in the cSH2 Domain
The free energy of gating of the cSH2 domain specificity pocket was calculated using the thermodynamic integration method. (A complete theoretical derivation is provided in the supplemental material.) That is,
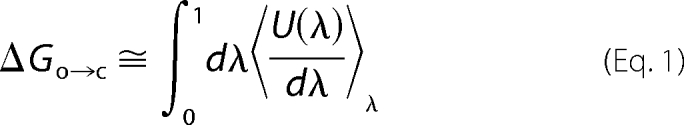 |
The potential energy function U(λ) was defined in terms of the r.m.s.d. of the N-peptide bond dihedral angles φi that describe the conformational change of the loop. That is,
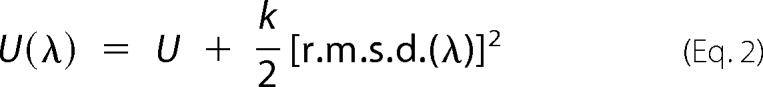 |
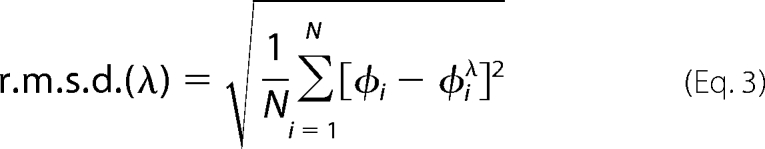 |
Here, φiλ denotes the set of intermediate values that each dihedral adopts in between the two end points, φi,o and φi,c. That is,
The free-energy simulations were carried out following a stepwise protocol, initially including 21 intermediate λ steps, or windows; to resolve the energy barriers more precisely, 2–5 intermediate steps were added as necessary. The sampling time for each window was 4 ns following a preliminary 1-ns equilibration period. The initial configurations of the molecular system for each λ-window were either extracted from unbiased simulations of the cSH2 domains or produced from biased simulations targeting a specific value of λ. A force constant (k) of 2,400 kcal mol−1 radian−2 was used. Calculations with values of k between 600 and 5,000 kcal mol−1 radian−2 show that the results do not significantly depend on this choice. To better resolve the energetics of the open-to-closed gating transition, the calculation was actually carried out in two stages. First, the phi (φ) and psi (ψ) angles of residue 226 were varied to yield ΔG(φ226, ψ226). Subsequently, ψ224 and φ225 and were varied, whereas φ226 and ψ226 were constant, to obtain ΔG(φ225, ψ224). These stages correspond to supplemental Fig. S1, Transition #1 and Transition #2, respectively. The reference angles for the closed state EF-loop are those in the ZAP-70 apo crystal structure (φ225 = −84.8°, ψ224 = −24.1°, φ226 = 95.1°, ψ226 = −0.9°). The reference angles for the open state are those in the ITAM-bound crystal structure of ZAP-70 (φ225 = 67.2°, ψ224 = 121.0°, φ226 = −103.7°, ψ226 = −170.2°) or Syk (φ225 = 67.4°, ψ224 = 124.4°, φ226 = −107.0°, ψ226 = −169.4°).
Conformational Free-energy Landscapes of the ZAP-70 SH2 Tandem
Two-dimensional umbrella-sampling simulations and the weighted histogram analysis method (as in Ref. 14) were employed to calculate the free-energy landscape or potential of mean force underlying the transition from the apo to the ITAM-bound conformations of the ZAP-70 SH2 tandem. The bias potentials ξ1 and ξ2 were defined in terms of a difference of r.m.s.d. values, relative to the structure of either state of the tandem. That is,
The r.m.s.d. functions in ξ1 and ξ2 thus refer to two different sets of atoms; it is assumed that, collectively, these two sets are representative of the conformational change. The first set describes the relative arrangement of the SH2 domains, represented by their core secondary structure regions (supplemental Fig. S2); this includes the backbone atoms of residues 33–38, 45–51, 56–62, and 69–70 (nSH2 domain); residues 121–142 (inter-SH2 linker); and residues 186–191, 197–203, 208–214, and 221–222 (cSH2 domain). The second set describes the fold of the α-helix connecting the inter-SH2 domain to the cSH2 domain, namely the backbone atoms of residues 142–160. The bias potential is therefore
The index j represents each of the umbrella-sampling simulations, or windows, in which the two-dimensional landscape is partitioned. ξ1,j0 and ξ2,j0 denote the reference values of the difference r.m.s.d. potentials in each of these windows. These were spaced in each dimension by 0.5 Å. Each window was simulated for 5 ns, from 3 to 5 times, using independent initial configurations of the simulation system. These configurations were extracted from unbiased sampling (initiated from both apo and ITAM-bound states) or generated from intervening 1-ns targeted simulations from adjacent umbrella-sampling windows. Three complete free-energy landscapes were derived and compared to estimate the statistical error (supplemental Fig. S5).
RESULTS
Conformational Gating of the cSH2 Domain-binding Pocket
It has been shown that the cSH2 domain of ZAP-70 has a weaker affinity for ITAM phosphopeptides than that of Syk (8). However, ZAP-70 cSH2 binds plain Tyr(P) (6), indicating that its reduced affinity for ITAM is not related to Tyr(P) coordination. Instead, it appears to be due to the so-called specificity pocket, which is tuned to recognize a Leu or Ile three residues C-terminal of Tyr(P). Indeed, the crystal structure of the cSH2 domain in the apo tSH2 from ZAP-70 (6) reveals a subtle conformational change in the EF-loop (residues 223–227, between the βE and βF strand) that seems to close this pocket when compared with the ITAM-bound state (Fig. 3, A and B). We noted that in many SH2 domains (15), including the cSH2 of Syk, this pocket contains a tyrosine (Tyr239 in Syk) (Fig. 3C) that is replaced by Leu in ZAP-70 (Fig. 3B). We reasoned that this substitution might be important in determining the plasticity of the binding site, and hence, might influence ITAM recognition.
FIGURE 3.

Gating of the EF-loop in cSH2ZAP and cSH2Syk. A, the ZAP-70 cSH2 domain is shown in its ITAM-bound (cyan) and apo (ice blue) states (6, 9), with the specificity pocket closed or open, respectively, depending on the EF-loop conformation (highlighted in red). In B and C, simulation snapshots of the open state of the specificity pocket are shown for ZAP-70 and Syk. Long-lived hydrogen-bonding networks between water molecules and key backbone atoms are indicated in color, and conserved hydrophobic side chains are gray. D, calculated free-energy cost associated with EF-loop closure, for ZAP-70 (gray) and Syk (black).
To assess this hypothesis, we carried out free-energy simulations of the proposed gating process in the cSH2 domains of ZAP-70 and Syk (cSH2ZAP and cSH2Syk) (Fig. 3D). In ZAP-70, closure of the cSH2 specificity pocket appears to entail no significant energy cost (ΔG = 0.2 ± 0.3 kcal mol−1), suggesting that both open and closed states are populated in solution and at room temperature. These are separated by small kinetic barriers in the order of a few kBT, consistent with NMR measurements (6). Significantly, two structural water molecules that form a hydrogen-bonding network with main-chain atoms (9) are readily expelled upon pocket closure (Fig. 3B).
In contrast, closure of the EF-loop in the canonical cSH2 domain of Syk is energetically disfavored (ΔG = 4.2 ± 0.7 kcal mol−1) (Fig. 3D). In the open state, the conserved Tyr239 mediates a protein-water hydrogen-bonding network not unlike that in ZAP-70 (Fig. 3C). However, the seemingly greater stability of this interaction network, conferred by the Tyr hydroxyl group, hinders the closure of the pocket (supplemental Fig. S1). The corresponding ratio of closed to open states, about 1:1,000, implies that the Syk cSH2 is likely to be constitutively open, in contrast to that of ZAP-70, which is much more prone to adopt a closed state. This conformational gating provides a rationale for the observation that the cSH2 domain of ZAP-70 binds phosphorylated ITAM fragments with significantly lower affinity than the nSH2 domain, in contrast to Syk, for which the affinity of either domain is comparable (6).
Distinct Plasticity of the SH2 Tandems of Syk and ZAP-70
Having established that the regulatory units in Syk and ZAP-70 differ at the single-domain level, we now compare the structural flexibility of the SH2 tandems. A differentiation in this regard has been proposed to explain their distinct specificity in regulatory stimuli (6, 7, 9), but its molecular basis is presently unclear. A large reorientation of the SH2 domains is indeed associated with ITAM binding in ZAP-70 (6, 9) (Fig. 1B), but for Syk, only the bound-state structure is known (10).
To assess whether the inherent plasticity of the Syk and ZAP-70 SH2 tandems differ, we carried out a series of molecular dynamics simulations of these tandems, both in the ITAM-bound state (tSH2ZAPITAM and tSH2SykITAM) and upon release of the ITAM peptide (tSH2ZAPΔITAM and tSH2SykΔITAM). For the bound-state simulations, the ∼20-residue ITAM peptides derive from the T-cell receptor CD3-ζ and -ϵ chain, respectively. A total of 250 ns of simulation time were computed for each construct, split into 10 trajectories to diversify the exploration of conformational space.
Taken individually, the nSH2, cSH2, and inter-SH2 domains were structurally constant in this time scale, in both Syk and ZAP-70 and for both bound and unbound states. For example, the r.m.s.d. of their core secondary structure regions remained within ∼0.5 Å of the corresponding crystal structures (supplemental Fig. S2). In tandem, the ITAM-bound constructs were also markedly stable (Fig. 4, a and c, and supplemental Fig. S2); we observed only small concerted motions of the SH2 domains, comparable with those seen in the unit cell of the Syk crystal, which contains four copies of the tandem (10) (supplemental Fig. S3B). This stability correlates with the persistence of the Tyr(P) coordination network, which involves conserved side chains from both SH2 domains (Fig. 5A and supplemental Fig. S4A). Of particular interest is a lysine residue (Lys242 in ZAP-70, Lys227 in Syk) from the cSH2 domain, which provides a coordination contact for the nSH2-bound Tyr(P) at the SH2-SH2 interface (9). According to our simulations, this interaction is unequivocally stable in both Syk and ZAP-70 (Fig. 5A and supplemental Fig. S4A). Therefore, we cannot concur with the notion that differences in Tyr(P) coordination at the SH2-SH2 interface underlie the more promiscuous regulation of Syk (10).
FIGURE 4.

Structural dynamics of the SH2 tandem in different constructs. The conformational variability of the tandem is represented as a probability density in two dimensions, namely the r.m.s.d. relative to the apo (x axis) or ITAM-bound (y axis) structures determined by crystallography. The location of the actual crystal structures in this representation is indicated by green circles as follows: ITAM-bound ZAP-70 (9) in a, b, e, and f; ITAM-bound Syk (10) in c and d; apo ZAP-70 (6) in a; and kinase-bound ZAP-70 (11) in g. In panel c, additional experimental structures of the corresponding SH2 tandem from the same crystal are indicated. Green dashed lines indicate the approximate direction of motion between apo and ITAM-bound states of ZAP-70, as observed in the simulations of the tSH2ZAPApo (a) and tSH2ZAPΔITAM (b) constructs. Correlation coefficients between selected probability density distributions from this figure are shown in Table 1. w.r.t., with respect to.
FIGURE 5.

Representative simulation snapshots illustrating key interactions between ITAM and the SH2 tandem and between the SH2 domains (see also supplemental Fig. S4). A, long-lived interactions between the ITAM phosphopeptide and side chains from the nSH2 and cSH2 domains of ZAP-70 and Syk, in the ITAM-bound state. For clarity, the peptide is omitted, and only the C-terminal Tyr(P) (C-pY) phosphate is shown. Long-lived H-bonds during the simulations are indicated by dashed lines. B, as in A, for the N-terminal Tyr(P) (N-pY) of ITAM and the cSH2 domain. C, as in A, after simulated relaxations upon removal of ITAM from the ITAM-bound state. D, proposed complete phosphate coordination by the nSH2 domain of ZAP-70, in the apo conformation of the tSH2.
Instead, our simulations indicate that it is in the ITAM-free state that the SH2 tandem dynamics differ the most. Upon disengagement of ITAM, the SH2 domains in the ZAP-70 tandem became rapidly uncoupled, leading to a concerted rearrangement toward the apo state (Fig. 4, a and b, and supplemental Fig. S3C). This is consistently observed in independent simulations, indicating that the conformational changes associated with ITAM binding and release likely follow a narrowly defined pathway. In contrast, the SH2 domains of Syk show a clear propensity to remain associated (Fig. 4, c and d, and supplemental Fig. S3D), driven by interdomain salt bridges and H-bonds (e.g. Arg40-Glu237 and Glu18-Lys242/Tyr241), not available in ZAP-70 because of polar-to-hydrophobic substitutions (Glu18 → Ala, Arg40 → Leu, and Tyr241 → Leu) (Fig. 5, A and C). Previous thermodynamic studies have indeed suggested that the unbound Syk tandem exists in equilibrium between two states, both competent for binding ITAM (7), and recent NMR data indicate that the ensemble of unliganded conformations is rather compact (16). Our simulations show that this ensemble does indeed comprise a range of conformations in which the Syk SH2 domains remain associated, which may facilitate the recognition of a greater variety of ligands. This, combined with a constitutively open specificity pocket, likely contributes to a greater promiscuity in the regulation of the Syk kinase when compared with ZAP-70.
SH2 Tandem Dynamics upon Stepwise Engagement of ITAM
That the structure and dynamics of the ZAP-70 SH2 tandem are influenced by ITAM binding is not unexpected; more informative, however, is the role of the individual Tyr(P) motifs in ITAM. To address this question, we calculated two series of simulations, amounting to 250 ns each, in which the ZAP-70 tandem starts in the ITAM-bound conformation, in complex with either an N-terminal (tSH2ZAPnITAM) or a C-terminal (tSH2ZAPcITAM) ITAM fragment.
We observed a striking contrast in the tandem dynamics between these systems (Fig. 4, e and f). The tSH2ZAPnITAM construct, in which an ITAM fragment is bound to the cSH2 domain, exhibited a concerted shift toward the apo state (Fig. 4e) despite the fact that the nITAM fragment remains stably bound (supplemental Fig. S4C). Indeed, this conformational trend is very much comparable with that seen in the tSH2ZAPΔITAM simulations discussed above (Fig. 4b and Table 1). This observation suggests that partial binding of ITAM to the cSH2 domain is unlikely to affect the collective dynamics of the tandem, relative to the apo state. In contrast, the conformational sampling of tSH2ZAPcITAM construct (Fig. 4f) closely resembled that of the ITAM-bound state (Fig. 4a and Table 1), indicating that the interactions mediated by the C-terminal Tyr(P) at the interface between the SH2 domains (supplemental Fig. S4C) are sufficient to stabilize the ITAM-bound conformation.
TABLE 1.
Correlation coefficients of the probability-density data in Fig. 4
For clarity, only correlations employed in the discussion of the results are provided. Kin, kinase domain.
| tSH2ZAPITAM | tSH2ZAPApo+Kin | |
| tSH2ZAPApo | 0.02 | 0.56 |
| tSH2ZAPΔITAM | tSH2SykITAM | |
| tSH2ZAPITAM | 0.03 | 0.45 |
| tSH2SykΔITAM | 0.22 | 0.50 |
| tSH2ZAPcITAM | tSH2ZAPnITAM | |
| tSH2ZAPITAM | 0.96 | 0.16 |
| tSH2ZAPΔITAM | 0.10 | 0.88 |
Interestingly, the network of C-terminal Tyr(P) interactions in the ITAM and cITAM constructs is identical (Arg17, Arg37, Lys242, Tyr238), except that in the latter, Arg41 (from nSH2ZAP) competes with Lys242 and Tyr238 (from cSH2ZAP) for interactions with the ligand (Fig. 5A and supplemental Fig. S4C). This suggests that Arg41 may form a transient interaction with the Tyr(P) motif in a state in which the C-terminal half of ITAM binds to the N-terminal SH2 domain prior to any conformational change in the tandem. To explore this hypothesis further, we simulated the ZAP-70 SH2 tandem in its apo state, after docking a cITAM fragment onto its nSH2 domain (tSH2ZAPApo+cITAM). In this conformation, the Lys242 side chain of the cSH2 domain is too far displaced to contribute to Tyr(P) coordination. However, we now observed a consistent reconfiguration of Arg41 in all simulations, from a solvent-facing side-chain orientation in the apo state to one in which the guanidinium group completes the missing Tyr(P) coordination site (Fig. 5D).
Taken together with our analysis of the isolated cSH2 domain, these observations suggest a model in which ITAM binding to ZAP-70 proceeds via initial tethering to the nSH2 domain, with a complete Tyr(P)-binding site transiently formed by this domain alone. Binding of this first Tyr(P) at the SH2-SH2 interface would then promote a second conformation of the tandem, in which new interactions with the cSH2 domain (Lys242, Tyr238) replace some previously contributed by nSH2 (Arg41). Finally, the localization of ITAM to the tandem would effectively raise the local concentration of the second Tyr(P) motif, accelerating the kinetics of binding to the subset of open states of the cSH2 domain. Incidentally, this mechanism of avidity implies precise constraints on the length of the inter-Tyr(P) linker in ITAM, particularly for ZAP-70, consistent with experiments (17, 18).
Influence of cITAM on the SH2 Tandem Energy Landscape
To quantitatively substantiate this microscopic model of conformational regulation of the SH2 tandem, a large-scale computational effort was made to calculate the free-energy landscape that underlies the transition between apo and ITAM-bound conformations, both in the absence and in the presence of a cITAM peptide fragment. To compute these landscapes, we used two-dimensional umbrella sampling. In this method, biasing potentials acting on specific structural variables are introduced to enhance the sampling of all available conformational space, while recording the response (in equilibrium) of the molecular system. As a first variable, we chose the difference in backbone r.m.s.d. relative to apo and ITAM-bound states for the tandem core regions; this is meant to reflect the rigid body motion of the domains. As a second variable, we chose the analogous quantity for the α-helix that connects the inter-SH2 linker to the cSH2 domain (referred to as αL2-αL3); this α-helix undergoes partial refolding and reorientation upon ITAM binding (6, 9, 11). The computational cost required for each of these landscapes exceeded 3 μs of simulation time (Fig. 2).
This quantitative analysis provides further support to the mechanism outlined above. Two main energy minima are observed in each free-energy landscape (Fig. 6). A deep minimum (Fig. 6, A and B, bottom left) in the presence or absence of the cITAM fragment corresponds to the ligand-free conformation, close to the apo state crystal structure. That this is a broad minimum indicates significant conformational flexibility; this is in accord with an additional set of unbiased molecular dynamics simulations of the apo state, again summing up to 250 ns (Fig. 2), which reveal concerted domain-domain fluctuations in the direction of the ITAM-bound state (Fig. 4a). The second minimum corresponds to the ITAM-bound conformation (Fig. 6, A and B, top right). In the absence of cITAM, this is only a shallow, local minimum and is less stable by ∼7 kcal mol−1. Thus, in the absence of ITAM ligands, this conformation will be populated only marginally. In clear contrast, this region of the free-energy landscape becomes a much deeper energetic minimum when the cITAM fragment is bound to the tandem, comparable in magnitude with that of the apo state. These calculations thus indicate that initial binding of the C-terminal end of ITAM, with its Tyr(P) near the SH2-SH2 interface, would increase the probability of the alternate, up-regulating tandem conformation by several orders of magnitude. Subsequent binding of the second half of the ITAM peptide to an open state of the cSH2 domain would serve to secure this conformation by hindering the unbinding of the first half.
FIGURE 6.

Conformational free-energy landscape of the ZAP-70 SH2 tandem, in the absence (A) or presence (B) of a C-terminal ITAM peptide fragment. The tandem conformation is described via two differences in the r.m.s.d. (Δr.m.s.d.) with respect to apo (6) and ITAM-bound (9) crystal structures; one represents the orientation of the domains (inset, green), and the other represents the fold of the inter-SH2 linker helix (inset, red) (see ”Computational Procedures“). Contours indicate changes of 2 kcal/mol. The statistical uncertainty estimated from repeat calculations is <1.5 kcal mol−1 (supplemental Fig. S5).
Intrinsic Conformational Bias in αL2-αL3 Region
From these free-energy landscapes (Fig. 6), it is also worth noting that reorientation of the αL2-αL3 region and refolding of the αL3 helix are required to begin to escape the deep energetic minimum centered around the apo (or kinase-bound) conformation. Indeed, experimental evidence supports that a conformational change in the linker may be coupled to ITAM binding (6, 19). To further investigate the role of this structural element, we performed additional simulations of the isolated helix (each in five sets of 50-ns trajectories), starting from the folded (i.e. ITAM-bound) or unfolded (i.e. apo) crystal structure. In each prefolded simulation, the helix was stable over 50 ns, whereas in four of the five unfolded sets, spontaneous refolding of the helix was observed within 20–50 ns (Fig. 7). By contrast, refolding was rarely observed in a comparable series of control simulations, in which the central Thr-Thr-Ala-His sequence of the helix was scrambled to all possible permutations (Fig. 7).
FIGURE 7.

Final snapshots of the LinkerZAP simulations. The linker sequence in each system is LIA-X4-HER, with the central X4 sequence indicated. Simulations were initiated from the folded ITAM-bound (9) or unfolded apo (6) wild-type sequence or from the 11 unfolded mutant linkers.
Thus, the sequence of the αL3 segment (conserved across Syk family kinases) appears to be predisposed to fold as an α-helix; therefore, it partially compensates for the energy cost of the conformational change in the tSH2 required for ITAM binding (i.e. in addition to the SH2-ITAM interactions). Interestingly, this segment is located in close proximity to the densely packed kinase interface in the autoinhibited complex (11) (Fig. 1A). Therefore, it is plausible that the refolding propensity of the αL2-αL3 element is subtly impaired by the kinase domain interface; as a result, the assembly equilibrium of the tSH2-kinase complex would be shifted toward the disassembled state in the context of available ITAM ligands.
Disassembly of Tandem-Kinase Complex Facilitates ITAM Recognition
To examine the disassembly hypothesis from a thermodynamic standpoint, we recomputed the free-energy landscape of the ZAP-70 tSH2, now in association with the kinase domain (Fig. 8). That is, we assessed the degree to which the conformational change that occurs upon ITAM binding is favored or disfavored in the context of the tSH2-kinase interface, relative to the isolated tandem. We specifically focused on the change in the inter-SH2 domain (αL2-αL3 region) and the initial reorientation of the cSH2 domain (Fig. 6A, left side); as mentioned, further changes are not expected to be influenced by the catalytic domain.
FIGURE 8.

Conformational free-energy landscape of the ZAP-70 SH2 tandem in the autoinhibited ZAP-70 tandem-kinase complex. The free-energy surface was computed analogously to those shown Fig. 6 and is plotted identically.
As expected, this partial landscape reveals a minimum around the apo-like conformation, consistent with the x-ray structure of the auto-inhibited complex and with the isolated tSH2 landscape (Fig. 6A). In contrast to the latter, however, the free-energy gradient around this minimum is noticeably steeper; that is, the initiation of the conformational change in the tSH2 required for ITAM binding is energetically more costly if the tSH2 is docked onto the kinase domain. From this result, it follows that the structural dynamics of the tSH2, isolated or in the context of the kinase, should differ. Indeed, conventional simulations in the 250-ns time scale reveal more restricted flexibility in the context of the kinase (Fig. 4g versus Fig. 4a and supplemental Fig. S6). This increased order in the tandem seems to be imposed through the inter-SH2 domain, by both the kinase domain and the tandem-kinase linker, which form a dense network of stabilizing interactions. Interestingly, this interface buries several residues that are essential for regulation via phosphorylation (11), including key tyrosines such as Tyr315/Tyr319 in the SH2-kinase linker and Tyr597/598 in the kinase C-lobe (Fig. 1A).
Taken together, our results support the notion that full engagement of ITAM will be mediated preferentially by SH2 tandems that are transiently dissociated from the catalytic domain, as a result of a subtle conformational bias within the inter-SH2 region. This bias provides a mechanism whereby the population of ZAP-70 kinases susceptible to additional phosphorylation, and thus complete activation, is increased in the vicinity of the membrane receptor.
DISCUSSION AND CONCLUSIONS
We have presented a comprehensive computational study that provides novel microscopic insights into the mechanism of regulation of Syk tyrosine kinases, and particularly ZAP-70. First, we have shown that the conformational dynamics of the regulatory SH2 modules of Syk and ZAP-70 differ, as a result of specific sequence substitutions. In particular, the so-called specificity pocket in the cSH2 domain is constitutively open in Syk, whereas in ZAP-70, it tends to adopt a close conformation in the unliganded state. This is consistent with the observation that phosphopeptides hardly bind to the ZAP-70 cSH2, whereas phosphate does so readily, in contrast to the cSH2 domain of Syk and to the nSH2 domains of both proteins (6, 8, 20, 21). Structural flexibility is thought to relate to ligand diversity in other SH2 domains (22). For example, a similar mechanism of SH2 gating was recently suggested to regulate SHIP2 (23), differentiating it functionally from the closely related phosphatase SHIP1. Thus, modulation of the conformational dynamics of SH2 domain specificity pockets appears to provide one mechanism for fine-tuning signaling pathways. In addition, we have shown that Syk and ZAP-70 also differ in the dynamic range of their SH2 tandems, albeit not in the ITAM-bound state, as was previously inferred (10). Instead, this differentiation arises in the apo state, as a result of sequence substitutions at the SH2-SH2 interface; these control the degree of domain-domain association, which is greater in Syk than ZAP-70. This kind of conformational differentiation is reminiscent of the SH3-SH2 regulatory tandems in members of Src tyrosine kinases. These differ mainly in the sequence of the interdomain connector, where a handful of amino acids is believed to modulate the conformational free-energy landscape of the tandem, and thus, the assembly equilibrium of the tandem-kinase complex (14). In the present case, we propose that substitutions at the SH2-SH2 interface underlie the broader target specificity of Syk, relative to ZAP-70. These appear to enable the Syk tSH2 to adopt a range of preorganized conformations even in the unliganded state, in accord with previous biophysical and thermodynamic measurements (7, 16).
Second, we have dissected in detail the mechanism by which ITAM recognition modulates the structure and dynamics of the ZAP-70 SH2 tandem. Our analysis indicates that binding of the C-terminal half of ITAM to the tandem transiently involves stabilizing interactions with the nSH2 domain only. Thus, contrary to previous views (6), but consistent with the thermodynamic data (20, 21), we have shown that the nSH2 domain can form a complete site for phosphopeptide binding. Nevertheless, this is a transient state; as our free-energy calculations demonstrate, the bound phosphotyrosine has a profound influence on the conformational free-energy landscape of the SH2 tandem. Specifically, it increases the likelihood of the ligand-bound conformation by relieving the energetic strain associated with this protein conformation. Microscopically, this entails a partial reconfiguration of the nSH2-phophotyrosine interaction network to form alternative interactions with the cSH2 domain. We argue that it is in this alternate tandem conformation that binding of the N-terminal ITAM motif to the cSH2 domain can proceed. Although this domain is only half-competent, binding would be enhanced, for entropic reasons, by the preceding engagement of the C-terminal phosphotyrosine, i.e. by an increase in the effective concentration of ITAM. Thus, our analyses lend support and provide microscopic detail to the proposal sketched in Ref. 6, rather than that in Ref. 2.
Like other signaling complexes, the ZAP-70 tandem-kinase complex naturally exists in an equilibrium between assembled and disassembled states; although the former is autoinhibited, the latter is susceptible to phosphorylation of sites otherwise unexposed, and thus, full activation (2). Considering the physiological function of ZAP-70 and its dependence on ITAM, it is logical to deduce that ITAM engagement causes, or somehow promotes, the disassembly of the autoinhibited ZAP-70 complex (11). Nonetheless, despite its appeal, this model is not entirely consistent with single-particle electron microscopy studies of Syk, which reportedly reveal much less noticeable changes upon activation by phosphorylation (24). Moreover, the mechanism of disassembly is not self-evident from comparison of the crystal structures of the ZAP-70 tSH2 in complex with either ITAM or the kinase because the rearrangement of the SH2 domains upon ITAM binding is not sterically hindered by the catalytic domain (supplemental Fig. S7).
On the other hand, recent biochemical studies of ZAP-70 in vitro are supportive of the disassembly mechanism in that mutants presumed to either stabilize or destabilize the tSH2-kinase interface are seen to bind ITAM with apparent affinities that are weaker or stronger, respectively, than that of the wild type (25). This proves an interdependence between tSH2 recognition of ITAM and the equilibrium of assembly and disassembly of the tandem-kinase complex. This notwithstanding, the variation in the apparent Kd of the tSH2-ITAM interaction is 5-fold at most, demonstrating that the influence of ITAM binding on this equilibrium is very subtle. This is not entirely surprising in view of the atomic structure of the autoinhibited complex (which, notably, carries the aforementioned stabilizing mutations) because the tSH2-kinase interface is seen to involve the inter-SH2 domain only.
To shed light on the important question of ITAM-mediated activation, we extended our conformational free-energy calculations to include the full-length kinase complex. In summary, this microscopic analysis is supportive of the hypothesis that ITAM binding contributes to the up-regulation of ZAP-70 by favoring the disassembled state (11). In particular, our calculations reveal that the interdomain region is intrinsically disposed to facilitate the structural changes that full ITAM engagement entails. However, association with the kinase domain appears to cause a subtle but significant conformational restriction of this region. Thus, ITAM binding would be energetically less costly, and thus more probable, for kinase-dissociated SH2 tandems.
From the signal transduction viewpoint, the implication of this finding is that activated antigen receptors would preferably recruit uninhibited ZAP-70 complexes to the vicinity of the membrane (Fig. 9). Through cis-phosphorylation, and thus full activation, this local accumulation of uninhibited ZAP-70 would eventually contribute to altering the balance between activating and inhibitory enzymes at the immune synapse. This molecular mechanism of selective recruitment and amplification, therefore, resonates with emerging themes in intercellular communication, such as spatial organization and collective signaling (26). It is also in keeping with modern views on molecular recognition and allostery, based upon the notions of population equilibria and conformational selection (27, 28).
FIGURE 9.

Proposed mechanism of recruitment and enhanced activation of ZAP-70 in the vicinity of the antigen-receptor membrane complex. Double phosphorylation of ITAM motifs on the intracellular side of the T-cell receptor (TCR) complex provides a recognition platform for the SH2 tandem of ZAP-70. Transiently disassembled ZAP-70, coexisting in equilibrium with the dominant autoinhibited state, preferentially engages ITAM, for energetic reasons related to the conformational preferences of the SH2 tandem. ITAM recognition first entails binding of nSH2 to the C-terminal half of ITAM. The nSH2 domain is fully able to bind phosphopeptide, in contrast to the cSH2 domain, whose specificity pocket tends to be closed. A rearrangement of the SH2 tandem allows the cSH2 domain to co-participate in coordinating the C-terminal ITAM motif. Complete ITAM binding may now proceed as the avidity effect associated with the divalent SH2/ITAM interaction prevails over the propensity for closure of the cSH2 domain. Localized, disassembled ZAP-70 exposes key phosphorylation sites to neighboring, activated kinases; this modification precludes reassembly of the SH2 tandem (i.e. inhibition) as the dehydration energy cost involved is excessive. ZAP-70 is thus constitutively active, even after ITAM release. Iterations of this cycle of conformational selection of the uninhibited state and phosphorylation result in an accumulation of activated ZAP-70 kinases in the proximity of the immunological synapse. pY, Tyr(P). pMHC, peptide-MHC complex.
Supplementary Material
Acknowledgments
We thank L. R. Forrest, S. Khalid, and F. Marinelli for helpful discussions.
This work was supported in part by the Deutsche Forschungsgemeinschaft (DFG) Cluster of Excellence ”Macromolecular Complexes“ and by the Leibniz Supercomputer Center.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Methods and Figs. S1–S7.
- tSH2
- SH2 tandem
- nSH2
- N-terminal SH2
- cSH2
- C-terminal SH2
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. van Oers N. S., Weiss A. (1995) Semin. Immunol. 7, 227–236 [DOI] [PubMed] [Google Scholar]
- 2. Au-Yeung B. B., Deindl S., Hsu L. Y., Palacios E. H., Levin S. E., Kuriyan J., Weiss A. (2009) Immunol. Rev. 228, 41–57 [DOI] [PubMed] [Google Scholar]
- 3. Mócsai A., Ruland J., Tybulewicz V. L. (2010) Nat. Rev. Immunol. 10, 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ottinger E. A., Botfield M. C., Shoelson S. E. (1998) J. Biol. Chem. 273, 729–735 [DOI] [PubMed] [Google Scholar]
- 5. Bu J. Y., Shaw A. S., Chan A. C. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 5106–5110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Folmer R. H., Geschwindner S., Xue Y. (2002) Biochemistry 41, 14176–14184 [DOI] [PubMed] [Google Scholar]
- 7. Grucza R. A., Fütterer K., Chan A. C., Waksman G. (1999) Biochemistry 38, 5024–5033 [DOI] [PubMed] [Google Scholar]
- 8. Narula S. S., Yuan R. W., Adams S. E., Green O. M., Green J., Philips T. B., Zydowsky L. D., Botfield M. C., Hatada M., Laird E. R., et al. (1995) Structure 3, 1061–1073 [DOI] [PubMed] [Google Scholar]
- 9. Hatada M. H., Lu X., Laird E. R., Green J., Morgenstern J. P., Lou M., Marr C. S., Phillips T. B., Ram M. K., Theriault K., et al. (1995) Nature 377, 32–38 [DOI] [PubMed] [Google Scholar]
- 10. Fütterer K., Wong J., Grucza R. A., Chan A. C., Waksman G. (1998) J. Mol. Biol. 281, 523–537 [DOI] [PubMed] [Google Scholar]
- 11. Deindl S., Kadlecek T. A., Brdicka T., Cao X., Weiss A., Kuriyan J. (2007) Cell 129, 735–746 [DOI] [PubMed] [Google Scholar]
- 12. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiorkiewicz-Kuczera J., Yin D., Karplus M. (1998) J. Phys. Chem. B. 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 14. Faraldo-Gómez J. D., Roux B. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 13643–13648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee C. H., Kominos D., Jacques S., Margolis B., Schlessinger J., Shoelson S. E., Kuriyan J. (1994) Structure 2, 423–438 [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y., Oh H., Burton R. A., Burgner J. W., Geahlen R. L., Post C. B. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 11760–11765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gauen L. K., Zhu Y., Letourneur F., Hu Q., Bolen J. B., Matis L. A., Klausner R. D., Shaw A. S. (1994) Mol. Cell Biol. 14, 3729–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vély F., Nunès J. A., Malissen B., Hedgecock C. J. (1997) Eur. J. Immunol. 27, 3010–3014 [DOI] [PubMed] [Google Scholar]
- 19. Grazioli L., Germain V., Weiss A., Acuto O. (1998) J. Biol. Chem. 273, 8916–8921 [DOI] [PubMed] [Google Scholar]
- 20. Isakov N., Wange R. L., Burgess W. H., Watts J. D., Aebersold R., Samelson L. E. (1995) J. Exp. Med. 181, 375–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Labadia M. E., Jakes S., Grygon C. A., Greenwood D. J., Schembri-King J., Lukas S. M., Warren T. C., Ingraham R. H. (1997) Arch. Biochem. Biophys. 342, 117–125 [DOI] [PubMed] [Google Scholar]
- 22. Taylor J. D., Ababou A., Fawaz R. R., Hobbs C. J., Williams M. A., Ladbury J. E. (2008) Proteins 73, 929–940 [DOI] [PubMed] [Google Scholar]
- 23. Zhang Y., Wavreille A. S., Kunys A. R., Pei D. (2009) Biochemistry 48, 11075–11083 [DOI] [PubMed] [Google Scholar]
- 24. Arias-Palomo E., Recuero-Checa M. A., Bustelo X. R., Llorca O. (2009) Biochim. Biophys. Acta 1794, 1211–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deindl S., Kadlecek T. A., Cao X., Kuriyan J., Weiss A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 20699–20704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manz B. N., Groves J. T. (2010) Nat. Rev. Mol. Cell Biol. 11, 342–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsai C. J., Ma B., Nussinov R. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 9970–9972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boehr D. D., Nussinov R., Wright P. E. (2009) Nat. Chem. Biol. 5, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.