

Abstract
Polycyclic aromatic hydrocarbons (PAH) are environmental and tobacco carcinogens. Metabolic activation of intermediate PAH trans-dihydrodiols by aldo-keto reductases (AKRs) leads to the formation of electrophilic and redox-active o-quinones. We investigated whether O-methylation by human recombinant soluble catechol-O-methyltransferase (S-COMT) is a feasible detoxication step for a panel of structurally diverse PAH-catechols produced during the redox-cycling process. Classes of PAH non-K-region o-quinones (bay region, methylated bay region, and fjord region o-quinones) produced by AKRs were employed in the studies. PAH o-quinones were reduced to the corresponding catechols by dithiothreitol under anaerobic conditions and then further O-methylated by human S-COMT in the presence of S-[3H]adenosyl-l-methionine as a methyl group donor. The formation of the O-methylated catechols was detected by HPLC-UV coupled with in-line radiometric detection, and unlabeled products were also characterized by LC-MS/MS. Human S-COMT was able to catalyze O-methylation of all of the PAH-catechols and generated two isomeric metabolites in different proportions. LC-MS/MS showed that each isomer was a mono-O-methylated metabolite. 1H NMR was used to assign the predominant positional isomer of benzo[a]pyrene-7,8-catechol as the O-8-monomethylated catechol. The catalytic efficiency (kcat/Km) varied among different classes of PAH-catechols by 500-fold. The ability of S-COMT to produce two isomeric products from PAH-catechols was rationalized using the crystal structure of the enzyme. We provide evidence that O-8-monomethylated benzo[a]pyrene-7,8-catechol is formed in three different human lung cell lines. It is concluded that human S-COMT may play a critical role in the detoxication of PAH o-quinones generated by AKRs.
Keywords: Carcinogenesis, Enzymes, Mass Spectrometry (MS), Oxidative Stress, Reactive Oxygen Species (ROS), Aldo-Keto Reductases, Catechol-O-methyltransferase, Catechols, Polycyclic Aromatic Hydrocarbons
Introduction
Polycyclic aromatic hydrocarbons (PAH)2 are ubiquitous environmental pollutants that are products of fossil fuel combustion and found in tobacco smoke and are suspect lung carcinogens (1, 2). PAH are considered to be procarcinogens and require metabolic activation to elicit their deleterious effects. Benzo[a]pyrene (B[a]P) is a representative PAH and widely used to study the metabolic activation of PAH (3, 4).
There are three major pathways for the activation of B[a]P. In the first pathway, P450 peroxidases catalyze the generation of radical cations (5), which lead to the formation of depurinating adducts and result in G to T transversions (6). The other two pathways share the same proximate carcinogen intermediate, (−)-trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene (B[a]P-7,8-trans-dihydrodiol), which is formed by the sequential actions of P450s and epoxide hydrolase. B[a]P-7,8-trans-dihydrodiol is then transformed either to (+)-anti-7,8-dihydroxy-9α,10α-epoxy-7,8,9,10-tetra-hydrobenzo[a]pyrene (anti-B[a]PDE) by P450 1A1/P450 1B1 (4, 7, 8) or to B[a]P-7,8-dione by aldo-keto reductases (AKR1A1 and AKR1C1 to -1C4) (9–11) (Scheme 1).
SCHEME 1.

Metabolic activation of B[a]P and detoxication of B[a]P-7,8-dione by COMT.
PAH o-quinones, produced by AKRs are electrophilic and highly reactive to endogenous nucleophiles. PAH o-quinones can readily form conjugates with cellular thiols to yield l-cysteine, N-acetyl-l-cysteine, and GSH conjugates, leading to their elimination (12, 13). PAH o-quinones can also react with DNA to form both stable and depurinating adducts, which may result in mutagenesis (14–16). PAH o-quinones are also able to undergo nonenzymatic/enzymatic reduction to reform catechols at the expense of NADPH and establish futile redox cycles that amplify the generation of reactive oxygen species (ROS). ROS can cause DNA damage resulting in the formation of 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxo-dGuo) lesions and can contribute to G to T transversions in ras and p53 (17, 18). PAH o-quinones were found to be more mutagenic than diol-epoxides in an in vitro p53 mutagenesis assay provided they redox-cycled and a linear correlation was observed between mutagenic efficiency and the presence of 8-oxo-dGuo in the p53 DNA (19–21). More recently, the metabolic activation of B[a]P-7,8-trans-dihydrodiol to B[a]P-7,8-dione was demonstrated in human lung adenocarcinoma (A549) cells, which show high constitutive expression of AKRs. This metabolic activation led to the formation of ROS and 8-oxo-dGuo lesions in cellular DNA. Importantly, the amount of ROS and 8-oxo-dGuo formed in these experiments was increased in the presence of a COMT inhibitor (22). This suggested that COMT could intercept the catechol and prevent redox cycling.
Whether human COMT catalyzes the detoxication of PAH o-quinones by O-methylation of the corresponding catechol remains to be directly demonstrated. Catechol-O-methyltransferase (COMT; EC 2.1.1.6) catalyzes transfer of a methyl group from S-adenosyl-l-methionine (AdoMet) to the hydroxyl group of a variety of catechols, including catecholamine neurotransmitters and the catechol estrogens (23–25). There are two major COMT isoforms in humans, the soluble cytosolic form (S-COMT) and the membrane-bound endoplasmic reticulum form (MB-COMT) encoded by a single gene at 22q11.2 (26, 27). The two isoforms share identical amino acid sequences, except the MB-COMT contains an NH2-terminal extension of 50 amino acids to serve as a hydrophobic anchor to the membrane (28). COMT is widely distributed among various organs in the body, including lung, where high COMT activity was found (29). Except in brain, S-COMT is the predominant form in most tissues (30–32). In the present study, we have expressed and purified human recombinant S-COMT and investigated whether O-methylation of PAH-catechols by human COMT is one of the detoxication pathways for PAH o-quinones. We have used the available crystal structure of S-COMT to rationalize our findings. We provide evidence that the O-8-monomethylated B[a]P-7,8-catechol is produced in three different human lung cells.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Pyrocatechol, guaiacol, and (AdoMet) chloride, were purchased from Sigma. Naphthalene-1,2-dione (NP-1,2-dione (1)), chrysene-1,2-dione (C-1,2-dione (2)), chrysene-3,4-dione (C-3,4-dione (3)), 5-methyl-chrysene-1,2-dione (5MC-1,2-dione (4)), benz[a]anthracene-3,4-dione (BA-3,4-dione (5)), 7-methylbenz[a]anthracene-3,4-dione (7MBA-3,4-dione (6)), 12-methylbenz[a]anthracene-3,4-dione (12MBA-3,4-dione (7)), 7,12-dimethylbenz[a]anthracene-3,4-dione (DMBA-3,4-dione (8)), benzo[c]phenanthrene 3,4-dione (B[c]Ph-3,4-dione (9)), benzo[a]pyrene-7,8-dione (B[a]P-7,8-dione (10)), and benzo[g]chrysene-11,12-dione (B[g]C-11,12-dione (11)) (Scheme 2) were obtained from either the NCI, National Institutes of Health, Chemical Carcinogen Standard Reference Repository (Midwest Research Institute, Kansas City, MO) or synthesized by published methods (33, 34). All solvents were HPLC grade, and all other chemicals used were of the highest grade available. S-Adenosyl-l-[methyl-3H]methionine ([3H]AdoMet; 82–84 Ci/mmol) was purchased from American Radiolabel Chemical, Inc. (Arlington Heights, IL). Porcine COMT was purchased from MP Biomedical LLC (2460 units/mg).
SCHEME 2.

Chemical structures of PAH o-quinones. The curly arrow denotes the presence of a bay region (2, 3, 5, 6, and 10), a methylated bay region (4, 7, and 8), or a fjord region (9 and 11). Ring numbering in the terminal benzo-ring is shown.
Standard Radiometric Assay for COMT Activity
Assays were conducted in 1.5-ml microcentrifuge tubes containing 50 mm KPO4 buffer of pH 7.8, 1.0 mm dithiothreitol, 1.0 mm MgCl2, 50 μm [3H]AdoMet (100 cpm/pmol), 1.0 mm pyrocatechol, human recombinant COMT, in a final volume of 0.1 ml. Reactions were initiated by the addition of [3H]AdoMet, incubated at 37 °C for 10 min, and terminated by the addition of 200 μl of 1 m HCl containing 1 μl/ml guaiacol as a carrier. The reaction mixtures were then extracted twice with 0.8-ml aliquots of toluene. After the addition of toluene, each sample was vortexed vigorously for 30 s and then centrifuged at 13,000 × g to aid in the formation of a distinct interface between the aqueous and organic phases. The organic layers were carefully pipetted from the aqueous layer, combined, and then backwashed with 1.0 ml of l m HCl by vigorous vortexing. The organic phase was again pipetted from the aqueous phase and mixed with scintillation fluid. The radioactivity was measured on a Tri-Carb 2100TR scintillation counter (machine efficiency for tritium is 65%), and the corrected cpm were converted to pmol of guaiacol formed using the specific radioactivity of AdoMet.
Construction of pET19b-COMT (WT) Expression Vectors
The vectors pET19b-COMT (D51G/S60F/K162R) and pET12a-COMT (K162R) were kindly provided by Prof. Zhu (Department of Pharmacology, Toxicology, and Therapeutics, University of Kansas Medical Center, Kansas City, KS). To obtain the pET12a-COMT (WT), the pET12a-COMT (K162R) construct was used as a template by conducting site-directed mutagenesis using the QuikChange method following the manufacturer's protocol. The following forward and reverse primers were used (where the underlined nucleotides indicate the mutation introduced): 5′-dGGCCTGCTGCGGAAGGGGACAGTGCTACTG-3′ and 5′-dCCAGTAGCACTGTCCCCTTCCGCAGCAGGCC-3′. The introduction of the wild-type sequence into the pET12a-COMT (K162R) construct was verified by dideoxysequencing. The pET12a-COMT (WT) vector was then digested with NdeI and BamHI to excise the COMT (WT) cDNA insert. The cDNA insert was then subcloned into the pET19b vector obtained by digesting pET19b-COMT (D51G/S60F/K162R) with NdeI and BamHI.
Expression and Purification of COMT
The pET19b-COMT expression vector was transformed into competent Escherichia coli C41(DE3) cells. The cells were grown in 1-liter cultures of Luria-Bertani medium at 37 °C (containing 100 μg/ml ampicillin). Upon reaching A600 = 0.6, 1 mm isopropyl-1-thio-β-d-galactopyranoside was added to induce enzyme expression, and cells were further cultured for 5 h. The culture was centrifuged for 10 min at 10,000 × g at 4 °C. Harvested cells were washed with saline, and the pellets were resuspended in 20 mm Tris-HCl (pH 7.5), 20 mm 2-mercaptoethanol, 0.2 m NaCl, Sigma protease inhibitor (1:100 (v/v); catalog no. P8340) and 5 mm imidazole. Resuspensions were lysed by sonication and centrifuged twice for 10 min at 10,000 × g at 4 °C. The lysate was loaded onto a nickel-Sepharose column equilibrated with 20 mm Tris-HCl (pH 7.5), 20 mm 2-mercaptoethanol, 0.2 m NaCl, and 5 mm imidazole, and the column was washed with the same buffer. Bound protein was eluted with a linear gradient of 100–400 mm imidazole. Active fractions containing COMT were identified using the standard COMT assay conditions, measurement of A280 nm, and visualization of the protein content of each fraction by SDS-PAGE. Peak fractions were pooled and dialyzed overnight in 20 mm Tris-HCl (pH 7.5) containing 1 mm EDTA, 10 mm DTT, 20% glycerol. COMT was obtained in homogeneous form by this procedure, as judged by SDS-PAGE, and had a final specific activity of 345 nmol of guaiacol formed/min/mg. The purified enzyme was stored in 20 mm Tris-HCl (pH 7.5) containing 1 mm EDTA, 10 mm DTT, 20% glycerol at −80 °C for future use. Under these storage conditions, the enzyme was found to be stable for 9 months.
Kinetic Studies on O-Methylation of PAH-catechols
Experiments were conducted anaerobically in a glove box purged with argon. All the solvent and aqueous solutions were degassed by freeze-pump-thaw cycling five times and stored in sealed containers filled with argon. The reactions were performed in 1.5-ml amber glass vials with polytetrafluoroethylene/silicone septa closures. The reaction system contained 10 mm KPO4 buffer of pH 7.8, 1.0 mm dithiothreitol, 1.0 mm MgCl2, 50 μm [3H]AdoMet (100 cpm/pmol), 0–20 μm PAH o-quinone in a final volume of 0.2 ml. The reactions were initiated by the addition of 0.225, 0.45, or 1.35 μg of human recombinant COMT at 25 °C or 2 μg of porcine COMT at 37 °C. The amount of enzyme used was always in the linear range as determined by plots of initial velocity versus enzyme concentration. The reactions were quenched by the addition of 50 μl of ice-cold 1% formic acid and were placed on ice. The reaction mixtures were then extracted twice with 0.7-ml aliquots of toluene, and the toluene layer was backwashed with 0.5 ml of H2O by vigorous vortexing. The organic phase was then dried by a SpeedVac concentrator (Thermo Scientific). The residue was dissolved in 100 μl MeOH, and an aliquot of 50 μl was analyzed by scintillation counting or by HPLC analysis. The initial velocity was estimated by the slope of the linear portion of the progress curve over 10 min. Kinetic analyses were performed by fitting the data using the program Grafit using non-linear regression to the Michaelis-Menten equation,
When substrate inhibition was observed, initial velocity data were fit in a similar manner to the following equation,
where v is the initial velocity of the reaction, [S] is the molar concentration of the substrate, and Km is the Michaelis-Menten constant for the substrate. Because of the iterative fits to the equations, each method provided estimates of the kinetic parameters as mean ± S.E. Dividing Vmax by the molar concentration of the enzyme gave kcat. Ki is the dissociation constant for the substrate from the enzyme inhibitor complex.
Metabolism of B[a]P-7,8-dione in Human Lung Cells
A549 (human lung adenocarcinoma cells) were obtained from American Type Culture Collection (ATCC number CCL-185) and maintained in F-12K nutrient mixture (Kaighn's modification) with 10% heat-inactivated FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. H358 (human bronchoalveolar cells) were obtained from American Type Culture Collection (ATCC number CRL-5807) and maintained in RPMI 1640 nutrient mixture with 10% heat-inactivated FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. HBEC-KT (immortalized human bronchial epithelial cells) originated from a patient without lung cancer were a gift from Dr. John Minna at University of Texas Southwestern Medical Center and maintained in keratinocyte-serum free medium medium with 0.1–0.2 ng/ml recombinant EGF, 20–30 μg/ml bovine pituitary extract, and 2 mm l-glutamine. Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2 and were passaged every 3 days at a 1:6 dilution. Cultured cells with a passage number of 10–20 were used in the experiments to reduce variability during cell culture.
The cells (5 × 106) were treated with B[a]P-7,8-dione (2 μm, 0.2% DMSO) in HBSS buffer containing 1 mm sodium pyruvate. The culture media were collected at 0 and 24 h, respectively, and subsequently acidified with 0.1% formic acid before extraction with 2 × 1.5-fold volume of cold H2O-saturated ethyl acetate. The organic phases of culture media were combined and dried under vacuum. The residue was redissolved in 100 μl of methanol. A 20-μl aliquot was analyzed by LC-MS/MS.
Identification of O-Methylated PAH-catechols by HPLC-RAM-UV and LC-MS/MS
The O-methylated metabolites generated with [3H]AdoMet were analyzed by a Waters Alliance 2695 chromatographic system (Waters Corp., Milford, MA) in tandem with a Waters 996 photodiode array detector and a β-RAM inline radiometric detector (IN/US Systems Inc., Tampa, FL). Chromatographic separation was achieved on a reverse-phase column (Zorbax-ODS C18, 5 μm, 4.6 × 250 mm, DuPont) eluted with the following linear gradient of H2O (0.1% formic acid; solvent A)/MeOH (solvent B) at a flow rate of 0.5 ml/min. For gradient 1, solvent B was changed from 80 to 98% (v/v) over 15 min, kept at 98% over 5 min, changed from 98 to 80% over 1 min, and kept at 80% for 4 min (for B[g]C-11,12-dione (11)). For gradient 2, solvent B was changed from 50 to 90% (v/v) over 5 min, kept at 90% over 15 min, changed from 90 to 50% over 1 min, and kept at 50% for 4 min (for NP-1,2-dione (1)). For gradient 3, solvent B was changed from 80 to 98% (v/v) over 5 min, kept at 98% over 15 min, changed from 98 to 80% over 1 min, and kept 80% for 4 min (for all other o-quinones). Eluates from the column were introduced into the inline radiometric detector following mixture of the scintillant with the HPLC effluent (IN/US Systems Inc.) at a flow rate 1.5 ml/min. The O-methylated B[a]P-7,8-catechols in cell extracts were separated on the same reversed phase column, but the mobile phase consisted of 5 mm ammonium acetate and 0.1% trifluoroacetic acid (TFA) in H2O (solvent A) and 5 mm ammonium acetate and 0.1% TFA in acetonitrile (solvent B) at a flow rate of 0.5 ml/min. Gradient conditions were as follows: 50–65% B over 30 min, 65–95% B over 10 min, followed by an isocratic hold at 95% B for another 10 min. At 50 min, the mobile phase was returned to 50% B in 1 min, and the column was equilibrated for 9 min before the next injection. The total run time for each injection was 60 min.
LC-MS/MS identification of the O-methylated catechols was conducted with a Finnigan TSQ Quantum Ultra spectrometer (Thermo Fisher, San Jose, CA) equipped with an atmospheric pressure chemical ionization source. The mass spectrometer was operated in the positive ion mode with the following parameters: discharge current (6 μA), vaporizer temperature (400 °C), sheath gas pressure (23 arbitrary units), auxiliary gas pressure (8 arbitrary units), capillary temperature (300 °C), tube lens offset (38 V), source CID (−14 V). The masses of the metabolites were obtained by detecting the molecular ion from Q1 full scan, and the corresponding mass spectrum of each metabolite was obtained from a Q3 full scan of the product ions of the molecular ion. The chromatography conditions for the metabolites were identical to those used for HPLC-RAM-UV detection.
Identification of the Major O-Methylated Metabolite of B[a]P-7,8-catechol by 1H NMR Analysis
To generate sufficient O-methylated metabolite of B[a]P-7,8-catechol for characterization, the O-methylation reactions were performed on a large scale. Metabolites were synthesized in a 100-ml system containing 10 mm KPO4 buffer of pH 7.8, 1.0 mm dithiothreitol, 1.0 mm MgCl2, 1.25 mm AdoMet, and 20 μm B[a]P-7,8-dione under anaerobic conditions. The reaction was started by the addition of 1.13 mg of human recombinant COMT and incubated for 90 min at 37 °C. The reaction was quenched by the addition of 250 μl of formic acid. Metabolites were extracted with 1 × 100 ml of ethyl acetate followed by 1 × 50 ml of ethyl acetate. The pooled extracts were evaporated to complete dryness. The residues were redissolved in 4 ml of methanol and subjected to semipreparative HPLC column (Whatman Partisil 10 ODS-3) chromatography. The column was eluted at a flow rate of 2 ml/min using a gradient of H2O (solvent A) and methanol (solvent B). Solvent B was changed from 30 to 80% (v/v) over 15 min and kept at 80% for 20 min, changed from 80 to 98% over 10 min and kept at 98% for 15 min, and changed from 98 to 30% over 1 min and kept at 30% for 9 min. The O-methylated catechols were collected on the basis of their characteristic UV spectra. The pooled elutes were evaporated to complete dryness. 1H NMR data were collected on a Bruker DMX 360 spectrometer operating at 360 MHz, using CDCl3 as the solvent.
RESULTS
O-Methylation of B[a]P-7,8-catechol by Porcine COMT
A pilot study was conducted to examine the ability of porcine COMT to detoxify B[a]P-7,8-dione by forming O-methylated B[a]P-7,8-catechols. Under anaerobic conditions, the o-quinone was completely reduced to PAH-catechol by dithiothreitol and then further O-methylated by porcine COMT in the presence of [3H]AdoMet as methyl group donor. As a result, two O-methylated metabolites were detected by HPLC with an inline β-RAM radioactive detector, which eluted close together from the reverse phase column (data not shown). The experiment was replicated with cold AdoMet, and the molecular ions of both metabolites were detected to be m/z 299 by LC/MS, indicating them to be two isomeric O-methylated B[a]P-7,8-catechols. With this system established, we next chose to study the ability of human recombinant COMT to O-methylate a series of PAH-catechols derived from their corresponding o-quinones.
Expression and Purification of COMT
We developed a simple, rapid purification of human recombinant His-tagged S-COMT to obtain this enzyme in high yield. SDS-polyacrylamide gel electrophoresis analysis gave a single band with an Mr of 27,000 demonstrating that homogeneous recombinant enzyme had been obtained for our studies. The specific activities of COMT using pyrocatechol and epicatechin as substrates under standard enzyme assay conditions were 345 and 339 nmol of O-methylated catechol formed/min/mg, respectively, and these values were about 20-fold higher than those previously reported for the same substrates (35). We then proceeded to conduct PAH-catechol conjugation with this enzyme.
Identification of O-Methylated PAH-catechols by HPLC-RAM and LC-MS/MS
A series of structurally diverse PAH o-quinones with differences in aromatic ring number and arrangement (starting from two rings of naphthalene-1,2-dione (1) to five rings of benzo[a]pyrene-7,8-dione (10)) were examined as substrates. Qualitatively, it was found that both porcine and human recombinant COMT were able to detoxify PAH o-quinone via O-methylation of the corresponding PAH-catechols. In most cases, two monomethyl isomers of PAH-catechols were formed by human COMT, as detected by both HPLC-RAM-UV (Fig. 1) and LC-MS/MS (Fig. 2). Based on the radiochromatograph peak area, the ratio of two isomeric O-methylated metabolites was estimated. PAH-catechols from naphthalene-1,2-dione (NP-1,2-dione (1)), chrysene-3,4-dione (C-3,4-dione (3)) and B[a]P-7,8-dione (10) formed one predominant metabolite accounting for over 80% of the product. Other PAH-catechols generated comparable amounts of the two isomeric metabolites (Table 2). The ratio of the two isomeric metabolites formed did not vary with the substrate concentration or over the time course of the reaction. MS/MS spectra provided detailed structure information on the isomers. The fragmentation patterns of all products under collision-induced dissociation were similar and demonstrated that the first cleavage resulted in the loss of 32 atomic mass units from the molecular ion, which corresponded to -OCH3. Rearrangement at the remaining phenolic group probably resulted in a change from -C-OH to -C=O. Subsequent cleavage then takes place at the -C=O group, resulting in a loss 28 atomic mass units. O-Methylated products from bay region methylated catechols (e.g. O-methylated 12MBA-3,4-catechol and O-methylated DMBA-3,4-catechol) also showed the loss of -CH3 (15 atomic mass units) from -OCH3 (Table 1). Our LC-MS/MS analyses confirmed that the metabolites in the reaction system were O-monomethyl PAH-catechols, but this method gave no information on the position of O-methylation.
FIGURE 1.

HPLC-RAM identification of the O-methylated metabolites (M1 and M2) of the representative PAH-catechols produced by human recombinant COMT. The reactions were conducted under anaerobic conditions in 10 mm KPO4 buffer, pH 7.8, 1.0 mm dithiothreitol, 1.0 mm MgCl2, 50 μm [3H]AdoMet, 20 μm PAH o-quinone, and 0.225 or 0.45 μg of human recombinant COMT at 25 °C. The reactions were quenched by 1% formic acid and extracted with toluene. The organic phase was then dried and dissolved in MeOH for HPLC-UV-RAM analysis. A and B, HPLC-UV-RAM chromatogram of O-methylated B[a]P-7,8-catechols; C and D, HPLC-UV-RAM chromatogram of O-methylated 5MC-1,2-catechols; E and F, HPLC-UV-RAM chromatogram of O-methylated B[g]C-11,12-catechols. The red triangles indicate the beginning and end of peak integration in the chromatograms. AU, absorbance units.
FIGURE 2.

LC-MS/MS identification of the O-methylated metabolites (M1 and M2) of the representative PAH-catechols containing a bay region (B[a]P-7,8-catechol), a methylated bay region (5MC-1,2-catechol), and a fjord region (B[g]C-11,12-catechol). The reactions were conducted under anaerobic conditions in 10 mm KPO4 buffer, pH 7.8, 1.0 mm dithiothreitol, 1.0 mm MgCl2, 50 μm unlabeled AdoMet, 20 μm PAH o-quinone, and 0.225 or 0.45 μg of human recombinant COMT at 25 °C. The reactions were quenched by 1% formic acid and extracted with toluene. The organic phase was then dried and dissolved in MeOH for LC/MS/MS analysis. A–C, ion chromatogram of O-methylated B[a]P-7,8- catechols, MS2 of isomer 1, and MS2 of isomer 2; D–F, ion chromatogram of O-methylated 5MC-1,2- catechols, MS2 of isomer 1 and MS2 of isomer 2; G–I, ion chromatogram of O-methylated B[g]C-11,12-catechols, MS2 of isomer 1, and MS2 of isomer 2.
TABLE 2.
Kinetic constants of o-methylation of PAH catechols
Kinetic parameters are given as mean values ± S.E.
| Quinone | Vmax | kcat | Km | Ki | kcat/Km | M1a | M2b |
|---|---|---|---|---|---|---|---|
| nmol/min/mg | min−1 | μm | μm | min−1 μm−1 | % | % | |
| NP-1,2-dione (1) | 114 ± 3.9 | 3.1 ± 0.1 | 0.6 ± 0.1 | 4.9 | 100 | - | |
| C-1,2-dione (2) | 159 ± 44 | 4.3 ± 1.2 | 2.5 ± 1.0 | 5.4 ± 2.5 | 1.7 | 62 | 38 |
| C-3,4-dione (3) | 16.4 ± 1.6 | 0.5 ± 0.04 | 26.4 ± 4.0 | 0.02 | 83 | 16 | |
| 5MC-7,8-dione (4) | 174 ± 25 | 4.7 ± 0.7 | 0.5 ± 0.2 | 3.9 ± 1.0 | 10.1 | 55 | 45 |
| BA-3,4-dione (5) | 118 ± 22 | 3.2 ± 0.6 | 0.8 ± 0.3 | 13.1 ± 5.7 | 4.0 | 59 | 41 |
| 7MBA-3,4-dione (6) | 326 ± 86 | 8.9 ± 2.3 | 5.6 ± 1.9 | 3.4 ± 1.4 | 1.6 | 53 | 47 |
| 12MBA-3,4-dione (7) | 188 ± 35 | 5.1 ± 1.0 | 0.6 ± 0.2 | 2.1 ± 0.7 | 9.0 | 62 | 38 |
| DMBA-3,4-dione (8) | 191 ± 47 | 5.2 ± 1.3 | 0.8 ± 0.3 | 3.1 ± 1.2 | 6.8 | 32 | 68 |
| B[c]Ph-3,4-dione (9) | 117 ± 3 | 3.2 ± 0.1 | 0.9 ± 0.1 | 3.5 | 67 | 34 | |
| B[a]P-7,8-dione (10) | 54.2 ± 3.0 | 1.5 ± 0.1 | 2.1 ± 0.4 | 0.7 | 90 | 10 | |
| B[g]C-11,12-dione (11) | 143 ± 64 | 3.9 ± 1.7 | 0.5 ± 0.4 | 1.6 ± 1.0 | 8.0 | 36 | 64 |
| Pyrocatechol | 345 ± 6 | 9.4 ± 0.6 | 46 ± 3 | 0.2 |
a Percentage of product as isomer 1.
b Percentage of product as isomer 2.
TABLE 1.
Molecular ions and product ions of o-methylated PAH catechols detected by LC/MS/MS
| O-Methylated catechols of quinones | Molecular ions m/z, [M + H]+ | Product ions m/z |
||
|---|---|---|---|---|
| [M + H-15]+ | [M + H − 32]+ | [M + H − 32 − 28]+ | ||
| NP-1,2-dione (1) | 175 | 143 | 115 | |
| C-1,2-dione (2) | 275 | 243 | 215 | |
| C-3,4-dione (3) | 275 | 243 | 215 | |
| 5MC-7,8-dione (4) | 289 | 257 | 229 | |
| BA-3,4-dione (5) | 275 | 243 | 215 | |
| 7MBA-3,4-dione (6) | 289 | 257 | 229 | |
| 12MBA-3,4-dione (7) | 289 | 274 | 257 | 229 |
| DMBA-3,4-dione (8) | 303 | 288 | 271 | 243 |
| B[c]Ph-3,4-dione (9) | 275 | 243 | 215 | |
| B[a]P-7,8-dione (10) | 299 | 267 | 239 | |
| B[g]C-11,12-dione (11) | 325 | 293 | 265 | |
NMR Analysis of O-Methylated B[a]P-7,8-catechols
To further confirm the structure of the major metabolite and position of O-methylation that occurred with B[a]P-7,8-catechol, an adequate amount of metabolite was enzymatically synthesized and analyzed by [1H]NMR. Integration of the NMR spectrum showed that there were 14 protons in the metabolite (Fig. 3). Protons at 4.16 ppm (s, 3H) were assigned to be ArOCH3, a broad peak at 5.95 ppm was assigned to ArOH, and the remaining 10 protons between 7.55 and 8.98 ppm were assigned to ArH. A minor peak with a peak area much less than 1 at 4.00 ppm was assigned to be the ArOCH3 of the minor isomer (where the underlined protons are the ones assigned). Based on literature precedent, the major O-methylated isomer was assigned as O-8-monomethyl-B[a]P-7,8-catechol (see “Discussion”).
FIGURE 3.

[1H] NMR spectrum of O-8-monomethylated B[a]P-7,8-catechol recorded at 360 MHz in CDCl3.
Kinetic Evaluation of O-Methylation of PAH-catechols
The kinetic profiles of PAH-catechols were different among different classes of PAH-catechols studied (bay region, methylated bay region, fjord region) (Fig. 4 and Table 2). Preference of O-methylation of PAH-catechols by COMT was ranked by comparing their catalytic efficiency (kcat/Km), which varied greatly among the classes of PAH-catechols studied (Table 2). Generally, O-methylation of most of the PAH-catechols, except C-3,4-catechol, was much more efficient (kcat/Km = 0.7–10 min−1 μm−1) than that of pyrocatechol, which is the simplest catechol (kcat/Km = 0.2 min−1 μm−1). O-Methylation of 5MC-1,2-catechol was most efficient. C-3,4-catechol seemed to be an exception among all PAH-catechols and was the least efficient, with a kcat/Km value that was 1–2 orders of magnitude lower than the other substrates. Km values of most of PAH-catechols were in the low micromolar range, which was much lower than that of pyrocatechol (Km = 46 μm). O-Methylation of catechols of NP-1,2-dione (1), C-3,4-dione (3), B[c]Ph-3,4-dione (9), and B[a]P-7,8-dione (10) followed normal Michaelis-Menten kinetics. For PAH-catechols of the chrysene and benz[a]anthracene series, O-methylation was accompanied by marked substrate inhibition. Non-linear regression analysis of the kinetic data fit to the Michaelis-Menten equation, and the equation for substrate inhibition clearly distinguished between these two kinetic patterns. This was most noticeable when the bay region was either methylated or occluded by a fjord region. For example in catechols of the chrysene series, inhibition with 5MC-1,2-catechol, which contains a methylated bay region, or B[g]C-11,12-catechol, which contains a fjord region, was pronounced. By contrast, chrysene-3,4-catechol followed normal Michaelis-Menten kinetics. In the benz[a]anthracene series, the methylated structure in 7MBA-3,4-catechol, 12MBA-3,4-catechol, and DMBA-3,4-catechol, produced marked inhibition relative to that of BA-3,4-catechol.
FIGURE 4.

Kinetic characterization of O-methylation of representative PAH-catechols containing a bay region (B[a]P-7,8-catechol), a methylated bay region (5MC-1,2-catechol), and a fjord region (B[g]C-11,12-catechol). The initial velocity was estimated by the slope of a linear portion of the progress curve over 10 min. Kinetic analyses were performed by fitting the data to the Michaelis-Menten equation or the substrate inhibition equation. Reactions contained 10 mm KPO4 buffer, pH 7.8, 1.0 mm dithiothreitol, 1.0 mm MgCl2, 50 μm unlabeled AdoMet, 0–20 μm PAH o-quinone, and 0.225 or 0.45 μg of human recombinant COMT at 25 °C. A, velocity versus [S] curve for B[a]P-7,8-catechol; B, velocity versus [S] curve for 5MC-1,2-catechol; C, velocity versus [S] curve for B[g]C-11,12-catechol.
S-Adenosyl-l-homocysteine (SAH) is a COMT inhibitor that is produced from AdoMet after donating the methyl group during O-methylation of catechol substrates (36). To determine whether SAH was responsible for the inhibition profiles observed during the O-methylation of PAH-catechols, we found the IC50 of the SAH for COMT to be about 30 μm. Because the highest substrate concentration used in these studies was 20 μm, it is unlikely that the formation of SAH was responsible for the inhibition observed. Therefore, the inhibition profiles for O-methylation of some PAH-catechols may be ascribed to substrate (catechol) inhibition.
Metabolism of B[a]P-7,8-dione in Human Lung Cells Treated with 2 μm B[a]P-7,8-dione
One mono-O-methylated B[a]P-7,8-catechol was formed in A549, H358, and HBEC-tk cell lines after incubating the cells with 2 μm B[a]P-7,8-dione for 24 h, and this was detected by LC-MS/MS (Fig. 5). The metabolite had the same retention time and the same reaction transition m/z 299→239, described in Table 1, as the O-8-monomethyl-B[a]P-7,8-catechol authentic standard that was enzymatically synthesized with human recombinant COMT. No significant amount of the other isomeric O-methylated catechol was detected in the cells.
FIGURE 5.
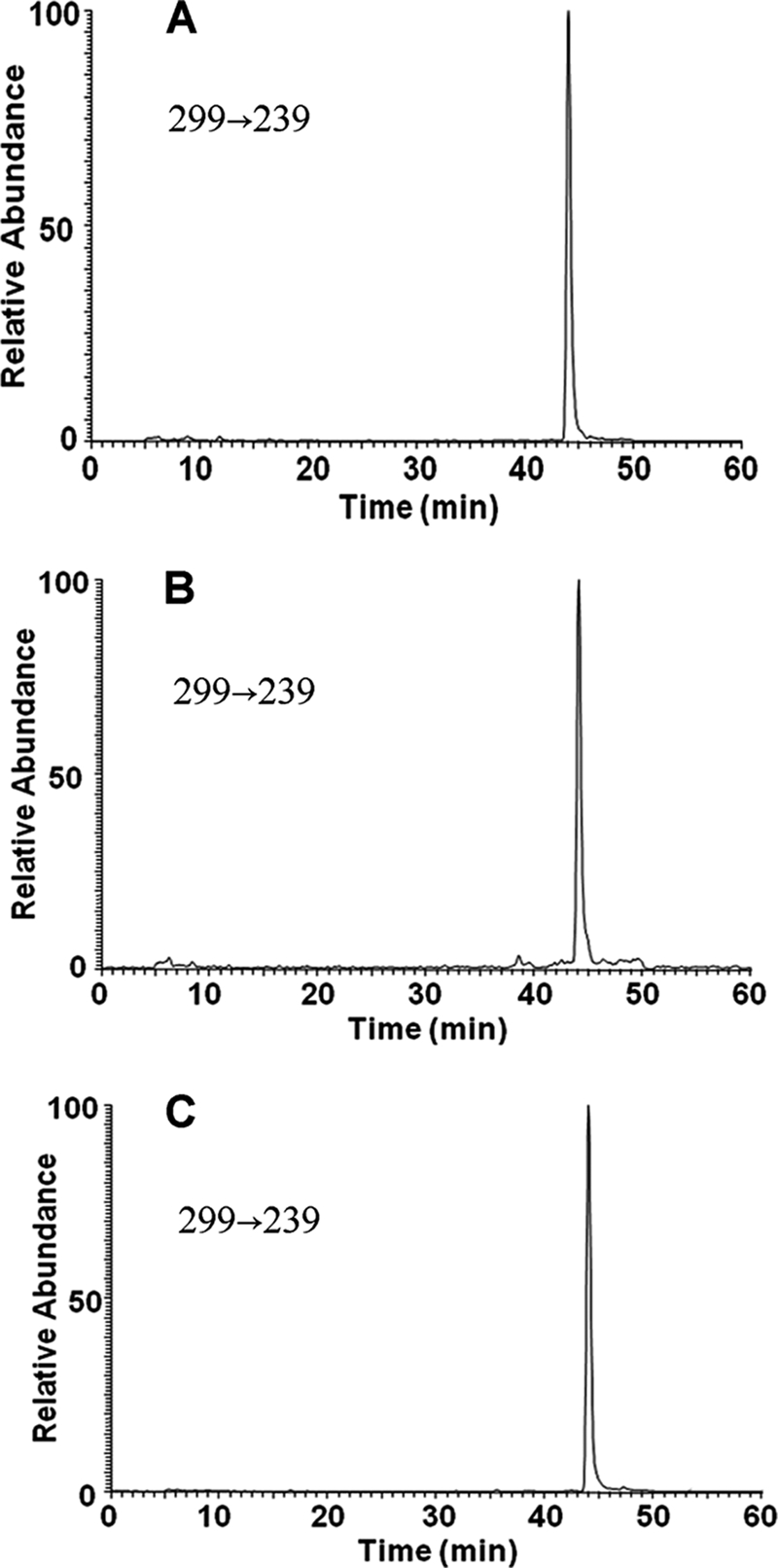
LC-MS/MS detection of O-8-monomethylated B[a]P-7,8-catechol in the extracted samples from A549 (A), H358 (B), and HBEC-KT (C) cells at 24 h. B[a]P-7,8-dione (2 μm, 0.2% DMSO) in HBSS buffer were incubated with lung cells and collected at 0 and 24 h, respectively. The culture media were extracted with ethyl acetate. The organic phases were dried under vacuum and redissolved in methanol. O-Methylated B[a]P-7,8-catechol was analyzed with LC-MS/MS by monitoring reaction transition m/z 299→239 ([M + H]+ → [M + H − 32 − 28]+). The peak had the same retention time and mass transition as authentic O-8-monomethylated B[a]P-7,8-cactechol.
DISCUSSION
This study demonstrates that PAH o-quinones produced by the AKR pathway of PAH activation can be intercepted at the level of the catechol by O-methylation catalyzed by human COMT. The o-quinones employed in these studies are the AKR products of the most representative PAH-trans-dihydrodiols. They are highly electrophilic Michael acceptors and redox-active compounds. O-Methylation of PAH-catechols catalyzed by COMT provides a possible pathway to eliminate the deleterious properties of PAH o-quinones.
To conduct this study, we first verified that O-methylation of B[a]P-7,8-catechol was possible with commercially available porcine COMT. We then purified human recombinant S-COMT from E. coli to conduct a thorough study. O-Methylation by COMT only took place at one of the two PAH-catechol hydroxyl groups generating mono-O-methylated catechols. Human S-COMT was more position-specific for catechols of NP-1,2-dione (1), C-3,4-dione (3), and B[a]P-7,8-dione (10) because one dominant product was formed. The predominant O-methylated B[a]P-7,8-catechol was chosen as a representative product for structure elucidation by [1H] NMR to identify the position mono-O-methylated. Previous studies assigned the chemical shift of the methoxy protons at the C1 and C2 positions of phenanthrene and naphthalene catechol. Although they were closely related, they were well resolved as singlets at 3.98 and 4.02 ppm in [1H]NMR, respectively (37, 38). Based on the presence of the sharp singlet at 4.16 ppm and another minor singlet at 4.00 ppm observed in the ratio of 9:1, we assigned the structure as O-8-monomethyl-B[a]P-7,8-catechol, which is equivalent to the 2-position of phenanthrene and naphthalene.
We next tried to rationalize why two positional isomers could form from the PAH-catechols. Based on the crystal structure of the human COMT 3,5-dinitrocatechol complex (39) (Protein Data Bank code 3BWY), we predict that Mg2+, AdoMet, and PAH-catechols would all bind in the shallow groove on the COMT surface. Mg2+ is octahedrally coordinated by Asp141, Asp169, Asn170, the two catechol hydroxyl groups, and a water molecule, thereby controlling the orientation of the catechol moiety for O-methylation. Because the positions of the two hydroxyl groups of 3,5-dinitrocatechol are invariant and fixed by coordination to Mg2+, we superimposed the B[a]P-7,8-catechol groups on those for this substrate (Scheme 3A). When we accommodate all of the PAH-catechols examined as substrates, we find that two productive binding modes are possible when the PAH ring systems are flipped (productive binding mode 1 and 2) (Scheme 3B). The active site can accommodate these larger ring structures in either binding mode without clashing with amino acid residue side chains, except with C-3,4-dione. Thus, two orientations for catechols exist at the active site, resulting in two isomeric methylated catechols observed. The positional specificity for O-8-methylation of B[a]P-7,8-catechol might be explained if it assumes one preferred orientation in COMT. For other PAH-catechols that produce almost equal amounts of each positional isomer, superimposition studies would suggest that neither productive binding mode is preferred over the other (Scheme 3B).
SCHEME 3.

Molecular docking of PAH-catechols in the active site of human COMT. A, human COMT containing 2,5-dinitrocatechol (DNC) and B[a]P-7,8 catechol modeled into the active site. Carbon atoms are colored in green for B[a]P-7,8-catechol, magenta for COMT, and gray for S-adenosyl-l-methionine (AdoMet) and DNC. All the other atoms are color-coded as follows: blue, nitrogen; red, oxygen; yellow, sulfur. Mg2+ and the coordinating water oxygen are shown in cyan and red, respectively. The interactions between the coordinating atoms and the metal ion are indicated by yellow dashed lines. The van der Waals surfaces of the residue atoms interacting with B[a]P-7,8-catechol are shown in dot representations. B, comparison of binding modes of different PAH-catechols. Two productive binding modes are shown (mode 1 and mode 2). The numbers are color-coded with the corresponding PAH ring systems as follows: C-1,2-dione (2, black); C-3,4-dione (3, blue); 7-MBA-3,4-dione (6, orange); B[c]Ph-3,4-dione (9, red); and B[a]P-7,8-dione (10, green). Note that in both binding modes the expanding ring systems of C-3,4-dione are close to residue Leu198 or Trp143, which may account for its low catalytic efficiency. To accommodate C-3,4-dione, both Leu198 and Trp143 must move.
In general, the O-methylation of PAH-catechols, except C-3,4-catechol, was much more efficient than that previously reported for other COMT substrates, such as catecholamines (kcat/Km = 0.07–0.18 min−1 μm−1) (40) and catecholestrogens (kcat/Km = 0.03–0.14 min−1 μm−1) (41). The low Km values of most PAH-catechols for COMT was the main contributor for the high catalytic efficiency. The decreased Km may reflect increased interaction between the ring systems of PAH-catechols and hydrophobic residues that are in the active site cavity.
There are some interesting structure-related trends that impact the catalytic efficiency of O-methylation of PAH-catechols. In the chrysene series, substitutions at the 5-position of C-1,2-catechol, such as 5MC-1,2-catechol, which contains a methylated bay region, and B[g]C-11,12-catechol, which contains a fjord region, seemed to enhance reaction efficiency. On the other hand, an additional benzene ring fused at the 10- and 11-positions of C-1,2-catechol (to yield B[a]P-7,8-catechol) reduces the reaction efficiency. In the benz[a]anthracene series, methyl group substitution at the 7-position of BA-3,4-catechol (7MBA-3,4-catechol) reduced reaction efficiency, whereas the methyl group at the 12-position, which forms a methylated bay region, increased the reaction efficiency. Dimethyl substitution at both the 7- and 12-positions reduced the catalytic efficiency of DMBA-3,4-catechol to a value that was intermediate between 7MBA-3,4-catechol and 12MBA-3,4-catechol. Based on these observations in the chrysene and benz[a]anthracene series, we speculate that substitution leading to a methylated bay region or a fjord region that results in steric clashing of bay region hydrogen atoms and a resultant bent planar structure enhances reaction efficiency of O-methylation. We also find that chrysene-3,4-catechol is an exceptionally poor substrate for COMT although two products are observed. Inspection of this catechol superimposed on 3,5′-dinitrocatechol in the active site of COMT indicates that the active site has to be significantly expanded to accommodate the two binding modes responsible for each product to form. Thus, both Leu198 and Trp143 would have to move (Scheme 3B).
Pronounced substrate inhibition was observed with many PAH-catechols, resulting in Michaelis-Menten plots that significantly deviate from being hyperbolic. Two trends were noteworthy in the PAH-catechols that caused pronounced substrate inhibition. One is that coincidentally they gave isomeric products in almost equal amounts, and the other is that they contained a bay region methyl or fjord region in their structure. However, the two binding modes that give rise to each positional isomer do not account for the substrate inhibition because each reaction would obey classical Michealis-Menten kinetics. Instead, the fit of the kinetic data to the substrate inhibition equation implies the formation of an abortive or nonproductive complex. In the absence of more structural information, we are unable to provide a model of this complex. The pronounced substrate inhibition observed with the methylated and fjord region PAH-catechols may have biological significance because, on the one hand, they yield high apparent catalytic efficiencies but, on the other hand, they inhibit the enzyme in the l.0–3.0 micromolar range. This suggests that at low micromolar concentrations, COMT may be inefficient in detoxifying these catechols, suggesting that these compounds may be more deleterious in producing ROS.
In addition to providing in vitro evidence for the O-methylation of PAH-catechols catalyzed by human COMT, we also demonstrated that B[a]P-7,8-dione was metabolized to O-methylated B[a]P-7,8-catechol in three human lung cell-based models. It seems that O-methylation by COMT is an important mechanism for the detoxication of structurally diverse PAH o-quinones, especially at low concentrations, and protection of the lung against PAH insult.
The human gene encoding COMT contains a common G to A polymorphism that results in valine to methionine substitution at residue 108 for the S-COMT or residue 158 for MB-COMT. The Met/Met homozygous COMT activity in red blood cells was less than half of wild type, and Met/Val heterozygous COMT showed intermediate activity (42). The low activity of COMT mutant may associate with its poor thermostability at physiological temperature and not with its different kinetic properties. Because the endogenous substrates of COMT include catecholamine neurotransmitters and catechol estrogens, it has been suggested that the COMT V108M polymorphism is related to increased risk of psychotic disorders and breast cancer (43, 44). This SNP in the COMT gene has also been readily associated with an increased risk of lung cancer (45, 46). Because COMT served as a detoxication enzyme for a series of structurally diverse PAH-catechols, the present study provides evidence that these polymorphic variants that reduce COMT activity may increase susceptibility to lung cancer caused by smoking and air pollution.
Acknowledgment
We thank Dr. Michael Byrns for conducting NMR analysis.
This work was supported, in whole or in part, by National Institutes of Health Grants P30-ES-013508, R01-CA39504, and PA-DOH4100038714.
- PAH
- polycyclic aromatic hydrocarbon
- B[a]P
- benzo[a]pyrene
- ROS
- reactive oxygen species
- 8-oxo-dGuo
- 7,8-dihydro-8-oxo-2′-deoxyguanosine
- COMT
- catechol-O-methyltransferase
- S-COMT and MB-COMT
- soluble and membrane-bound COMT, respectively
- AdoMet
- S-adenosyl-l-methionine
- NP-1,2-dione
- naphthalene-1,2-dione
- C-1,2-dione
- chrysene-1,2-dione
- C-3,4-dione
- chrysene-3,4-dione
- 5MC-1,2-dione
- 5-methyl-chrysene-1,2-dione
- BA-3,4-dione
- benz[a]anthracene-3,4-dione
- 7MBA-3,4-dione
- 7-methylbenz[a]anthracene-3,4-dione
- 12MBA-3,4-dione
- 12-methylbenz[a]anthracene-3,4-dione
- DMBA-3,4-dione
- 7,12-dimethylbenz[a]anthracene-3,4-dione
- B[c]Ph-3,4-dione
- benzo[c]phenanthrene-3,4-dione
- B[a]P-7,8-dione
- benzo[a]pyrene-7,8-dione
- B[g]C-11,12-dione
- benzo[g]chrysene-11,12-dione.
REFERENCES
- 1. Grimmer G., Böhnke H. (1975) J. Assoc. Off. Anal. Chem. 58, 725–733 [PubMed] [Google Scholar]
- 2. Hecht S. S. (1999) J. Natl. Cancer Inst. 91, 1194–1210 [DOI] [PubMed] [Google Scholar]
- 3. Gelboin H. V. (1980) Physiol. Rev. 60, 1107–1166 [DOI] [PubMed] [Google Scholar]
- 4. Conney A. H. (1982) Cancer Res. 42, 4875–4917 [PubMed] [Google Scholar]
- 5. Cavalieri E. L., Rogan E. G. (1995) Xenobiotica 25, 677–688 [DOI] [PubMed] [Google Scholar]
- 6. Sagher D., Strauss B. (1983) Biochemistry 22, 4518–4526 [DOI] [PubMed] [Google Scholar]
- 7. Shimada T., Hayes C. L., Yamazaki H., Amin S., Hecht S. S., Guengerich F. P., Sutter T. R. (1996) Cancer Res. 56, 2979–2984 [PubMed] [Google Scholar]
- 8. Shimada T., Gillam E. M., Oda Y., Tsumura F., Sutter T. R., Guengerich F. P., Inoue K. (1999) Chem. Res. Toxicol. 12, 623–629 [DOI] [PubMed] [Google Scholar]
- 9. Burczynski M. E., Harvey R. G., Penning T. M. (1998) Biochemistry 37, 6781–6790 [DOI] [PubMed] [Google Scholar]
- 10. Palackal N. T., Burczynski M. E., Harvey R. G., Penning T. M. (2001) Biochemistry 40, 10901–10910 [DOI] [PubMed] [Google Scholar]
- 11. Palackal N. T., Lee S. H., Harvey R. G., Blair I. A., Penning T. M. (2002) J. Biol. Chem. 277, 24799–24808 [DOI] [PubMed] [Google Scholar]
- 12. Murty V. S., Penning T. M. (1992) Bioconjug. Chem. 3, 218–224 [DOI] [PubMed] [Google Scholar]
- 13. Murty V. S., Penning T. M. (1992) Chem. Biol. Interact. 84, 169–188 [DOI] [PubMed] [Google Scholar]
- 14. Shou M., Harvey R. G., Penning T. M. (1993) Carcinogenesis 14, 475–482 [DOI] [PubMed] [Google Scholar]
- 15. McCoull K. D., Rindgen D., Blair I. A., Penning T. M. (1999) Chem. Res. Toxicol. 12, 237–246 [DOI] [PubMed] [Google Scholar]
- 16. Balu N., Padgett W. T., Nelson G. B., Lambert G. R., Ross J. A., Nesnow S. (2006) Anal. Biochem. 355, 213–223 [DOI] [PubMed] [Google Scholar]
- 17. Kasai H., Crain P. F., Kuchino Y., Nishimura S., Ootsuyama A., Tanooka H. (1986) Carcinogenesis 7, 1849–1851 [DOI] [PubMed] [Google Scholar]
- 18. Cheng K. C., Cahill D. S., Kasai H., Nishimura S., Loeb L. A. (1992) J. Biol. Chem. 267, 166–172 [PubMed] [Google Scholar]
- 19. Yu D., Berlin J. A., Penning T. M., Field J. (2002) Chem. Res. Toxicol. 15, 832–842 [DOI] [PubMed] [Google Scholar]
- 20. Park J. H., Troxel A. B., Harvey R. G., Penning T. M. (2006) Chem. Res. Toxicol. 19, 719–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shen Y. M., Troxel A. B., Vedantam S., Penning T. M., Field J. (2006) Chem. Res. Toxicol. 19, 1441–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park J. H., Mangal D., Tacka K. A., Quinn A. M., Harvey R. G., Blair I. A., Penning T. M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 6846–6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Axelrod J., Tomchick R. (1958) J. Biol. Chem. 233, 702–705 [PubMed] [Google Scholar]
- 24. Axelrod J. (1966) Pharmacol. Rev. 18, 95–113 [PubMed] [Google Scholar]
- 25. Ball P., Knuppen R., Haupt M., Breuer H. (1972) J. Clin. Endocrinol. Metab. 34, 736–746 [DOI] [PubMed] [Google Scholar]
- 26. Tenhunen J., Salminen M., Lundström K., Kiviluoto T., Savolainen R., Ulmanen I. (1994) Eur. J. Biochem. 223, 1049–1059 [DOI] [PubMed] [Google Scholar]
- 27. Grossman M. H., Emanuel B. S., Budarf M. L. (1992) Genomics 12, 822–825 [DOI] [PubMed] [Google Scholar]
- 28. Ulmanen I., Lundström K. (1991) Eur. J. Biochem. 202, 1013–1020 [DOI] [PubMed] [Google Scholar]
- 29. Männistö P. T., Kaakkola S. (1999) Pharmacol. Rev. 51, 593–628 [PubMed] [Google Scholar]
- 30. Tenhunen J., Ulmanen I. (1993) Biochem. J. 296, 595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jeffery D. R., Roth J. A. (1984) J. Neurochem. 42, 826–832 [DOI] [PubMed] [Google Scholar]
- 32. Grossman M. H., Creveling C. R., Rybczynski R., Braverman M., Isersky C., Breakefield X. O. (1985) J. Neurochem. 44, 421–432 [DOI] [PubMed] [Google Scholar]
- 33. Fu P. P., Cortez C., Sukumaran K. B., Harvey R. G. (1979) J. Org. Chem. 44, 4265–4271 [Google Scholar]
- 34. Harvey R. G., Dai Q., Ran C., Penning T. M. (2004) J. Org. Chem. 69, 2024–2032 [DOI] [PubMed] [Google Scholar]
- 35. Bai H. W., Shim J. Y., Yu J., Zhu B. T. (2007) Chem. Res. Toxicol. 20, 1409–1425 [DOI] [PubMed] [Google Scholar]
- 36. Ueland P. M. (1982) Pharmacol. Rev. 34, 223–253 [PubMed] [Google Scholar]
- 37. Bolchi C., Catalano P., Fumagalli L., Gobbi M., Pallavicini M., Pedretti A., Villa L., Vistoli G., Valoti E. (2004) Bioorg. Med. Chem. 12, 4937–4951 [DOI] [PubMed] [Google Scholar]
- 38. Kim Y. H., Moody J. D., Freeman J. P., Brezna B., Engesser K. H., Cerniglia C. E. (2004) J. Ind. Microbiol. Biotechnol. 31, 507–516 [DOI] [PubMed] [Google Scholar]
- 39. Rutherford K., Le Trong I., Stenkamp R. E., Parson W. W. (2008) J. Mol. Biol. 380, 120–130 [DOI] [PubMed] [Google Scholar]
- 40. Lotta T., Vidgren J., Tilgmann C., Ulmanen I., Melén K., Julkunen I., Taskinen J. (1995) Biochemistry 34, 4202–4210 [DOI] [PubMed] [Google Scholar]
- 41. Dawling S., Roodi N., Mernaugh R. L., Wang X., Parl F. F. (2001) Cancer Res. 61, 6716–6722 [PubMed] [Google Scholar]
- 42. Syvänen A. C., Tilgmann C., Rinne J., Ulmanen I. (1997) Pharmacogenetics 7, 65–71 [DOI] [PubMed] [Google Scholar]
- 43. Nolan K. A., Volavka J., Czobor P., Cseh A., Lachman H., Saito T., Tiihonen J., Putkonen A., Hallikainen T., Kotilainen I., Räsänen P., Isohanni M., Järvelin M. R., Karvonen M. K. (2000) Psychiatr. Genet. 10, 117–124 [DOI] [PubMed] [Google Scholar]
- 44. Goodman J. E., Jensen L. T., He P., Yager J. D. (2002) Pharmacogenetics 12, 517–528 [DOI] [PubMed] [Google Scholar]
- 45. Zienolddiny S., Campa D., Lind H., Ryberg D., Skaug V., Stangeland L. B., Canzian F., Haugen A. (2008) Carcinogenesis 29, 1164–1169 [DOI] [PubMed] [Google Scholar]
- 46. Cote M. L., Yoo W., Wenzlaff A. S., Prysak G. M., Santer S. K., Claeys G. B., Van Dyke A. L., Land S. J., Schwartz A. G. (2009) Carcinogenesis 30, 626–635 [DOI] [PMC free article] [PubMed] [Google Scholar]