

Abstract
The ATP binding cassette (ABC) proteins make up a large superfamily with members coming from all kingdoms. The functional form of the ABC protein nucleotide binding domain (NBD) is dimeric with ATP binding sites shared between subunits. The NBD is defined by six motifs: the Walker A, Q-loop, Signature, Walker-B, D-loop, and H-loop. The D-loop contains a conserved aspartate whose function is not clear but has been proposed to be involved in cross-talk between ATP binding sites. Structures of various ABC proteins suggest an interaction between the D-loop aspartate and an asparagine residue located in Walker A loop of the opposing subunit. Here, we evaluate the functional role of the D-loop using a bacteriophage T4 ABC protein, Rad50 (gp46). Mutation of either the D-loop aspartate or the Walker A asparagine results in dramatic reductions in ATP affinity, hydrolysis rate, and cooperativity. The mutant proteins bind Mre11 (gp47) and DNA normally, but no longer support the ATP-dependent nuclease activities of Mre11. We propose that the D-loop aspartate functions to stabilize the Walker A asparagine in a position favorable for catalysis. We find that the asparagine is crucially important to the mechanism of ATP hydrolysis by increasing the affinity for ATP and positioning the γ-phosphate of ATP for catalysis. Additionally, we propose that the asparagine acts as a γ-phosphate sensor and, through its interaction with the conserved D-loop aspartate, transmits conformational changes across the dimer interface to the second ATP binding site.
Keywords: DNA Enzymes, DNA Recombination, DNA Repair, Enzyme Kinetics, Enzyme Mechanisms, ABC Protein, DNA Exonuclease, SMC Protein
Introduction
Rad50 and Mre11 form a heterotetrameric complex (Mre112/Rad502, referred to as the MR2 complex) that is involved in DNA double-strand break (DSB) repair (1). DSBs commonly occur and are caused by both internal and external agents. External DSB-inducing agents include ionizing radiation, ultraviolet light, and genotoxic chemicals (mutagens) that are found in the environment (2). Internal DSB-inducing agents include metabolic by-products (reactive oxygen species), tightly bound proteins that have the potential to stall the replication fork, and enzyme-catalyzed programmed DSBs such as those that occur during meiosis or the generation of antibody diversity (3–5). If not properly repaired DSBs have the potential to cause genomic rearrangements, which can lead to carcinogenesis or cellular death (6). There are three main pathways for repairing DSBs: homologous recombination (HR), non-homologous end-joining, and microhomology mediated end-joining (7). The MR complex plays a direct role in all three of these pathways but its role in HR is the best understood. HR is a multistep pathway involving dozens of proteins (8). The MR complex is involved in the first step of HR, which is the generation of a 3′ single-stranded DNA in a process referred to as DSB resection (the 5′ strand is resected). The precise role of the MR complex in DSB resection has only recently been revealed using the Saccharomyces cerevisiae model system (9–12). In eukaryotes, DSB resection occurs in two stages (13). In the first stage, the MR complex, along with Xrs2 and Sae2 in S. cerevisiae, removes 50–100 nucleotides from the 5′ end of the DSB. In the second stage, either Sgs1/Top3/RmiI, Dna2, and RPA or ExoI and RPA bind to the DSB that has been processed by the MR complex and carry out an extensive resection of the DSB (>500 nucleotides). After DSB resection, the recombinase (RecA or Rad51 ortholog) catalyzes strand invasion of the 3′ single-stranded DNA into a homologous DNA template creating a D-loop that is used as a primer for DNA synthesis (14). Finally, the extended strand can be dissociated from the homologous template through the action of a DNA helicase or the Holliday junction can be processed with a Holiday junction nuclease and DNA ligase.
Eukaryotes, Archaea, and several bacteriophages use the MR complex for DSB resection (1). Although bacteria contain an MR homolog (the SbcCD complex), it appears that it is not involved in DSB resection but instead functions to prevent of cruciform structures that have the potential to block the replication fork (15). In bacteria, the well characterized RecBCD helicase/nuclease carries out DSB resection (16). The T4 MR (gp46/47) complex has been shown in vivo to be required for homologous recombination and DSB repair and it is thought that the T4 and eukaryotic MR complexes perform similar functions (17).
The ATP binding cassette protein (ABC) superfamily is a diverse set of proteins that are found in all kingdoms of life (18). The majority of ABC proteins are transmembrane domain-containing transporters but a significant fraction are soluble proteins involved in DNA repair, chromosome condensation, RNA-protein remodeling, and protein translation (19). The common feature of ABC proteins is the nucleotide binding domain (NBD), which contains six conserved motifs: the Walker A, Q-loop, Signature, Walker-B, D-loop, and H-loop. Various crystal structures of ABC proteins have demonstrated that all six motifs are necessary to form a complete ATPase active site (19). The first crystal structure depicting what is the relevant dimeric form of the ABC protein NBD was Pyrococcus furiosus (Pfu) Rad50 (20). The structure revealed ATP-bound dimer arranged in a head-to-tail fashion so that each subunit presents its conserved motifs at the dimeric interface. The Walker A, Walker B, Q-loop, and H-loop motifs from one monomer form one-half of the ATP binding site with the Signature and the D-loop motifs from the adjacent monomer completing the second half. Rad50 proteins have an additional conserved motif, the CXXC motif that is located in the middle of a coiled-coil that splits the N- and C-terminal subdomains of the NBD (1). The CXXC motif has been shown in dimerize with a second CXXC motif to bind a Zn2+ cation in a tetrathiolate linkage similar to zinc-finger proteins (21). The exact function of this linkage is unclear but it is thought to mediate Rad50-dependent tethering of DNA ends (1, 22).
The physiological function of the MR complex is a DNA nuclease, which is carried out by the Mre11 subunit. Mre11 is a member of the protein phosphatase superfamily, which require divalent cations for activity (23). Rad50 is always required for the activity of T4 Mre11 but ATP is only activating when repetitive exonuclease activity is assayed, suggesting that ATP hydrolysis is involved in the translocation of the MR complex along the DNA substrate (24). Consistent with this, removal of the first two nucleotides during the 3′ to 5′ exonuclease reaction is ATP-independent, as is the observed single-stranded DNA endonuclease activity (24). The canonical divalent cation for Mre11 is Mn2+, but recent work on Mre11 from Pfu and T4 indicate that Mg2+ also supports nuclease activity (24, 25). Interestingly, Mg2+ appears to favor a potentially more physiologically relevant double-stranded DNA endonuclease activity that produces a 3′ single-stranded DNA overhang that is an intermediate in the HR pathway (25).
Although ATP is not necessary for all types of nuclease activity that can be carried out by the MR complex, mutagenesis studies have shown that, in vivo, ATP binding and hydrolysis are absolutely essential. The mechanism of ATP hydrolysis by the ABC protein superfamily, and the function of its six conserved motifs, has been an area of intense investigation. The functional roles of the classic ATPase motifs such as Walker A and Walker B are fairly well understood. The Walker A lysine stabilizes the hydrolysis transition state by counteracting the build-up of negative charge that occurs. The Walker B aspartate is virtually always involved in binding of the Mg2+ cation and the glutamate that follows the aspartate is believed to be the catalytic base that abstracts a proton from the attacking water molecule. The roles of the other four motifs that define the ABC protein superfamily are less understood. Crystal structures indicate that the Q-motif glutamine interacts with the Mg2+ cation and has been proposed to be involved in allosteric signal transmission to protein domains that are either covalently or tightly associated with the ABC protein NBD (e.g. Mre11 in the MR complex) (19). The Signature motif forms part of the ATP active site in trans and recent work from our laboratory indicates roles in the catalytic mechanism for ATP hydrolysis, ATP cooperativity, and allosteric signal transmission between the ATP and DNA binding sites (37). The H-loop histidine is somehow involved in catalysis, but its specific role is not known. It has been proposed that it may act as the catalytic base (instead of the Walker B glutamate) or as part of a “catalytic dyad” with the Walker B glutamate residue (26). The function of the D-loop is perhaps the least well understood of the six conserved ABC superfamily motifs. In various crystal structures of the ABC protein NBDs, the D-loop aspartate is found at the dimer interface, near the ATP binding site, but not making direct contacts with the bound ATP. Like the Signature motif, the D-loop motif acts in trans and therefore has been proposed to be involved in cross-talk between the two subunits of the NBD (20).
To reveal the functional importance of the ABC protein D-loop motif, we employed T4 Rad50 and site-directed mutagenesis. The D-loop aspartate and glutamate residues, along with their putative interaction partners, the Walker A asparagine and arginine residues (Fig. 1A), were mutated and their effects on ATP affinity, hydrolysis, and cooperativity were evaluated. Additionally, to clarify the role of ATP on the nuclease activity of Mre11, we examined the effects of these mutations within the context of the MR complex. Mutation of the D-loop glutamate and Walker A arginine has little effect on the kinetic parameters for Rad50 ATP hydrolysis or the nuclease activity of Mre11. The driving force behind the absolute conservation of the D-loop glutamate in Rad50 proteins ranging from human to phage is therefore not apparent from the numerous assays we carried out. In contrast, mutation of either the D-loop aspartate or the Walker A loop asparagine results in drastic reduction in kcat-ATP, a large inflation in both Km-ATP and Kd-ATP, and a reduction in the cooperativity of ATP hydrolysis. Although these mutations do not affect the ATP-independent nuclease activity of Mre11, the addition of ATP severely inhibits repetitive nuclease activity, suggesting that the complex is stalled in the ATP-bound form and cannot translocate or dissociate from the DNA substrate. These results, combined with the x-ray crystal structure of PfuRad50, strongly suggest that one of the roles of the D-loop aspartate is to stabilize and position the Walker A loop asparagine. The Walker A asparagine hydrogen bonds with the γ-phosphate of ATP, thereby enhancing the affinity of the enzyme for ATP and orienting its γ-phosphate for nucleophillic attack by the catalytic water molecule. Additionally, because of the effects these mutations have on cooperativity and DNA activation, we proposed that the Walker A asparagine acts as a γ-phosphate sensor that communicates the presence of ATP across the subunit interface through its interactions with the D-loop aspartate.
FIGURE 1.

A, ribbon structure of the dimeric PfuRad50 nucleotide binding domain. The subunits of the dimer (PDB code 1F2U) are colored green and cyan with the ATP molecules located at the dimer interface. The Walker A and D-loop motifs from opposing subunits that form a single ATP active site are indicated. B, multiple amino acid sequence alignment of the Walker A and D-loop motifs from selected Rad50 and ABC proteins. The protein names are preceded by the organism abbreviations, which are as follows: T4, bacteriophage T4; Pfu, P. furiosus; hs, Homo sapiens; and ec, E. coli. The dark gray shading indicates the conservation of the residues mutated in this study.
EXPERIMENTAL PROCEDURES
Materials
Oligodeoxynucleotides used for mutagenesis were from the Iowa State University DNA Facility. Pfu DNA polymerase Ultra was from Agilent Technologies, Inc. DNA sequencing was performed at the Iowa State University DNA Facility. Cellulose phosphate (coarse mesh), coupling enzymes (pyruvate kinase and lactate dehydrogenase), dNTPs, and NADH were from Sigma. Phosphoenolpyruvate was purchased from Alfa Aesar. Adenosine 5′-triphosphate was purchased from U. S. Biochemical Corporation. General chemicals, buffers, and media components were purchased from Fisher Scientific. Wild-type Mre11 protein was purified as described (24).
Mutagenesis
The Stratagene QuikChange site-directed mutagenesis protocol was used to generate the desired mutations. The presence of the mutation and the integrity of the entire open reading frame were verified by DNA sequencing. The forward primer sequences (mutated codon underlined; mutated bases in bold letters) used in the mutagenic reactions are: R37A, 5′-CAAAAAACTCTTATTACTGGAGCAAATGGCGGTGGTAAGTCTAC-3′; N38A, 5′-CTCTTATTACTGGACGAGCGGGCGGTGGTAAGTCTACTATGC-3′; D512N, 5′-GTGTTTGATGGGTCATTTAACGCCGAAGGTATTAAAGGTGTAGC-3′; D512A, 5′-GTGTTTGATGGGTCATTTGCGGCCGAAGGTATTAAAGGTGTAGC-3′; E514Q, 5′-GGGTCATTTGATGCCCAAGGTATTAAAGG-3′; E515A, 5′-GGGTCATTTGATGCCGCAGGTATTAAAGG-3. The reverse mutagenic primers were the reverse complement of the forward.
Protein Expression and Purification
Chemically competent Escherichia coli BL21(λDE3) Rosetta cells were transformed with the pET28 plasmid that encodes either T4 wild-type (WT) or mutant Rad50 containing an N-terminal His6 affinity tag. The transformed cells were plated onto LB agar containing 34 μg/ml of kanamycin and incubated overnight at 37 °C. 20 ml of LB-kanamycin were inoculated with a single colony and incubated overnight at 37 °C. 10 ml of this culture was used to inoculate two 1-liter LB-kanamycin flasks. The liquid cultures were shaken at 37 °C until the optical density reached 0.8. The cultures were then cooled to 18 °C and induced with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside. These induced cultures were incubated, shaking for ∼20 h at 18 °C, and harvested by centrifugation at 6000 × g.
Bacterial cell pellets containing expressed Rad50 (10 g wet cell paste) were resuspended in 100 ml of buffer containing 20 mm Tris-Cl, 500 mm NaCl, 5 mm imidazole, 10% glycerol (v/v), pH 8.0 (4 °C). Lysis was achieved by homogenization using an EmulsiFlex-C5 (Avestin, Inc.) at ∼12 k.p.s.i. The lysate was clarified by centrifugation at ∼32,500 × g and the supernatant was loaded onto ∼ 3 ml of nickel-agarose resin. The column was washed with 150 ml of lysis buffer, followed by 50 ml of lysis buffer containing 20 mm imidazole then 100 ml of lysis buffer containing 1 m NaCl. Prior to elution, the salt concentration was lowered to 200 mm by washing the column with 50 ml of 20 mm Tris-Cl, 200 mm NaCl, 10% glycerol (v/v), pH 8.0 (4 °C). The Rad50 protein was eluted in buffer containing 20 mm Tris-Cl, 200 mm NaCl, 150 mm imidazole, 10% glycerol (v/v), pH 8.0 (4 °C). The collected protein was loaded onto a 10-ml cellulose phosphate column equilibrated with 20 mm Tris-Cl, 200 mm NaCl, 10% glycerol (v/v), pH 8.0 (4 °C). The column was washed with 100 ml of equilibration buffer and then the Rad50 protein was eluted with 20 mm Tris-Cl, 400 mm NaCl, 20% glycerol (v/v), pH 8.0 (4 °C). The eluted protein was concentrated using an Amicon Ultra 50K concentrator. The final concentration was determined spectrophotometrically using an extinction coefficient (ϵ280 = 33,140 m−1 cm−1) calculated from the deduced protein composition.
Circular Dichroism (CD) Spectroscopy
CD studies on wild-type and mutant Rad50 were done at 22 °C on a Jasco J710 CD spectrometer in a 1-cm cell using a protein concentration of 1.0 to 0.5 mg/ml. Spectra were collected from 200 to 260 nm in increments of 1 nm. Each spectrum was blank-corrected and normalized to the ellipticity of the WT protein at 220 nm to correct for differences in protein concentration.
Mre11 Pull-down Assay
Purified Mre11 (10 μg) and purified WT or mutant Rad50 proteins (10 μg) were incubated for 10 min at 22 °C in 100 μl of pull-down buffer (20 mm Tris-Cl, 500 mm NaCl, 5 mm imidazole, 10% glycerol (v/v), pH 8.0). After the incubation period, 10 μl of nickel-agarose was added, followed by gentle rocking for 10 min to facilitate binding to the nickel-agarose. Next, 1 ml of pull-down buffer was added and the microcentrifuge tube was centrifuged at 3000 × g for 5 min. The supernatant was carefully removed and the nickel-agarose was washed with another 1 ml of pull-down buffer. The washing/centrifugation process was repeated three times. After the third wash, 50 μl of pull-down buffer plus 150 mm imidazole was added to the pelleted nickel-agarose, incubated for 5 min, and then centrifuged at 12,000 × g for 5 min. The 25 μl of the supernatant was removed and analyzed via SDS-PAGE.
Steady-state ATPase Kinetics
A standard coupled enzyme assay was used to measure steady-state ATP hydrolysis kinetics (27). Initial velocities were determined by coupling the production of ADP to the oxidation of NADH (50 μm) with pyruvate kinase/lactate dehydrogenase (1.8 units of protein kinase, 3 units of lactate dehydrogenase/assay). All assays were done at 30 °C in the presence of 50 mm Tris-Cl, 50 mm KCl, 5 mm MgCl2, and 0.1 mg/ml of BSA, pH 7.6, in a volume of 300 μl. ATP hydrolysis was assayed fluorometrically using an excitation wavelength of 340 nm and the rate of change in NADH fluorescence was monitored at an emission wavelength of 460 nm on a Cary Eclipse spectrofluorometer (Varian). All assays were started with the addition of Rad50 or preassembled MR complex. To determine estimates of maximum velocity (Vmax), Michaelis constant (Km), and Hill coefficient, the reaction velocities at various ATP concentrations were fitted to the Hill equation using Sigmaplot 10.0/Enzyme Kinetics Module 1.3 (Systat Software, Inc.),
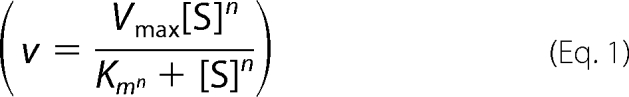 |
where v is the velocity at a given substrate (S) concentration, Vmax is the maximum velocity at saturating S, Km is the Michaelis constant, and n is the Hill coefficient. Steady-state kinetic constants were determined for both Rad50 alone and the Rad50·Mre11·DNA (MR-D) complex. The Mre11 concentration was always held in excess over the Rad50 concentration (5% molar excess). The MR-D assay contained a 1.5-fold excess of DNA over the MR complex and was well above the Kactivation-DNA (activation constant for DNA, i.e. concentration of DNA that gives 50% activation) (24).
The sequence of the oligonucleotides used to create the DNA substrate were the following, MR_DNA_ds50-F, 5′-CTCTTGGTGATTATGATGGTTGCAATACATTTAATTTCATTATCAATAAG-3′; MR_DNA_ds50-R, 5′-CTTATTGATAATGAAATTAAATGTATTGCAACCATCATAATCACCAAGAG-3′. The oligonucleotides were dissolved in sterile water and quantitated by measuring the absorbance at 260 nm (ϵMR_DNA_ds50-F, 487,300 m−1 cm−1, ϵMR_DNA_ds50-R, 515,100 m−1 cm−1). The oligonucleotides were mixed in equal molar ratios and annealed by heating to 80 °C for 5 min and slowly cooled to room temperature.
Determination of the Equilibrium Dissociation Constant for ATP
All fluorescence measurements were performed using a Cary Eclipse spectrofluorometer (Varian). Intrinsic tryptophan fluorescence of the Rad50 mutants (1 μm) was measured at 30 °C in 3.0 ml of 50 mm Tris-Cl, 50 mm KCl, 10 mm MgCl2, pH 8.0. Samples were excited at 295 nm and the emission was monitored at 335 nm. The measured values were first corrected for dilution and then for inner filter effect using the following equation:
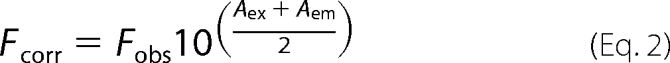 |
where Fcorr is the corrected fluorescence, Fobs is the observed fluorescence, Aex is the absorbance at 295 nm, and Aem is the absorbance at 335 nm. The corrected fluorescence data were plotted against ATP concentrations and fit to a simple equilibrium binding mechanism using Dynafit software (Biokin, Ltd.). The Kd estimates reflect the average of at least three separate titrations.
Steady-state Nuclease Activity
Nuclease activity was estimated by monitoring the removal of an internal fluorescent 2-aminopurine (2-AP) deoxyribonucleotide at either the 2nd or 17th position relative to the 3′ end of the substrate. Oligonucleotides used to create the 2nd position substrate were as follows (the 2-AP deoxyribonucleotide position is denoted by the bold X): MR_DNA_ds50-F(3′2AP), 5′-CTCTTGGTGATTATGATGGTTGCAATACATTTAATTTCATTATCAATTXG-3′; MR_DNA_ds50-R(3′2AP), 5′-CTAATTGATAATGAAATTAAATGTATTGCAACCATCATAATCACCAAGXG-3′. Oligonucleotides used to create the 17th position DNA substrate were as follows: MR_DNA_ds50-F(3′17AP), 5′-CTCTTGGTGATTATGATGGTTGCAATACATTTAXTTTCATTATCAATTAG-3′; MR_DNA_ds50-R(3′17AP), 5′-CTAATTGATAATGAAATTAAATGTATTGCAACCXTCATAATCACCAAGAG-3′. The oligonucleotides were dissolved in sterile water and quantitated by measuring the absorbance at 260 nm (ϵMR_DNA_ds50-F(3′2AP/17AP), 474,300 m−1 cm−1, ϵMR_DNA_ds50-R(3′2AP/17AP), 501,500 m−1 cm−1). The oligonucleotides were mixed in equal molar ratios and annealed by heating to 80 °C for 5 min and slowly cooled to room temperature. All assays were done at 30 °C in the presence of 50 mm Tris-Cl, 50 mm KCl, 5 mm MgCl2, 0.3 mm MnCl2, and 0.1 mg/ml of BSA, pH 7.6. The concentration of Mre11 was always in slight excess to Rad50. The concentrations of both the 2nd and 17th position DNA substrates were held at 1.3 μm. Samples were excited at 310 nm and the rate of change in fluorescence at 375 nm was monitored. Initial rates were determined for the MR complex (50 and 100 nm complex) on the 2nd position DNA substrate over a 5–10 min period in the absence of ATP. For the 17th position DNA substrate, rates were determined over the initial 5 min of the reaction for the MR complex (400 nm complex) in both the absence and presence of ATP.
RESULTS
Protein Expression, Purification, and Interaction with Mre11
The mutant Rad50 proteins behaved identically to the WT protein with very similar levels of expression (20 mg/liter of LB) and purity (supplemental Fig. S1). Like many DNA-binding proteins, T4 Rad50 binds to cellulose phosphate resin. All six mutants behave identically with regards to cellulose phosphate binding and elution (at 200 and 400 mm, respectively), suggesting that the overall structure of the mutant proteins is unchanged compared with the WT protein. Consistent with this, the circular dichroism spectra of WT and the Rad50 mutants are very similar (supplemental Fig. S2). To assess the ability of the Rad50 mutants to interact with Mre11, we performed a nickel-agarose pull-down assay. WT and the mutant Rad50 proteins are purified via a N-terminal His6 tag, whereas Mre11 is purified through a chitin binding domain and intein-mediated self-cleavage. Because Mre11 lacks a His tag, its retention on nickel-agarose relies on a tight interaction with Rad50 and indeed, a pull-down assay performed without Rad50 indicates that Mre11 does not non-specifically interact with the nickel-agarose resin (supplemental Fig. S3, lane 4). The pull-down assay indicates that WT and all six mutant Rad50 proteins interact with Mre11 (supplemental Fig. S3, lanes 5–11). The interaction between Mre11 and Rad50 survives extensive washing over a period of ∼30 min, suggesting that the interaction between Rad50 and Mre11 is very tight (i.e. a slow rate of dissociation). This result is consistent with the co-purification of Mre11 and Rad50 that is observed in human, S. cerevisiae, and Pfu systems (22, 28, 29).
Determination of Mg·ATP Dissociation Constants for Mutant Rad50 Proteins
T4 Rad50 contains two tryptophan residues, one of which is located in a region that is thought to undergo a significant structural rearrangement upon binding of ATP (20). Consistent with this, ATP binding to WT Rad50 results in an ∼30% quenching of tryptophan fluorescence, which enables the straightforward determination of Kd-ATP (9.4 μm for WT). To evaluate the effect of the Walker A and D-loop mutations on ATP affinity we determined the Kd-ATP for each Rad50 mutant (Table 1). The ATP affinity of R37A, E514Q, and E514A is slightly weaker compared with WT Rad50 (2.2–2.9-fold increases in Kd-ATP). In contrast, the N38A, D512N, and D512A Rad50 mutants have much weaker ATP affinity (22–36-fold increases in Kd-ATP). The degree of fluorescent quenching at saturating concentrations of ATP (30%) is similar for all of the Rad50 mutants studied here (data not shown), suggesting that the mutations have not affected the ability of Rad50 to adopt a WT-like ATP-bound conformation.
TABLE 1.
Kinetic constants for wild-type and mutant T4 Rad50 ATP hydrolysis activity
| Protein | kcat | Km-ATP | Hill coefficient | Kd-ATP |
|---|---|---|---|---|
| s−1 | μm | μm | ||
| WTa | 0.15 ± 0.01 | 16 ± 1 | 1.4 ± 0.1 | 9.4 ± 1.2 |
| R37A | 0.11 ± 0.01 | 27 ± 1 | 1.4 ± 0.1 | 24 ± 2.2 |
| N38A | 0.0025 ± 0.0001b | NDc | NDc | 280 ± 32 |
| D512N | 0.0024 ± 0.0003b | NDc | NDc | 340 ± 21 |
| D512A | 0.0037 ± 0.0003b | NDc | NDc | 220 ± 24 |
| E514Q | 0.12 ± 0.002 | 25 ± 1 | 1.4 ± 0.1 | 27 ± 1.7 |
| E514A | 0.12 ± 0.005 | 30 ± 3 | 1.2 ± 0.1 | 21 ± 2.1 |
a Herdendorf et al. (24).
b Specific activity determined with 2 mm ATP. Error represents the standard deviation of 3 measurements.
c ND, not determined.
Steady-state Kinetic Characterization of the ATPase Activity for Mutant Rad50 Proteins and MR-D Complexes
WT Rad50 has a kcat of 0.15 s−1, a Km-ATP of 16 μm, and a Hill coefficient of 1.4 (24). The kinetic parameters for R37A, E514Q, and E514A are relatively unchanged compared with WT Rad50 (Table 1). There are slight increases in Km-ATP (1.5 to 1.9-fold), very minor decreases in kcat-ATP (1.3–1.2-fold), and essentially no change in ATP cooperativity (within the standard error of the estimated constant). On the other hand, large reductions in kcat-ATP are observed for N38A, D512N, and D512A (40–60-fold). The activity of these three mutants is reduced to a level that makes determination of the kinetic parameters Km-ATP and Hill coefficient difficult (i.e. detection of the ATPase activity at ATP concentrations 5-fold below the Kd-ATP of the mutant requires impractically long time courses). For this reason, the reported kcat-ATP values given in Table 1 are formally specific activities determined at saturating ATP concentrations (as determined by their Kd-ATP).
We have previously shown that Mre11 and double-stranded DNA are allosteric effectors of WT Rad50, causing a 20-fold increase in kcat-ATP, a 3-fold increase in Km-ATP, and an increase in ATP cooperativity from 1.4 to 2.4 (24). To determine how the Walker A and D-loop mutations affect this allosteric activation, we determined the kinetic parameters for the mutant MR-D complexes. Due to the increase in kcat-ATP in the presence of Mre11 and DNA, we were able to determine the kinetic constants for all of the Rad50 mutants (Table 2). The results for R37A, E514Q, and E514A mirrored those of the mutant Rad50 proteins alone with 2-fold increases in Km-ATP, very minor decreases in kcat-ATP (less than 1.4-fold), and essentially no change in ATP cooperativity. The kcat-ATP for the N38A, D512N, and D512A MR-D complexes were reduced compared with the WT MR-D complex by 103-, 188-, and 119-fold, respectively. These fold-changes are ∼2–3 times larger than those observed for these Rad50 mutants alone, which results in 2–3-fold decreases in the level of DNA activation. The Km-ATP for the N38A, D512N, and D512A MR-D complexes are elevated 6-, 10-, and 8-fold, respectively, as compared with the WT MR-D complex. Similar to the reductions in DNA activation, all three low activity mutants have reduced ATP cooperativity with Hill coefficients ranging from 1.5 to 1.6.
TABLE 2.
Kinetic constants for wild-type and mutant Mre11·Rad50·DNA ATP hydrolysis activity
| Protein | kcat | Km-ATP | Hill coefficient | Mre11/DNA activation |
|---|---|---|---|---|
| s−1 | μm | |||
| WTa | 3.2 ± 0.1 | 49 ± 2 | 2.4 ± 0.2 | 21.9 |
| R37A | 2.3 ± 0.04 | 110 ± 3 | 2.3 ± 0.1 | 20.4 |
| N38A | 0.031 ± 0.003 | 290 ± 27 | 1.6 ± 0.2 | 12.4 |
| D512N | 0.017 ± 0.002 | 500.0 ± 95 | 1.6 ± 0.3 | 7.1 |
| D512A | 0.027 ± 0.002 | 410 ± 51 | 1.5 ± 0.2 | 7.3 |
| E514Q | 2.6 ± 0.08 | 60 ± 5 | 2.2 ± 0.1 | 21.4 |
| E514A | 2.3 ± 0.05 | 120 ± 4 | 2.1 ± 0.1 | 19.3 |
a Herdendorf et al. (24).
Steady-state Exonuclease Activity for WT and Mutant MR-D Complexes
The steady-state 3′ to 5′ exonuclease activity of the MR complex was followed by the release of the fluorescent nucleotide analog, 2-AP. The fluorescence of 2-AP is severely quenched when incorporated into DNA due to base-stacking interactions (30). Excision of 2-AP from the DNA results in a ∼200-fold increase in fluorescence (31). The mutant MR complexes were assayed for exonuclease activity using a 50-base pair DNA substrate with the 2-AP probe located at either the 2nd or 17th position relative to the 3′ end. The rate of excision of the 2-AP probe in the 1st and 2nd positions is ATP independent, therefore we initially assayed the MR complex mutants in the absence of ATP using the 2nd position DNA substrate (24). As seen in Fig. 2, all of the Rad50 mutants activate the exonuclease activity of Mre11 to similar degrees with less than 1.5-fold changes in kcat-exo2 (the superscript above exo refers to the position of the 2-AP probe). The exonuclease activity of Mre11 is highly dependent on the ability to form the MR complex (60-fold decrease in activity in the absence of Rad50), indicating that all six mutant Rad50 proteins bind Mre11 normally, consistent with the results of the Mre11 pull-down assay. Additionally, the nearly equivalent nuclease rates on the 2nd position DNA substrate indicates that DNA binding is not significantly affected by any of the six Rad50 point mutations. Similar to the 2nd position DNA substrate, in the absence of ATP, the exonuclease activity of the Rad50 mutants are relatively unchanged compared with the WT MR complex when the 2-AP probe is moved to the 17th position (Fig. 3). The kcat-exo17 values for R37A, D512N, and E514A are ∼2-fold higher than WT, whereas the kcat-exo17 values for N38A, D512A, and E514Q are near normal compared with the WT enzyme.
FIGURE 2.

WT and mutant steady-state exonuclease activity on the 2nd position 2-AP DNA substrate. Exonuclease activity was measured by the release of the fluorescent nucleotide analog, 2-AP, as a function of time. The assay consisted of Rad50 (50 or 100 nm), Mre11 (53 or 105 nm), and 1.3 μm DNA using buffer conditions described under “Experimental Procedures.” The dash indicates the nuclease activity of Mre11 in the absence of Rad50. The 2-AP probe is located at the 2nd position relative to the 3′ end of the DNA substrate. The value given above the bars is the specific activity estimate averaged from four independent measurements (two measurements at each protein concentration). Error bars represent the S.D. of the averaged values.
FIGURE 3.

WT and mutant steady-state exonuclease activity on the 17th position 2-AP DNA substrate. Exonuclease activity was measured by the release of the fluorescent nucleotide analog, 2-AP, as a function of time. The assay consisted of 400 nm Rad50, 420 nm Mre11, and 1.3 μm DNA substrate using buffer conditions described under “Experimental Procedures.” The dash indicates the nuclease activity of Mre11 in the absence of Rad50. A nuclease rate of 0.001 min−1 represents the limit of detection. The 2-AP probe is located at the 17th position relative to the 3′ end of the DNA substrate. The light and dark gray bars represent the average specific activity estimates in the presence and absence of ATP, respectively. The concentration of ATP, when included, was held at a concentration of 2 mm. The value given above the bars is the specific activity estimate averaged from three independent measurements and the error bars represent the S.D. of the averaged values.
In the absence of ATP, the steady-state exonuclease activities for WT and the mutant Rad50 proteins are reduced by ∼50-fold when the 2-AP probe is moved from the 2nd position to the 17th position (compare rates from Figs. 2 and 3). The addition of a saturating concentration of ATP to the assay increases the nuclease rate of the WT enzyme by ∼7-fold. The level of ATP activation for the R37A, E514Q, and E514A Rad50 mutants are very similar to the WT protein (8-fold), which results in a 2-fold higher kcat-exo17 for R37A and E514A (because their ATP-independent rate was also ∼2-fold higher). Interestingly, the addition of ATP to N38A, D512N, and D512A results in reduced exonuclease activity compared with their activity in reactions that lack ATP (5–11-fold reductions). Compared with the WT enzyme, the ATP-dependent kcat-exo17 for these three mutants is reduced between 35- and 43-fold.
DISCUSSION
Given the ubiquitous nature of ABC protein superfamily members and their known roles in human disease, it is surprising that the functional role of the D-loop is so poorly understood. A naturally occurring mutation of the D-loop aspartate residue (to histidine) in the cystic fibrosis conductance regulator is a known cause of cystic fibrosis, but the affect the mutation has on the cystic fibrosis conductance regulator is unknown (32). The D-loop is named for the absolutely conserved aspartate (D) but it has also been referred to as the “dimerization loop” because it is located at the dimer interface. Crystal structures of several ABC protein NBDs (including PfuRad50) show that the D-loop aspartate from one subunit hydrogen bonds with the backbone amide of an asparagine that is four residues upstream from the Walker A lysine of the opposing subunit (Fig. 4A). The side chain amide of this asparagine in turn forms a hydrogen bond with the γ-phosphate oxygen of ATP. Additionally, the catalytic water interacts with the backbone carbonyl oxygen of a residue located upstream from the D-loop aspartate. Although the importance of the asparagine within the Walker A motif has been overshadowed by the absolutely conserved lysine, an examination of the Walker A motif from a wide-range of ABC proteins reveals that its hydrogen-bond donating functionality of this position is highly conserved (as Asn, Thr, or Ser; Fig. 1B). The conservation of this functionality also extends outside of the ABC superfamily to members of the myosin superfamily (myosin, kinesin, and actin). In the ATPase protein families where this position is not conserved as a hydrogen-bond donor, the backbone amide of this position often serves as the hydrogen-bond donor (refer to His554 in PDB code 1D2N for an example of this). Like the D-loop aspartate, the importance of the Walker A asparagine is underscored by the observation that its mutation to Ile in the cystic fibrosis conductance regulator protein has been shown to result in cystic fibrosis.
FIGURE 4.

A, a close up of the Walker A and D-loop residues from the T4 homology model generated using the Swiss model server and based on PDB code 1F2U. ATP, Mg2+, the putative catalytic water, and the residues relevant to this study are noted. The coloring is the same as shown in Fig. 1. Dashes represent potential hydrogen bonds. B, a structural alignment of the Walker A, D-loop, and Walker B motifs in the presence (green) and absence (cyan) of ATP. The alignment was generated with the iterative magic fit function of the DeepView software (36) using the apo and ATP-bound x-ray crystal structures of PfuRad50 (PDB codes 1F2T and 1F2U, respectively). The overall root mean square deviation for the alignment of all of the c-α carbons for the apo and ATP-bound structures was 1.2 Å. The positions of the relevant residues are indicated by black lines and the distance each residue moves between the apo and ATP-bound forms of the enzyme is indicated in the parentheses.
Here, we report the functional effects of mutating the D-loop aspartate (Asp512) and the Walker A motif asparagine (Asn38) using T4 Rad50, which contains a prototypical ABC protein NBD. Additionally, we examined the importance of a glutamate residue (Glu514) that is downstream of the conserved D-loop aspartate and an arginine (Arg37) that is adjacent to the Walker A loop asparagine. Mutation of Arg37 and Glu514 resulted in relatively minor effects on the binding affinity of ATP, the kinetic parameters for ATP hydrolysis, and on the ATP-dependent nuclease activity of Mre11. On the other hand, mutation of either the Asn38 or Asp512 resulted in large reductions in ATP affinity, the ATP hydrolysis rate, and in the ATP-dependent nuclease activities of Mre11. Additionally, the level of DNA activation and the cooperativity of ATP hydrolysis are reduced in these mutants as compared with the WT enzyme.
Based the functional changes in the ATPase and nuclease activities of the E514A and R37Q mutants, it is difficult to rationalize conservation of the D-loop glutamate (Glu514) among Rad50 proteins and other members of the ABC superfamily (Fig. 1). Our T4 homology model suggests that the carboxylic acid side chain of Glu514 may form a salt bridge with Arg37; however, the positively charged nature of the arginine side chain is not well conserved, even among Rad50 proteins (Fig. 1). This lack of conservation in the Arg37 position calls into question the presence of the salt bridge that is observed in the T4 homology model, which was generated from the PfuRad50 structure using the Swiss model server (33). In the ATP-bound PfuRad50 structure, the position of the D-loop glutamate (Glu831) differs by ∼3 Å between the two ATP active sites. In one active site, Glu831 hydrogen bonds with Gln31 across the dimeric interface (similar to the Glu514–Arg37 interaction observed in the T4 model) but in the other active site, Glu831 hydrogen bonds with Arg834 within the same subunit. In T4 Rad50, the effects of mutating Arg37 and Glu514 appears to correlate with each other (similar increases in Km- and Kd-ATP) but the magnitude of those effects (∼2-fold) are relatively minor, making their correlation rather tenuous. Overall, we conclude that the side chains of Arg37 and Glu514 play a relatively minor role in the structure and function of T4 Rad50.
In sharp contrast to Arg37 and Glu514, the side chains of Asn38 and Asp512 appear to play prominent roles in ATP binding affinity and the mechanism of ATP hydrolysis. Based on the crystal structure of PfuRad50 and all other ATP-bound ABC protein NBDs, it is clear that the side chain of the amino acid located four residues upstream from the Walker A lysine interacts with the γ-phosphate of ATP as a hydrogen bond donor. This interaction serves to increase the affinity of the enzyme for ATP by providing ∼2.4 kcal/mol in binding energy, which is near the range expected for a single hydrogen bond with the acceptor being ionic in character (34). In addition to increasing ATP affinity, the hydrogen bond between the asparagine amide and the γ-phosphate oxygen of ATP likely assists in positioning the γ-phosphate for nucleophillic attack by the catalytic water molecule. The N38A mutation reduced the kcat-ATP by 2 orders of magnitude compared with the wild-type enzyme. This drastic reduction in kcat-ATP is somewhat surprising, given the fact that the mutation of the Signature motif serine, which also donates a hydrogen bond to a γ-phosphate oxygen of ATP, results in only a 6-fold decrease in kcat-ATP.3 This larger than expected decrease in ATPase activity may indicate that the hydrogen bonds between Asn38, Asp512, and ATP serve to stabilize both the Walker A loop, which includes the lysine that has been shown to be essential for ATP hydrolysis and the D-loop, which includes a residue that interacts with the catalytic water (Ser510 in T4; Fig. 4A) (20, 35).
Mutation of the D-loop aspartate to either alanine or asparagine mimics the effects of the N38A mutation, suggesting that one of the roles of Asp512 is to orient Asn38 so that it can productively interact with the γ-phosphate of ATP. This is consistent with the arrangement show in Fig. 4A, where the side chain of the aspartate hydrogen bonds with the backbone amide of Asn38. It is somewhat surprising, however, that the D512N mutation is as functionally disrupted as the D512A mutation is, because D512N should be able to maintain its hydrogen bond with the backbone amide of Asn38 (albeit in a slightly weaker form due to loss of ionic character). We must conclude that the possible steric clash due to the introduction of a hydrogen bond donating group by the D512N mutation globally disrupts the hydrogen bond network shown in Fig. 4A.
The D-loop aspartate that interacts with Asn38 of one ATP active site is connected to the other ATP active site via a short loop that lies between the D-loop and the Walker B motif (i.e. six residues separate Asp512 and Glu505; Fig. 4B). It has been suggested that Glu505 acts as the base in the catalytic mechanism of ATP hydrolysis and our mutagenesis studies in T4 and human Rad50-NBD are consistent with this proposal.3 Because this is perhaps the most direct route between the two ATP active sites, it has been suggested that the D-loop may be involved in the positive cooperativity of ATP hydrolysis that is observed in the WT enzyme (25). The results of the experiments reported here are consistent with this proposal. The Hill coefficients for ATP hydrolysis are reduced from 2.4 for the WT MR complex to 1.5–1.6 for the Asn38 and Asp512 mutants. We propose that Asn38 acts as a “γ-phosphate sensor” that transmits the presence of bound ATP across the dimer interface via its interaction with Asp512. Asp512 then propagates this signal to the neighboring ATP site via its connection to the residues that make up the Walker B motif (Fig. 4B). Fig. 4B shows the relevant residues in a global alignment of the ATP-free and bound forms of PfuRad50 (20). As shown, upon binding of ATP, the D-loop aspartate moves over 5 Å toward the bound ATP. Similarly, the position of the Walker A asparagine moves by more than 3 Å into a position that is favorable for interacting with the γ-phosphate of ATP. The Walker B glutamate has only shifted by 1.2 Å in the ATP-bound structure, but given its potential role as a catalytic base, this difference may be at least partially responsible for the observed cooperativity in ATP hydrolysis. Additionally, it is important to note that the cooperativity of ATP hydrolysis is rather minimal in Rad50 alone and that the MR-D complex has significantly greater positive cooperativity. Therefore, it is possible that the movement of the Walker B glutamate is greater when Mre11 and DNA are bound to Rad50.
In addition to increasing the positive cooperativity of ATP hydrolysis, the binding of Mre11 and double-stranded DNA to WT Rad50 increases the rate of ATP hydrolysis. It is very possible that these two phenomena are directly connected. In the ATP-bound PfuRad50 structure (without Mre11 or DNA present), the putative catalytic base (Walker B Glu) is 3.8 Å away from the putative attacking water molecule (5 Å in the ATP-free enzyme), which is a suboptimal distance for proton abstraction. Based on the 22-fold activation of kcat-ATP upon binding of Mre11 and DNA, we propose that the Walker B glutamate (Glu505 in T4) repositions itself closer to the attacking water molecule, making proton abstraction more favorable. The mutation of Asn38 and Asp512 reduces both ATP cooperativity and DNA activation. Therefore, a possible mechanism for changing the position of the Walker B glutamate may involve altering the conformation of the residues that connect the D-loop to the Walker B motif.
The effect that mutating the Walker A asparagine and the D-loop aspartate has on the ATP-dependent nuclease activity of Mre11 is very similar to what is observed when a non-hydrolyzable ATP analog is used instead of ATP (24). It appears that the primary role of ATP hydrolysis in the exonuclease activity of the MR complex is to drive translocation of the complex following nucleotide excision and product release (20, 24). However, we have previously established that the rate-limiting step of the steady-state exonuclease reaction is productive substrate binding and that the actual rate of nucleotide excision/translocation is kinetically silent. In this situation, the increase in activity that is observed upon addition of ATP is due to an increase in processivity that reduces the number of slow-binding events that the MR complex must go through prior to reaching the 17th position. The potent inhibition that is observed in the N38A, D512N, and D512A mutant MR complexes is very likely due to their much reduced ATP hydrolysis activity, which slows their translocation along the DNA substrate while stabilizing the stalled MR-DNA complex (i.e. slowing down the dissociation from the DNA substrate). This slow dissociation rate must be at least partially rate-limiting for the N38A, D512N, and D512A mutant MR complexes.
In conclusion, we find although the Walker A asparagine has been largely overlooked, it plays a critical role in the mechanism of ATP hydrolysis by increasing the affinity of the enzyme for ATP and orienting the γ-phosphate for nucleophillic attack by the catalytic water molecule. The D-loop aspartate interacts with the asparagine at the dimer interface and likely functions to maintain the Walker A asparagine in the proper orientation for H-bonding with the γ-phosphate of ATP. We propose that the Walker A to D-loop connection acts as an ATP sensor that transmits the binding and/or hydrolysis of ATP to the second ATP active site via a short loop connecting the D-loop to the Walker B motif. This is likely one of the major allosteric pathways that leads to the ATP cooperativity that has been observed in Rad50 and several other ABC proteins. The high degree of conservation of these two residues strongly suggests that these conclusions are generally applicable to most members of the ABC protein superfamily.
Supplementary Material
Acknowledgments
We thank Dustin W. Albrecht for providing purified T4 Mre11 and Timothy J. Herdendorf for critically reading the manuscript.
This work was supported by Carver Trust Young Investigator Grant 10-3603 (to S. W. N.) and Iowa State University institutional support (to S. W. N.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
T. J. Herdendorf, D. W. Albrecht, E. E. Parrott, and S. W. Nelson, unpublished observations.
- MR
- Mre11/Rad50
- DSB
- double-strand break
- HR
- homologous recombination
- gp46
- T4 gene product 46
- gp47
- T4 gene product 47
- 2-AP
- 2-aminopurine deoxyribonucleotide
- MR-D
- Mre11·Rad50·DNA complex
- PDB
- Protein Data Bank
- NBD
- nucleotide binding domain.
REFERENCES
- 1. Connelly J. C., Leach D. R. (2002) Trends Biochem. Sci. 27, 410–418 [DOI] [PubMed] [Google Scholar]
- 2. Aguilera A., Gómez-González B. (2008) Nat. Rev. Genet. 9, 204–217 [DOI] [PubMed] [Google Scholar]
- 3. Hoeijmakers J. H. J. (2007) Mech. Ageing Dev. 128, 460–462 [DOI] [PubMed] [Google Scholar]
- 4. Borde V. (2007) Chromosome Res. 15, 551–563 [DOI] [PubMed] [Google Scholar]
- 5. Soulas-Sprauel P., Rivera-Munoz P., Malivert L., Le Guyader G., Abramowski V., Revy P., de Villartay J. (2007) Oncogene 26, 7780–7791 [DOI] [PubMed] [Google Scholar]
- 6. Povirk L. F. (2006) DNA Repair 5, 1199–1212 [DOI] [PubMed] [Google Scholar]
- 7. Bassing C. H., Alt F. W. (2004) DNA Repair 3, 781–796 [DOI] [PubMed] [Google Scholar]
- 8. San Filippo J., Sung P., Klein H. (2008) Annu. Rev. Biochem. 77, 229–257 [DOI] [PubMed] [Google Scholar]
- 9. Cejka P., Cannavo E., Polaczek P., Masuda-Sasa T., Pokharel S., Campbell J. L., Kowalczykowski S. C. (2010) Nature 467, 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Niu H., Chung W. H., Zhu Z., Kwon Y., Zhao W., Chi P., Prakash R., Seong C., Liu D., Lu L., Ira G., Sung P. (2010) Nature 467, 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mimitou E. P., Symington L. S. (2008) Nature 455, 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu Z., Chung W. H., Shim E. Y., Lee S. E., Ira G. (2008) Cell 134, 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mimitou E. P., Symington L. S. (2009) DNA Repair 8, 983–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Symington L. S. (2002) Microbiol. Mol. Biol. Rev. 66, 630–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Connelly J. C., de Leau E. S., Leach D. R. (1999) Nucleic Acids Res. 27, 1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dillingham M. S., Kowalczykowski S. C. (2008) Microbiol. Mol. Biol. Rev. 72, 642–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kreuzer K. N., Yap W. Y., Menkens A. E., Engman H. W. (1988) J. Biol. Chem. 263, 11366–11373 [PubMed] [Google Scholar]
- 18. Jones P. M., O'Mara M. L., George A. M. (2009) Trends Biochem. Sci. 34, 520–531 [DOI] [PubMed] [Google Scholar]
- 19. Hopfner K. P., Tainer J. A. (2003) Curr. Opin. Struct. Biol. 13, 249–255 [DOI] [PubMed] [Google Scholar]
- 20. Hopfner K. P., Karcher A., Shin D. S., Craig L., Arthur L. M., Carney J. P., Tainer J. A. (2000) Cell 101, 789–800 [DOI] [PubMed] [Google Scholar]
- 21. Hopfner K. P., Craig L., Moncalian G., Zinkel R. A., Usui T., Owen B. A., Karcher A., Henderson B., Bodmer J. L., McMurray C. T., Carney J. P., Petrini J. H., Tainer J. A. (2002) Nature 418, 562–566 [DOI] [PubMed] [Google Scholar]
- 22. Bhaskara V., Dupré A., Lengsfeld B., Hopkins B. B., Chan A., Lee J. H., Zhang X., Gautier J., Zakian V., Paull T. T. (2007) Mol. Cell 25, 647–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi L. (2004) Front. Biosci. 9, 1382–1397 [DOI] [PubMed] [Google Scholar]
- 24. Herdendorf T. J., Albrecht D. W., Benkovic S. J., Nelson S. W. (2011) J. Biol. Chem. 286, 2382–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hopkins B. B., Paull T. T. (2008) Cell 135, 250–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zaitseva J., Jenewein S., Jumpertz T., Holland I. B., Schmitt L. (2005) EMBO J. 24, 1901–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gilbert S. P., Mackey A. T. (2000) Methods 22, 337–354 [DOI] [PubMed] [Google Scholar]
- 28. Hopfner K. P., Karcher A., Shin D., Fairley C., Tainer J. A., Carney J. P. (2000) J. Bacteriol. 182, 6036–6041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee J. H., Paull T. T. (2006) Methods Enzymol. 408, 529–539 [DOI] [PubMed] [Google Scholar]
- 30. Frey M. W., Sowers L. C., Millar D. P., Benkovic S. J. (1995) Biochemistry 34, 9185–9192 [DOI] [PubMed] [Google Scholar]
- 31. Jean J. M., Hall K. B. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 37–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bareil C., Thèze C., Béroud C., Hamroun D., Guittard C., René C., Paulet D., Georges M., Claustres M. (2010) Hum. Mutat. 31, 1011–1019 [DOI] [PubMed] [Google Scholar]
- 33. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 34. Fersht A. R. (1988) Biochemistry 27, 1577–1580 [DOI] [PubMed] [Google Scholar]
- 35. Frelet A., Klein M. (2006) FEBS Lett. 580, 1064–1084 [DOI] [PubMed] [Google Scholar]
- 36. Guex N., Diemand A., Peitsch M. C. (1999) Trends Biochem. Sci. 24, 364–367 [DOI] [PubMed] [Google Scholar]
- 37. Herdendorf T. J., Nelson S. W. (2011) Biochemistry, in press [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.