

Abstract
ClpB is a hexameric chaperone that solubilizes and reactivates protein aggregates in cooperation with the Hsp70/DnaK chaperone system. Each of the identical protein monomers contains two nucleotide binding domains (NBD), whose ATPase activity must be coupled to exert on the substrate the mechanical work required for its reactivation. However, how communication between these sites occurs is at present poorly understood. We have studied herein the affinity of each of the NBDs for nucleotides in WT ClpB and protein variants in which one or both sites are mutated to selectively impair nucleotide binding or hydrolysis. Our data show that the affinity of NBD2 for nucleotides (Kd = 3–7 μm) is significantly higher than that of NBD1. Interestingly, the affinity of NBD1 depends on nucleotide binding to NBD2. Binding of ATP, but not ADP, to NBD2 increases the affinity of NBD1 (the Kd decreases from ≈160–300 to 50–60 μm) for the corresponding nucleotide. Moreover, filling of the NBD2 ring with ATP allows the cooperative binding of this nucleotide and substrates to the NBD1 ring. Data also suggest that a minimum of four subunits cooperate to bind and reactivate two different aggregated protein substrates.
Keywords: Allosteric Regulation, Chaperone Chaperonin, Heat Shock Protein, Nucleotide, Protein Folding, ClpB
Introduction
Yeast heat-shock protein 104 (Hsp104)5 and its bacterial homolog, caseinolytic peptidase B (ClpB), are molecular chaperones that in cooperation with the Hsp70/DnaK system reactivate most of the proteins that become aggregated after severe stress. They are specially required to increase survival when cells are exposed to extreme heat or other environmental stresses (1–4). These hexameric chaperones belong to the family of ATPases associated with diverse cellular activities (AAA+), a class of enzymes involved in protein quality control, membrane fusion, and DNA replication (5–11). There are two classes of AAA+ proteins that differ in the number of nucleotide binding domains; class I contains two domains, and class II contains one domain (12). As members of class I, Hsp100 and ClpB have two nucleotide binding sites per monomer that share the Walker A and B motifs with other nucleotide binding proteins (13) and, in contrast to other Hsp100 proteins, are separated by a unique large middle domain known as the M domain (14–16). The x-ray structure of Thermus thermophilus ClpB (ClpBTh) shows an N-terminal domain followed by a nucleotide binding domain (NBD-1), a middle (M) domain, and a second nucleotide binding domain (NBD-2) (17). The functional unit of these proteins is the hexamer that contains 12 nucleotide binding sites located in two stacked rings within the oligomeric particle.
Previous biochemical studies have revealed that both Hsp104 and ClpB show cooperativity between nucleotide binding sites that controls their ATPase activity (18, 19). Mutations at any of the NBDs that inhibit their ATPase activity also impair chaperone activity, indicating that ATP hydrolysis at both NBDs is required for productive substrate handling (20). The ATPase activity of different subunits is tightly coupled in the active oligomer, as incorporation of inactive monomers into the hexamer inhibits its chaperone activity (21–23). These studies demonstrate that the AAA+ domains of the same and of different subunits of the hexamer must work in a coordinated manner to reactivate protein aggregates. The following experimental evidences also prove the existence of communication between the two NBDs of Hsp100 proteins. Non-covalent interactions allow allosteric communication between the NBDs of ClpBTh, as mixtures of inactive fragments containing each one of the NBDs recover chaperone activity (24). Furthermore, the ATPase activity of Hsp104 NBD1 is also regulated by the hydrolysis cycle at NBD2 (18). ATP hydrolysis at NBD2 and, therefore, exchange of ATP by ADP increases the kcat of NBD1 activating its hydrolysis. A recent study has identified cis and trans protomer interactions that regulate the activity of Hsp104 (25). Intrasubunit communication has also been proposed for ClpBTh as the ATPase activity of NBD2 is stimulated after an M-domain conformational change triggered by ATP binding to NBD1 (26). However, how communication between NBDs of different or the same subunit is established within the functional oligomeric particle remains as yet largely unknown.
We have examined the nucleotide binding properties of WT ClpBEco and different variants in which one or both NBDs have been mutated to render them unable to bind or hydrolyze nucleotide (27). Substitution of the conserved lysine residues at the Walker A motif (Lys-212 and Lys-611 in NBD1 and NBD2) by alanine abolishes nucleotide binding (N, null mutation). Replacement of the glutamic acid residues at the Walker B motif (Glu-276 and Glu-678 at the NBD1 and NBD2) by alanine generates NBDs able to bind but not hydrolyze ATP (T, trap mutation) (27). The nomenclature of the ClpB variants used in this study and their biochemical properties are summarized in Table 1. Our results demonstrate that the affinity of NBD2 for ATP (Kd = 3–7 μm) is higher than that of NBD1 (Kd ≈ 60–250 μm). Binding of nucleotides to NBD2 is not affected by the oligomerization state of the protein, in contrast to NBD1-nucleotide complex formation that depends on hexamer stability, as found for ClpBTh (28, 29). ATP binding to NBD2 results in an increase in the affinity of NBD1 for this nucleotide and sets up the interactions at the NBD1 ring for the cooperative binding of ATP and protein substrates. However, the same enhancement in the affinity of NBD1 for ADP is not triggered by binding of this ligand to NBD2, suggesting that the allosteric communication route from NBD2 to NBD1 distinguishes ATP from ADP.
TABLE 1.
ClpB variants used in this work and their properties
| Variant | NBD1 | NBD2 | Mutation |
|---|---|---|---|
| WT | Binding and hydrolysis | Binding and hydrolysis | None |
| T1T2 | Binding | Binding | E276A/E678A |
| T1N2 | Binding | None | E276A/K611A |
| N1T2 | None | Binding | K212A/E678A |
| N1N2 | None | None | K212A/K611A |
| T1 | Binding | Binding and hydrolysis | E276A |
| T2 | Binding and hydrolysis | Binding | E678A |
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Wt ClpB and ClpB mutants were expressed in BB4561 Escherichia coli strain and purified as previously described (30). ClpB single point mutants were cloned using Stratagene QuikChange II kit, and mutations were confirmed by DNA sequencing. DnaK, DnaJ, and GrpE were obtained as previously reported (31, 32). Protein concentration was determined by the colorimetric Bradford assay (Bio-Rad). Malate dehydrogenase (MDH) and glucose-6-phosphate dehydrogenase (G6PDH) were purchased from Sigma, and lactate dehydrogenase and pyruvate kinase were from Roche Applied Science. Concentrations of ClpB refer to monomers unless otherwise stated.
Size-exclusion Chromatography
Wt ClpB or ClpB mutants (20–50 μm) were loaded on a Superose 6 PC 3.2/30 (GE Healthcare) equilibrated in 50 mm Tris-HCl, pH 7.5, 20 mm MgCl2, and different KCl concentrations (50, 150, or 500 mm). Experiments were performed at 25 °C.
Isothermal Titration Calorimetry (ITC)
Calorimetric measurements were performed in a VP-ITC Microcalorimeter (MicroCal LLC., Northampton, MA). Proteins were dialyzed against 50 mm Tris-HCl, pH 7.4, 150 mm KCl, 20 mm MgCl2. Nucleotides were freshly dissolved in dialysis buffer before ITC measurements. Typical titration experiments were performed with WT ClpB or the corresponding protein variant at a protomer concentration of 12 μm. Consecutive injections (6–10 μl) of a nucleotide solution (1.2–1.6 mm or 350–400 μm) were carried out after sample equilibration at 25 °C. The same nucleotide solution was injected into dialysis buffer to estimate the dilution enthalpy that was used to correct the binding enthalpy. Binding isotherms were analyzed with a one-site binding model using Origin 7.0 software (OriginLab Corp., Northampton, MA).
Binding of Fluorescent Nucleotide Analogs to ClpB
WT ClpB or the corresponding protein variant (5–10 μm) dissolved in 50 mm Tris-HCl, pH 7.4, 5 mm MgCl2, and different KCl concentrations (15, 50, 150, or 500 mm) were titrated with MANT-ADP or MANT-ATP (0.05–20 μm). After incubating the mixture for 5 min at 25 °C, the fluorescence spectra were recorded on a Fluorolog-3 spectrofluorimeter (HORIBA Jobin Yvon) with excitation at 356 nm. Excitation and emission slits were set at 2 nm. Control experiments to account for nonspecific interactions between the protein and labeled nucleotides were carried out with N1N2, a ClpB mutant unable to bind ATP at either NBD. Hydrolysis of MANT-ATP by ClpB was assayed measuring inorganic phosphate with the malachite green method (33).
Partial Proteolysis
WT ClpB or the corresponding protein variant (5 μm) was incubated at 30 °C in 50 mm Tris-HCl, pH 7.4, 20 mm MgCl2, 50 mm KCl for 5 min in the absence or presence of the desired nucleotide (ATPγS, ATP, or ADP) concentration. Proteolysis was immediately initiated upon the addition of thermolysin to a final chaperone/protease ratio (w/w) of 135. The kinetics of the proteolysis reaction was followed by SDS-PAGE (15%) analysis of the digestion products at different incubation times and nucleotide concentrations, as previously reported (20). The amount of full-length protein protected against proteolytic digestion was estimated by densitometry of the corresponding gel band with a gel scanner G-800 and the Quantity One software (Bio-Rad). The initial rates of full-length protein loss as a function of nucleotide concentration were analyzed with the Hill equation (see below) to estimate the apparent affinity of the NBD1 of each protein variants for ATP and ADP. Each data point is the average of at least two independent experiments.
MDH Aggregation
The ability of T1T2 or T1N2 variants to avoid aggregation of thermally denatured MDH was followed by measuring the turbidity at 550 nm of MDH samples (6 μm) at 47 °C in 50 mm Tris-HCl, pH 7.5, 20 mm MgCl2, 2 mm DTT, and 50 or 150 mm KCl. Experiments were performed at 2 mm ATP and increasing chaperone concentrations to estimate the Kd of both ClpB variants for denatured MDH and at increasing ATP concentrations and constant T1T2 (4 μm) and T1N2 (12 μm) concentration to determine the Kd of the nucleotide-dependent substrate association. Data were fitted to the Hill equation,
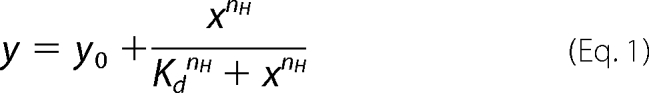 |
where y is the percentage of protection against MDH aggregation, x is the ATP concentration, nH is the Hill coefficient, and Kd is the dissociation constant. Kinetic measurements were performed in a Cary spectrophotometer (Varian).
Reactivation of MDH Aggregates
MDH (6 μm) was denatured and aggregated by incubating the protein 30 min at 47 °C in 50 mm Tris-HCl, pH 7.5, 150 mm KCl, 20 mm MgCl2, 2 mm DTT. Aggregates were diluted to 1 μm in 50 mm Tris-HCl, pH 7.5, 50 mm KCl, 20 mm MgCl2, and 2 mm DTT containing 3 μm WT ClpB, 2 μm DnaK, 0.6 μm DnaJ, and 0.4 μm GrpE. Reactivation was started after the addition of different ATP concentrations (0–2 mm) to the sample containing an ATP regenerating system (10 mm phosphoenolpyruvate and 20 ng/ml pyruvate kinase) at 30 °C. MDH activity was recorded as previously described (34).
Reactivation of G6PDH Aggregates
G6PDH (10 μm) was denatured at 70 °C for 30 min in 50 mm Tris-HCl, pH 7.5, 150 mm KCl, 20 mm MgCl2, and 10 mm DTT. Denatured and aggregated G6PDH was diluted to 0.4 μm in the same buffer containing 50 mm KCl. Reactivation was started after the addition of different ATP concentrations (0–2 mm) to the sample containing 0.7 μm DnaJ, 0.35 μm GrpE, 3.5 μm DnaK, 5 μm ClpB, and an ATP-regenerating system (10 mm phosphoenolpyruvate and 20 ng/ml pyruvate kinase). G6PDH activity was measured at 30 °C as previously described (35).
Reactivation of Denatured Luciferase by the K-system Alone
Luciferase (2.5 μm) was denatured for 45 min at 25 °C in 6 m guanidinium-HCl, 100 mm Tris, pH 7.5, 10 mm DTT. Denatured luciferase was diluted to 25 nm in buffer 50 mm Tris, pH 7.5, 15 mm MgCl2, 50 mm KCl, 5 mm DTT. Reactivation was carried out in the presence of 1 μm DnaK, 1 μm DnaJ, 1.2 μm GrpE, and the above-mentioned ATP regenerating system at 25 °C after adding different ATP concentrations (0.05, 0.1, and 2 mm). Luciferase activity was measured after 2 h of incubation using the luciferase assay system (Promega E1500) in a Sinergy HT (Biotek) luminometer.
RESULTS
The Nucleotide Binding Domains of ClpB Have Different Affinities for Nucleotides
We first examined nucleotide (ADP, ATP, or ATPγS) binding to WT ClpB or ClpB variants with one or both NBDs mutated to impair nucleotide binding or hydrolysis by ITC. As previously discussed for ClpB (36) and other related ATPases (37), data analysis is not straightforward due to the presence of 12 different nucleotide binding sites that might have distinct binding properties and might display cooperativity as a result of complex allosteric interactions between different binding sites. Although the value of the apparent thermodynamic parameters of nucleotide binding depends on the binding model used to analyze the data (dependent or independent binding sites, positive and/or negative cooperativity), the stoichiometry of the binding reaction can be estimated from titration experiments. The experimental conditions employed in this study (protein and KCl concentration) ensure that proteins are hexameric in their apo-form, i.e. before the addition of nucleotide that favors ClpB oligomerization (20), and therefore that the experimentally measured heat change is solely due to nucleotide binding and ligand-induced protein conformational changes. Data showed that only half of the available binding sites of WT ClpB are filled with ATPγS or ADP (n = 0.9 ± 0.1 and 0.83 ± 0.12, respectively; Fig. 1; Table 2) within the nucleotide concentration range 0–150 μm. A similar stoichiometry was obtained for ATP binding to two ClpB mutants, one with both NBDs able to bind but not to hydrolyze ATP (T1T2; n = 0.92 ± 0.14) and a second variant with the same mutation in NBD2 and an NBD1 unable to bind nucleotide (N1T2; n = 0.8 ± 0.06) (Fig. 2; Table 2). However, T1N2 did not bind ATP within the same nucleotide concentration range (0–50 μm; Fig. 2). These data suggest that nucleotide binding to NBD2 explains ligand interaction with hexamers containing both NBDs competent to bind nucleotides below 50 μm and that if binding to NBD1 occurs at higher nucleotide concentrations it is not detected under these experimental conditions. Because only one of the two available sites binds nucleotides, we have used a single-site binding model to fit the experimental data (Fig. 2B). The thermodynamic parameters obtained from the fittings are shown in Table 2. The assumption that the NBD2s behave as independent binding sites is further supported by the similar affinity of the NBD2 of monomeric and hexameric ClpB for nucleotides (see below). Overall, these results indicate that the affinity of the NBD2 of all variants for nucleotides is similar, the Kd values ranging from 3 to 7 μm (Table 2). They also show that nucleotide binding to this site is an exothermic process with a favorable enthalpy change, as already described for ADP and ATPγS binding to WT ClpB (36). As compared with ATPγS, ATP binding to T1T2 and N1T2 results in an ∼2-fold reduction in enthalpy and a favorable entropy change, suggesting that the analog does not fully mimic the effect of ATP.
FIGURE 1.

Half of the nucleotide binding sites of WT ClpB bind ATPγS or ADP below 50 μm nucleotide. Shown is the base-line-corrected instrumental response of successive injections of ATPγS into a solution of WT ClpB at 25 °C. 6 μl (1.6 mm nucleotide) were injected into a solution containing 28 μm WT ClpB. Experiments were performed in 50 mm Tris-HCl, pH 7.5, 150 mm KCl, 20 mm MgCl2. Inset, shown are the integrated data of WT ClpB titration with ATPγS (black squares) or ADP (white circles) and fit of the corresponding binding isotherms to a single-site binding model (solid lines).
TABLE 2.
Isothermal titration calorimetry analysis of nucleotide binding to WT ClpB and different protein variants
Experiments were repeated at least two times using two different protein batches. −, interaction was not detected.
| ClpB | n | kd | ΔH | −TΔS | ΔG |
|---|---|---|---|---|---|
| μm | kcal/mol | kcal/mol | kcal/mol | ||
| ADP | |||||
| wt | 0.8 ± 0.1 | 5.8 ± 1.7 | −10.8 ± 1.2 | 3.6 ± 1.0 | −7.2 ± 0.2 |
| ATP γS | |||||
| wt | 0.9 ± 0.2 | 3.8 ± 2.6 | −11.5 ± 3.1 | 4.1 ± 4.0 | −7.4 ± 0.3 |
| T1T2 | 0.7 ± 0.1 | 3.1 ± 0.2 | −13.1 ± 0.2 | 5.6 ± 0.1 | −7.5 ± 0.1 |
| ATP | |||||
| T1T2 | 1.0 ± 0.1 | 3.1 ± 0.3 | −4.9 ± 0.06 | −2.6 ± 0.1 | −7.5 ± 0.1 |
| T1N2 | |||||
| N1T2 | 0.8 ± 0.1 | 6.9 ± 2.0 | −4.2 ± 1.1 | −2.8 ± 0.9 | −7.0 ± 0.2 |
FIGURE 2.

The affinity of NBD2 for nucleotides is higher than that of NBD1. A, shown is the base-line-corrected instrumental response of successive injections of ATP into a solution of T1N2 (upper trace), N1T2 (middle trace), and T1T2 (bottom trace) at 25 °C. 8 μl of 350 μm ATP were injected into the calorimetric cell containing 10 μm (T1T2, T1N2) or 12 μm (N1T2) protein monomer. Experiments were performed in 50 mm Tris-HCl, pH 7.5, 150 mm KCl, and 20 mm MgCl2. Titrations were shifted for better viewing. B, integrated data of T1N2 (black squares), N1T2 (white circles), and T1T2 (black circles) titration with ATP and fit of the corresponding binding isotherms to a single-site binding model (solid lines).
To validate the assignment of this binding event to the filling of NBD2, the same protein variants were titrated with fluorescent nucleotide analogs (MANT-ATP or MANT-ADP). WT ClpB does not hydrolyze MANT-ATP under any of the experimental conditions employed (supplemental Fig. S2A). The ClpB variant N1N2, unable to bind nucleotides, is used as a control to account for unspecific interactions of the fluorescent ligands with the protein. Mixing of fluorescent nucleotides with N1N2 results in a fluorescence intensity increase (Fig. 3A) that is further enhanced when these ligands interact with protein variants competent to bind nucleotides at NBD2 (WT ClpB, T1T2, and N1T2) but not at NBD1 (T1N2) (Fig. 3A). Therefore, fluorescence results also show, in concordance with ITC data, that only NBD2 binds nucleotide below 20 μm MANT-ATP or MANT-ADP. The estimated Kd values, between 3.3 and 5 μm (Fig. 3B; Table 3), are in good agreement with those derived from ITC. To prove that the affinity of NBD2 for nucleotides did not rely on the oligomerization state of the protein, the same type of experiments were performed under experimental conditions that stabilized hexameric (low KCl concentration) or monomeric (lower protein concentration and 500 mm KCl) ClpB (supplemental Figs. S1 and S2). The Kd values are similar (Table 3), indicating that the affinity of NBD2 for nucleotides does not depend on the oligomerization state of ClpB and, therefore, that the assumption of independent and equivalent NBD2 sites made above is reasonable. It should be noted that the apparently lower affinity of NBD1 for nucleotides cannot be determined by these techniques because of sensitivity limits of ITC and the impossibility to use a higher concentration of fluorescent nucleotides due to the inner filter effect (38).
FIGURE 3.

Binding of fluorescent nucleotide analogs to different ClpB variants. A, emission spectra of MANT-ADP in the absence (short dashed line) or presence of different ClpB variants (WT, T1T2, T1N2, and N1N2) are shown. Nucleotide and protein concentrations were 10 μm and 20 μm, respectively. Experiments were carried out in 50 mm Tris-HCl, pH 7.5, 20 mm MgCl2, 50 mm KCl, pH 7.5. B, titrations of WT ClpB and protein variants (10 μm) with MANT-ADP or MANT-ATP in the buffer described above are shown. The intensity values obtained with mutant N1N2, coming from nonspecific interaction between the protein and the corresponding fluorescent-nucleotide analog, were subtracted from the values obtained with the other protein variants. Data are the means ± S.D. (n = 2). a.u., arbitrary units.
TABLE 3.
Binding of fluorescent nucleotides to wt ClpB and selected mutants.
Titrations were performed under experimental conditions that modify the oligomeric state of ClpB. Experiments were repeated at least two times. ND, not determined. −, binding of nucleotide analogs to T1N2 was not detected.
| ClpB variant | MANT-ATP |
MANT-ADP |
||
|---|---|---|---|---|
| Kd | nH | Kd | nH | |
| μm | μm | |||
| Wt | 3.6 ± 0.2a | 1.7 ± 0.1a | 3.4 ± 0.2b | 1.7 ± 0.1b |
| 4.6 ± 0.2b | 1.4 ± 0.1b | |||
| 3.2 ± 0.2c | 1.4 ± 0.1c | |||
| T1T2 | 3.3 ± 0.2a | 1.6 ± 0.1a | 3.2 ± 0.3b | 1.8 ± 0.2b |
| 4.4 ± 0.3b | 1.3 ± 0.1b | |||
| 4.9 ± 0.4c | 1.1 ± 0.1c | |||
| T1N2 | – | – | – | – |
| N1T2 | 3.3 ± 0.1a | 1.6 ± 0.1a | ND | ND |
| 4.4 ± 0.3b | 1.4 ± 0.1b | |||
| 3.3 ± 0.1c | 1.4 ± 0.1c | |||
a 10 μm protein in 15 mm KCl.
b 10 μm protein in 50 mm KCl buffer.
c 5 μm protein in 500 mm KCl buffer.
The Affinity and Cooperativity of Nucleotide Binding to NBD1 Depends on the Nucleotide Status of NBD2
To estimate the affinity of NBD1 for nucleotides, we have used two different methods that, provided that the protein remains hexameric, are sensitive to ligand binding to this site. The first one is partial proteolysis of ClpB by thermolysin (20). Nucleotide binding to WT ClpB protects the chaperone against proteolysis, and impairment of nucleotide binding to NBD1 abolishes this effect in N1T2 (supplemental Fig. S3A) and N1 (20) regardless of the ability of these protein variants to bind or hydrolyze ATP at NBD2. Therefore, protection of full-length hexameric ClpB against proteolytic degradation primarily reflects nucleotide binding to NBD1. The relative affinity of the NBD1 of different protein variants for nucleotides has been estimated by quantifying the digestion rates of their full-length forms at increasing nucleotide concentrations (Fig. 4A). Initial rates of proteolysis are determined from the linear part of the curves during the first minutes of the reaction (Fig. 4B). The values obtained for T1N2 are higher than those of WT ClpB and T1T2, indicating a higher susceptibility of that mutant to protease digestion (Fig. 4B). They decrease with increasing nucleotide concentration, following saturation kinetics, due to ligand binding to the NBD1 that protects the chaperone against protease digestion. Assuming that thermolysin has a similar affinity for the apo-forms of these protein variants, the nucleotide concentration dependence of the initial proteolysis rate would reflect the apparent affinity of their NBD1s for nucleotides. Analysis of the curves obtained in the presence of ATP (T1T2 and T1N2) or ATPγS (WT ClpB) with the Hill equation yields apparent Kd values of 160 and 60 μm for T1N2 and WT ClpB or T1T2, respectively and Hill coefficients close to 1 in all cases (Fig. 4C; Table 4). These data show that the affinity of NBD1 for nucleotides is 10–50 times lower than that of NBD2 at 50 mm KCl (Table 4). Conditions that favor oligomer dissociation (i.e. 300 mm KCl) result in a lack of protection against protease digestion, suggesting that nucleotide binding to the NBD1 requires protein oligomerization (supplemental Fig. S3), in contrast to NBD2, which binds nucleotide in the monomeric state of the protein (supplemental Fig. S2). The 2-fold increase of the Kd values observed by partial proteolysis at 150 mm KCl (not shown) might be due to the salt-induced destabilization of the protein hexamer (19, 39), which could hinder ligand binding to the NBD1. This interpretation agrees with the previously reported importance of nucleotide binding in hexamer stabilization (20). Interestingly, when nucleotide binding to the NBD2 is impaired (T1N2), the affinity of NBD1 for ATP dropped significantly (Fig. 4C; Table 4). These results indicate that ATP binding to NBD2 regulates the affinity of NBD1 for this ligand, which in turn reflects allosteric communication between the NBDs. However, a different picture was obtained for ADP (Fig. 4D; Table 4). The affinity of NBD1 for this ligand remains similar regardless of the ability of the protein variant to bind ADP at the NBD2, indicating that ADP does not promote the same allosteric communication between the NBDs, as compared with ATP. Therefore, allosteric signaling between both NBDs is sensitive to the nature of the bound nucleotide, as reported for ClpBTth (19) and Hsp104 (40).
FIGURE 4.

ATP, but not ADP, binding to NBD2 regulates the affinity of NBD1 for the corresponding nucleotide. A, shown are representative gels of partially digested T1T2 (upper) and T1N2 (lower) in the presence of increasing ATP concentrations, obtained after 10 (T1T2) and 4 (T1N2) min incubation with thermolysin at a chaperone/protease ratio (w/w) of 135. B, shown are kinetics of the proteolysis of 5 μm T1T2 (upper panel) or T1N2 (lower panel) by thermolysin at increasing ATP concentrations and a chaperone/thermolysin ratio (w/w) of 135. Samples were incubated at 25 °C, and the time dependence of the reaction was analyzed by 15% SDS-PAGE. The amount of full-length protein protected against proteolysis was estimated by densitometry using the value obtained in the absence of protease as 100%. C, shown is the initial proteolysis rate of WT ClpB (○) and mutants T1T2 (filled circle) and T1N2 (□) in the presence of increasing concentrations of ATPγS (WT ClpB) and ATP (T1T2 and T1N2). Analysis with the Hill equation (solid lines) gives the Kd and Hill coefficient values shown in Table 4. D, experimental procedures were as in C, in the presence of ADP. Experimental conditions were as detailed in B. The solid lines correspond to data analysis with the Hill equation. Data are the means ± S.D. (n = 2–3).
TABLE 4.
Estimation of the affinity for nucleotides of the NBD1 of WT ClpB and selected protein variants by partial proteolysis
Experiments were repeated at least three times.
| Nucleotide | ClpB | Kd | nH |
|---|---|---|---|
| μm | |||
| ATP γS | wt | 50 ± 6 | 1.0 ± 0.2 |
| ATP | T1T2 | 61 ± 5 | 0.9 ± 0.1 |
| T1N2 | 163 ± 9 | 1.1 ± 0.1 | |
| ADP | wt | 35 ± 4 | 1.2 ± 0.2 |
| T1T2 | 43 ± 4 | 0.9 ± 0.1 | |
| T1N2 | 39 ± 3 | 0.9 ± 0.1 |
As an alternative method to estimate the affinity of NBD1 for nucleotide, we have followed the formation of chaperone-MDH complexes. The study of the contribution of the two NBDs of ClpB (20) and Hsp104 (41) to substrate binding clearly demonstrates that formation of stable chaperone-substrate complexes requires ATP binding to NBD1 in the absence of nucleotide hydrolysis at either NBD. In agreement with these observations, our data show that only the ClpB variants T1T2 (27) and T1N2 are able to prevent aggregation of thermally denatured MDH, whereas protein variants that hydrolyze nucleotide at one or both NBDs (WT ClpB, T1, T2, and N2; Fig. 5A) cannot avoid it. Complex formation can also be achieved in the absence of nucleotide binding to NBD2 (T1N2), as found for a similar Hsp104 mutant that binds reduced and carboxymethylated α-lactalbumin (41). The estimated Kd values for the interaction of T1T2 and T1N2 with partially unfolded MDH (0.21 ± 0.02 and 0.78 ± 0.08 μm, respectively) point out a previously unnoticed 3–4-fold reduction of the chaperone affinity for denatured MDH when nucleotide binding to NBD2 is impaired (Fig. 5A). We next analyzed the affinity of the NBD1 of T1T2 and T1N2 for ATP by following complex formation at increasing ATP concentration. Protein concentration is fixed (4 and 12 μm for T1T2 AND T1N2, respectively) to compensate the different affinity of these ClpB mutants for the substrate. The protein variant T1T2 prevents MDH aggregation at lower ATP concentrations than T1N2 (Fig. 5B). Data analysis with the Hill equation gives Kd values of 61 ± 3 and 363 ± 12 μm ATP for the interaction of T1T2 and T1N2 with thermally denatured MDH, respectively, thus reinforcing the hypothesis that the affinity of NBD1 for ATP and, hence, for substrate proteins depends on the nucleotide status of NBD2 (Fig. 5C). The Hill coefficient, nH, is 3.7 ± 0.6 and 1.3 ± 0.1 for complex formation with T1T2 and T1N2, indicating that nucleotide binding to NBD2 allows the cooperative interaction of ATP and denatured MDH with NBD1. It is worth mentioning that this cooperative effect is not observed when ATP binding is analyzed in the absence of protein substrate by proteolysis experiments (see above). Because the affinity of ClpB for denatured MDH is higher than for ATP, nucleotide binding is the limiting step in ClpB-MDH complex formation. The different Kd values obtained in these experiments as compared with proteolysis might be due, among other things, to the effect that substrate could have on the nucleotide binding properties of NBD1.
FIGURE 5.

ATP binding to NBD2 allows cooperative binding of ATP to the NBD1 in the presence of a substrate protein. A, shown is the percentage of protection against aggregation of thermally denatured MDH (6 μm) by increasing concentrations of T1T2 and T1N2. Experiments were performed at 47 °C in 50 mm Tris-HCl, pH 7.5, 20 mm MgCl2, 150 mm KCl, 2 mm DTT, and 2 mm ATP. Data corresponding to WT ClpB, T1, T2, and N2 indicate that they cannot form stable complexes with denatured MDH. B, aggregation of 6 μm MDH in the presence of 4 μm T1T2 (solid lines) or 12 μm T1N2 (dashed lines) at increasing ATP concentration is shown. Experiments were carried out as in A. Scattering values are normalized to controls obtained without ATP. C, extent of protection against aggregation of 6 μm MDH by 4 μm T1T2 or 12 μm T1N2 as a function of ATP concentration is shown. Experiments were carried out as described in B. Analysis of the data with the Hill equation gives Kd values of 61 ± 3 and 363 ± 12 μm and nH values of 3.7 ± 0.6 and 1.3 ± 0.1 for T1T2 and T1N2, respectively. Data are the means ± S.D. (n = 3).
Dependence of the Chaperone Activity of ClpB on ATP Concentration
Finally, we asked how nucleotide filling of the two NBDs affects the chaperone activity of ClpB. Impairment of the ATPase activity at any of the two NBDs abolishes or severely reduces the chaperone activity of ClpB, indicating that coupling of the activity of both NBDs is required to extract and reactivate aggregated substrates (20). As mentioned above, reactivation activities are assayed at 50 mm KCl to stabilize the population of functional hexamers.
The bichaperone network used in these experiments fails to reactivate aggregated MDH below 100 μm ATP, when the NBD2 ring of ClpB is saturated and the NBD1 ring is partially occupied by the nucleotide (Fig. 6A). Failure is not due to a defective activity of the KJE system at this nucleotide concentration, as under the same experimental conditions it refolds guanidinium-HCl-denatured luciferase in the absence of ClpB (Fig. 6C), as can be expected from the affinity of DnaK for nucleotides (Kd values ranging from 7 to 150 nm) (42–46). The Km value for MDH reactivation is 230 ± 7 μm, and the Hill coefficient is 3.7 ± 0.3. Interestingly, a similar ATP dependence is observed for the reactivation of another aggregated substrate, G6PDH (Fig. 6B). The Hill analysis of G6PDH reactivation data gives a Km of 392 ± 26 μm and a Hill coefficient, nH, of 3.7 ± 0.3. Taken together these data suggest that at least four subunits of the protein hexamer cooperate to bind thermally denatured MDH and to efficiently process both substrates.
FIGURE 6.

ATP-dependent processing of aggregated MDH and G6PDH suggests the cooperation of at least four protein subunits. Shown is the percentage of MDH (A) and G6PDH (B) reactivation as a function of nucleotide concentration. Protein activity was recorded after 120 min of reactivation at increasing ATP concentrations. The solid line represents the best fit of the data to the Hill equation. The estimated Km and nH values are 230 ± 7 μm and 3.7 ± 0.3 for MDH and 392 ± 12 μm and 3.7 ± 0.3 for G6PDH. Similar values were obtained for 60- and 90-min reactivation. Data are the means ± S.D. (n = 3). Insets, shown is time dependence of MDH and G6PDH reactivation at increasing ATP concentrations. C, shown is reactivation of aggregated luciferase (25 nm) that was denatured with guanidinium-HCl by the DnaK system (1 μm DnaK, 1 μm DnaJ, and 1.2 μm GrpE) at different ATP concentrations. Experiments were carried out in 50 mm Tris-HCl, pH 7.5, 15 mm MgCl2, 50 mm KCl, and 5.5 mm DTT at 25 °C, and luciferase activity was measured after a 2-h reactivation.
DISCUSSION
The ATPase cycle of molecular chaperones regulates the transition between different conformations with distinct affinities for nucleotides and substrate proteins (26, 41, 47). This nucleotide-driven conformational transition includes allosteric interactions essential for the chaperone activity of these proteins. The functional hexamer of ClpB (16, 48) contains 12 nucleotide binding sites arranged in two-tiered rings, which can establish intra- and intersubunit allosteric communication (19–21, 23, 49). Understanding how this oligomeric machine couples ATP binding and hydrolysis with substrate remodeling and disaggregation is necessary to unravel its mechanism of action. The contribution of individual binding domains to the overall properties of the protein has been characterized to describe the complex allosteric interactions within the ClpB hexamer (18, 20–23, 49). Although this strategy has aided comprehension of how the disaggregase activity of ClpB and other members of the Hsp100 family (Hsp104 and ClpBTh) occurs, the process is still far from being understood at a molecular level due to the complexity of the protein and the difficulty to work with protein aggregates. Herein, we further explore allosteric communication in this complex protein particle.
The first finding of this work is that both NBDs of ClpB show a pronounced difference in affinity for nucleotides. One piece of experimental evidence supporting this finding is the binding stoichiometry found for WT ClpB and selected protein variants at low nucleotide concentration (below 50 μm), which indicates that only one of the two NBDs is occupied by nucleotides. The unambiguous assignment of this binding event to filling of NBD2 states that its affinity for nucleotides is higher than that of NBD1. The stoichiometry of nucleotide binding to ClpB seems to contradict a previous ITC study (50). This work suggested that full-length ClpB was able to bind only one molecule of ATPγS at the NBD2 of each subunit, whereas both NBDs could interact with ADP with affinities similar to those shown here. The reason for this discrepancy might be the presence of KCl in our experiments, which is known to modulate the population of functional protein oligomers (19, 51). As recently reported (36), the lack of a quantitative description of the properties of each of the 12 available nucleotide binding sites and of the inter- and intrasubunit signaling that might communicate them makes difficult the use of an appropriate binding model to fit the experimental data. The model used herein considers ligand binding to equivalent and independent NBD2 sites. The assumption of independent NBD2 sites is based on their ability to bind nucleotides with similar affinity in oligomeric and monomeric full-length ClpB. This finding is in agreement with the effective capacity of truncated, monomeric NBD2 of ClpBTh to bind nucleotides (47). Our data also show that the affinity of NBD2 for nucleotides can be 10–50 times (see below) higher than that of NBD1. A similar difference in Km values has also been described for the NBDs of Hsp104, based on analysis of the kinetics of ATP hydrolysis (18).
A recent molecular dynamic simulation of a hexameric model of ATP-bound ClpB might explain the different affinities of the NBDs for nucleotides (29). This study shows that the rim of the ATP binding pocket of NBD1 is negatively charged, building an interface that is complementary in shape and charge to the surface of a neighboring NBD1. The two neighbor monomers provide ATP-interacting residues. These properties would result in a low affinity of NBD1 for nucleotides due to unfavorable electrostatic interactions and in the observed sensitivity of nucleotide binding to the stability of the protein hexamer. A truncated monomeric variant of ClpBTh containing the NBD1 and the M domain of the protein also fails to bind nucleotides and recovers nucleotide binding competence in the oligomeric complex that forms when mixed with purified NBD2 (28). Although the NBD2 is also located at the interface between two adjacent subunits, only one of them seems to provide residues to interact with ATP (29). Our data support this model as the affinity for nucleotides of the NBD2 of full-length hexameric and monomeric ClpB is similar. A more hydrophobic NBD2 together with its positively charged entrance might explain its higher affinity for nucleotides.
The second interesting finding of this study is that the affinity of NBD1 for nucleotides is regulated by the nucleotide state of NBD2. ATP, but not ADP, binding to NBD2 enhances the affinity of NBD1 for the corresponding nucleotide. This effect reflects transmission of the allosteric signal generated upon ATP binding to NBD2 toward the nucleotide binding ring close to the substrate entrance, the NBD1 ring. Communication between the NBDs has been described for ClpBEco (20), Hsp104 (18), and ClpBTth (24, 26). However, only in the case of Hsp104 has it been demonstrated that the nucleotide bound at the NBD2 affects to the catalytic properties (18) and affinity (25) of NBD1 for nucleotides. If, as proposed for ClpB (52) and Hsp104 (53, 54), the substrate is threaded from the N to the C terminus of the central cavity, ATP binding to NBD2 would favor priming of the protein particle with substrates. The fact that ADP binding to NBD2 does not modify the affinity of NBD1 for this nucleotide points out that the ATP- and ADP-induced allosteric signal transmitted from NBD2 to NBD1 is different, and therefore, it could help to establish the asymmetric functioning of both NBDs in the hexamer. Functional asymmetry has been recently proposed for Hsp104, as the T<->R transition at both NBDs seems to be inversely linked (25). Data presented here also indicate that ATP binding to NBD2 induces an allosteric signal that promotes cooperative binding of ATP and substrates to the NBD1 ring. Thus, it seems that interplay between the NBDs is necessary for the coordinated translocation of substrates that would require the concerted action of a minimum number of subunits of the functional oligomeric assembly. The modulation of the cooperativity of nucleotide and substrate binding to the NBD1 ring by the nucleotide status of NBD2 was previously unnoticed. Because this cooperative effect is not observed in the absence of substrate, it might be caused by its nucleotide-dependent interaction with several chaperone monomers that must cooperate to process it. It is worth mentioning that the minimum number of subunits estimated to cooperate in substrate binding and processing is around 4, the same value obtained in a recent study using hybrid ClpB hexamers containing WT and mutant subunits (23). A similar number of active subunits has been estimated for the unfoldases ClpX (37, 55) and HslU (56). These related hexamers, which contain six nucleotide binding sites, appear to bind a maximum of four ATPs in solution under saturating nucleotide concentration, and based on the x-ray structures of several forms of ClpX, it was suggested that they could function asymmetrically (55). Similarly, the asymmetry that allosteric communication between the two NBD rings and between different monomers might promote could provide the structural basis of the unidirectional translocation of polypeptides through the central cavity from the N-terminal to the C-terminal exit of ClpB.
In summary, our data demonstrate that the two NBDs of ClpB display different affinity for nucleotides, that of NBD1 being regulated by the nucleotide state of NBD2. ATP, but not ADP, binding to the high affinity site, the NBD2, generates an allosteric signal that increases the affinity of NBD1 for ligands (ATP and substrates) and modifies intersubunit communication so that several subunits cooperate to bind and reactivate aggregated substrates.
Supplementary Material
Acknowledgment
We are grateful to Janire Castelo for technical assistance.
This work was supported by Ministerio de Ciencia e Innovación Grants BFU2007-64452 and BFU2010-15443, Universidad del País Vasco and Gobierno Vasco Grant IT-358-07, and Diputación Foral de Bizkaia Grant DIPE08/18.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- Hsp104
- heat-shock protein 104
- ClpB
- caseinolytic peptidase B
- T1
- ClpBE279A
- T1T2
- ClpBE279A/E678A
- T1N2
- ClpBE279A/K611A
- N1T2
- ClpBK212A/E678A
- N1N2
- ClpBK212A/K611A
- N1
- ClpB(K212A)
- T2
- ClpBE678A
- N2
- ClpBK611A
- ClpBEco
- ClpB from E. coli
- ClpBTh
- ClpB from T. thermophilus;
- MDH
- malate dehydrogenase
- G6PDH
- glucose-6-phosphate dehydrogenase
- MANT
- 2′(3′)-O-(N-methylanthraniloyl)-ADP
- NBD
- nucleotide binding domain
- ITC
- isothermal titration calorimetry
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
REFERENCES
- 1. Sanchez Y., Lindquist S. L. (1990) Science 248, 1112–1115 [DOI] [PubMed] [Google Scholar]
- 2. Squires C. L., Pedersen S., Ross B. M., Squires C. (1991) J. Bacteriol. 173, 4254–4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schirmer E. C., Glover J. R., Singer M. A., Lindquist S. (1996) Trends Biochem. Sci. 21, 289–296 [PubMed] [Google Scholar]
- 4. Sanchez Y., Taulien J., Borkovich K. A., Lindquist S. (1992) EMBO J. 11, 2357–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neuwald A. F., Aravind L., Spouge J. L., Koonin E. V. (1999) Genome Res. 9, 27–43 [PubMed] [Google Scholar]
- 6. Vale R. D. (2000) J. Cell Biol. 150, F13–F19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mogk A., Bukau B. (2004) Curr. Biol. 14, R78–R80 [DOI] [PubMed] [Google Scholar]
- 8. Hanson P. I., Whiteheart S. W. (2005) Nat. Rev. Mol. Cell Biol. 6, 519–529 [DOI] [PubMed] [Google Scholar]
- 9. Bukau B., Weissman J., Horwich A. (2006) Cell 125, 443–451 [DOI] [PubMed] [Google Scholar]
- 10. Erzberger J. P., Berger J. M. (2006) Annu. Rev. Biophys. Biomol. Struct. 35, 93–114 [DOI] [PubMed] [Google Scholar]
- 11. Mogk A., Haslberger T., Tessarz P., Bukau B. (2008) Biochem. Soc. Trans. 36, 120–125 [DOI] [PubMed] [Google Scholar]
- 12. White S. R., Lauring B. (2007) Traffic 8, 1657–1667 [DOI] [PubMed] [Google Scholar]
- 13. Walker J. E., Saraste M., Runswick M. J., Gay N. J. (1982) EMBO J. 1, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haslberger T., Weibezahn J., Zahn R., Lee S., Tsai F. T., Bukau B., Mogk A. (2007) Mol. Cell 25, 247–260 [DOI] [PubMed] [Google Scholar]
- 15. Doyle S. M., Wickner S. (2009) Trends Biochem. Sci 34, 40–48 [DOI] [PubMed] [Google Scholar]
- 16. Lee S., Choi J. M., Tsai F. T. (2007) Mol. Cell 25, 261–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee S., Sowa M. E., Watanabe Y. H., Sigler P. B., Chiu W., Yoshida M., Tsai F. T. (2003) Cell 115, 229–240 [DOI] [PubMed] [Google Scholar]
- 18. Hattendorf D. A., Lindquist S. L. (2002) EMBO J. 21, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schlee S., Groemping Y., Herde P., Seidel R., Reinstein J. (2001) J. Mol. Biol. 306, 889–899 [DOI] [PubMed] [Google Scholar]
- 20. Mogk A., Schlieker C., Strub C., Rist W., Weibezahn J., Bukau B. (2003) J. Biol. Chem. 278, 17615–17624 [DOI] [PubMed] [Google Scholar]
- 21. Schirmer E. C., Ware D. M., Queitsch C., Kowal A. S., Lindquist S. L. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 914–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Werbeck N. D., Schlee S., Reinstein J. (2008) J. Mol. Biol. 378, 178–190 [DOI] [PubMed] [Google Scholar]
- 23. del Castillo U., Fernández-Higuero J. A., Pérez-Acebrón S., Moro F., Muga A. (2010) FEBS Lett. 584, 929–934 [DOI] [PubMed] [Google Scholar]
- 24. Beinker P., Schlee S., Auvula R., Reinstein J. (2005) J. Biol. Chem. 280, 37965–37973 [DOI] [PubMed] [Google Scholar]
- 25. Franzmann T. M., Czekalla A., Walter S. G. (2011) J. Biol. Chem. 286, 17992–18001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Watanabe Y. H., Takano M., Yoshida M. (2005) J. Biol. Chem. 280, 24562–24567 [DOI] [PubMed] [Google Scholar]
- 27. Weibezahn J., Schlieker C., Bukau B., Mogk A. (2003) J. Biol. Chem. 278, 32608–32617 [DOI] [PubMed] [Google Scholar]
- 28. Werbeck N. D., Zeymer C., Kellner J. N., Reinstein J. (2011) Biochemistry 50, 899–909 [DOI] [PubMed] [Google Scholar]
- 29. Zietkiewicz S., Slusarz M. J., Slusarz R., Liberek K., Rodziewicz-Motowidło S. (2010) Biopolymers 93, 47–60 [DOI] [PubMed] [Google Scholar]
- 30. Woo K. M., Kim K. I., Goldberg A. L., Ha D. B., Chung C. H. (1992) J. Biol. Chem. 267, 20429–20434 [PubMed] [Google Scholar]
- 31. Mehl A. F., Heskett L. D., Neal K. M. (2001) Biochem. Biophys. Res. Commun. 282, 562–569 [DOI] [PubMed] [Google Scholar]
- 32. Zylicz M., Yamamoto T., McKittrick N., Sell S., Georgopoulos C. (1985) J. Biol. Chem. 260, 7591–7598 [PubMed] [Google Scholar]
- 33. Baykov A. A., Evtushenko O. A., Avaeva S. M. (1988) Anal. Biochem. 171, 266–270 [DOI] [PubMed] [Google Scholar]
- 34. Schlieker C., Tews I., Bukau B., Mogk A. (2004) FEBS Lett. 578, 351–356 [DOI] [PubMed] [Google Scholar]
- 35. Diamant S., Ben-Zvi A. P., Bukau B., Goloubinoff P. (2000) J. Biol. Chem. 275, 21107–21113 [DOI] [PubMed] [Google Scholar]
- 36. Nagy M., Wu H. C., Liu Z., Kedzierska-Mieszkowska S., Zolkiewski M. (2009) Protein Sci. 18, 287–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hersch G. L., Burton R. E., Bolon D. N., Baker T. A., Sauer R. T. (2005) Cell 121, 1017–1027 [DOI] [PubMed] [Google Scholar]
- 38. Leskovar A., Reinstein J. (2008) Arch Biochem. Biophys. 473, 16–24 [DOI] [PubMed] [Google Scholar]
- 39. Akoev V., Gogol E. P., Barnett M. E., Zolkiewski M. (2004) Protein Sci. 13, 567–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hattendorf D. A., Lindquist S. L. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 2732–2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bösl B., Grimminger V., Walter S. (2005) J. Biol. Chem. 280, 38170–38176 [DOI] [PubMed] [Google Scholar]
- 42. Theyssen H., Schuster H. P., Packschies L., Bukau B., Reinstein J. (1996) J. Mol. Biol. 263, 657–670 [DOI] [PubMed] [Google Scholar]
- 43. Farr C. D., Slepenkov S. V., Witt S. N. (1998) J. Biol. Chem. 273, 9744–9748 [DOI] [PubMed] [Google Scholar]
- 44. Russell R., Jordan R., McMacken R. (1998) Biochemistry 37, 596–607 [DOI] [PubMed] [Google Scholar]
- 45. Chesnokova L. S., Witt S. N. (2005) Biochemistry 44, 11224–11233 [DOI] [PubMed] [Google Scholar]
- 46. Taneva S. G., Moro F., Velázquez-Campoy A., Muga A. (2010) Biochemistry 49, 1338–1345 [DOI] [PubMed] [Google Scholar]
- 47. Werbeck N. D., Kellner J. N., Barends T. R., Reinstein J. (2009) Biochemistry 48, 7240–7250 [DOI] [PubMed] [Google Scholar]
- 48. Wendler P., Shorter J., Plisson C., Cashikar A. G., Lindquist S., Saibil H. R. (2007) Cell 131, 1366–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hoskins J. R., Doyle S. M., Wickner S. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 22233–22238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li J., Sha B. (2002) J. Mol. Biol. 318, 1127–1137 [DOI] [PubMed] [Google Scholar]
- 51. Kim K. I., Cheong G. W., Park S. C., Ha J. S., Woo K. M., Choi S. J., Chung C. H. (2000) J. Mol. Biol. 303, 655–666 [DOI] [PubMed] [Google Scholar]
- 52. Weibezahn J., Tessarz P., Schlieker C., Zahn R., Maglica Z., Lee S., Zentgraf H., Weber-Ban E. U., Dougan D. A., Tsai F. T., Mogk A., Bukau B. (2004) Cell 119, 653–665 [DOI] [PubMed] [Google Scholar]
- 53. Tessarz P., Mogk A., Bukau B. (2008) Mol. Microbiol. 68, 87–97 [DOI] [PubMed] [Google Scholar]
- 54. Wendler P., Shorter J., Snead D., Plisson C., Clare D. K., Lindquist S., Saibil H. R. (2009) Mol. Cell 34, 81–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Glynn S. E., Martin A., Nager A. R., Baker T. A., Sauer R. T. (2009) Cell 139, 744–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yakamavich J. A., Baker T. A., Sauer R. T. (2008) J. Mol. Biol. 380, 946–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.