

Abstract
Periventricular white matter injury (PWMI), is the leading cause of chronic neurologic injury among survivors of preterm birth. The hallmark of PWMI is hypomyelination and a lack of mature, myelinating oligodendrocytes. Oligodendrocytes undergo a well-characterized lineage progression from neural stem cell to mature oligodendrocyte. Oligodendrocyte precursors have increased susceptibility to oxidative and free radical-mediated injury compared to mature oligodendrocytes due to lower levels of anti-oxidant enzymes and free radical scavengers. In this study, we show that oxidative stress disrupts oligodendrocyte differentiation by two mechanisms. First, oxidizing agents decrease the expression of key genes which promote oligodendrocyte differentiation from neural stem cells and increase the expression of genes known to inhibit differentiation. Second, global histone acetylation persists under conditions of oxidative stress, further contributing to the prevention of oligodendrocyte differentiation. Both of these mechanisms result in the arrest of oligodendrocyte differentiation without an increase in cell death.
Introduction
Advances in neonatal intensive care have resulted in improved survival of very low birth weight (VLBW) infants (<1.5 kg), however a number of these survivors have long-term neurologic disabilities which include cerebral palsy, cognitive and learning disabilities, and vision and hearing loss (Martin et al, 2005; Wilson-Costello et al, 2005). Periventricular white matter injury (PWMI), a spectrum of brain injury that ranges from focal cystic necrotic lesions (periventricular leukomalacia) to diffuse demyelination, is the leading cause of chronic neurologic injury in this population (Volpe, 2001a; Volpe 2001b). Early stages of PWMI are characterized by white matter volume loss and the loss of oligodendrocytes, the cellular source of myelin in the central nervous system (CNS).
The pathogenesis of PWMI is complex and multifactorial. There is evidence linking PWMI with maternal and/or fetal infection (Hagberg et al, 2002; Dammann et al, 1997; DiSalvo 1998), hypoxia/ischemia (Yesilirmak et al, 2007), impaired regulation of cerebral blood flow (Fukuda et al, 2006), formation of free radicals (Haynes et al, 2005), impaired myelination due to oligodendrocyte injury/loss (Cai et al, 2000; Inder et al, 2000), apoptotic cell death (Kadhim et al, 2006), microglial activation (Volpe, 2001) and excitotoxicity (Follett et al, 2004). Despite growing literature detailing associations, very little detailed information exists about the cellular mechanisms by which PWMI occurs. Several investigators have suggested that proinflammatory cytokines and reactive oxygen species disrupt precursor cell maturation and lead to arrest of oligodendrocyte development resulting in hypomyelination.
The period of greatest vulnerability for PWMI in the developing fetus and premature infants occurs between 23 and 32 weeks postconceptional age (Volpe, 2001b). This corresponds to the developmental window when oligodendrocyte precursors and immature oligodendrocytes are the predominant cell types in the cerebral white matter (Back et al, 1996; Back et al, 2001). Several studies demonstrate that the oligodendrocyte lineage displays maturation-dependent vulnerability to cellular injury. Immature, developing oligodendrocytes display increased susceptibility to oxidative stress and free radical-mediated injury compared to mature, myelinating oligodendrocytes due to lower levels of anti-oxidant enzymes and free radical scavengers, such as glutathione (Back et al, 1998; Baud et al, 2004b; Fern et al, 2000) and higher concentrations of unsaturated fatty acids and high rate of oxygen consumption (Halliwell, 1992). Studies in perinatal rats and rodent cell culture confirm that reactive oxygen species injure oligodendrocyte progenitors, leading to precursor cell death with subsequent decreased numbers of mature oligodendrocytes and ultimately hypomyelination in the cerebral white matter (Levison et al, 2001).
Oligodendrocytes undergo a defined lineage progression from neural stem cell to mature oligodendrocyte which has been well characterized through the assessment of stage specific antigens (Miller, 2002). Early inhibition of oligodendrocyte development appears to be dependent on both inhibitory signaling and epigenetic regulation. During oligodendrocyte development, histone deacetylation is critical for differentiation in the developing brain by either repressing genes that inhibit differentiation or by repressing negative regulatory elements in oligodendrocyte gene promoters so that maturation of oligodendrocytes can occur (Marin-Husstege et al, 2002; Liu et al, 2007).
In the present study, we used an in vitro model of oxidative stress to examine changes in expression of genes important to oligodendrocyte differentiation and how altered epigenetic regulation may contribute to those changes in gene expression. We show that treatment of oligodendrocyte precursor cells with oxidizing agents decreases expression of genes important in promoting oligodendrocyte maturation, such as Shh, Sox10, HDAC3, Olig 1 and Olig 2, and increases expression of Id2 and Id4, genes which inhibit oligodendrocyte differentiation. Finally, we show that oligodendrocyte differentiation is arrested in the precursor stage without an associated increase in cell death after exposure to oxidative stress. These results suggest that oxidative stress leads to the disruption of oligodendrocyte differentiation by altering the regulation of key genes required for this process.
Materials and Methods
Cell Culture Generation of Primary Cultures and Immunoselection Procedures
Cultures of oligodendrocyte precursors were established from the forebrains of 0 and 1-day-old Sprague-Dawley rats and seeded in 100mm Petri dishes with serum-containing media as previously described (Grinspan et al, 1995). After 24 hours, the cells were switched to a serum-free growth medium consisting of DMEM with 100 μg/ml transferrin, 100 μg/ml bovine serum albumin, 16 μg/ml putrescine, 60 ng/ml progesterone, 40 ng/ml sodium selenite, 5 mg/ml N-acetyl cysteine, 1 mM sodium pyruvate, 10 ng/ml d-biotin, 12.5 μg/ml insulin (Sigma-Aldrich, St. Louis, MO), 10 ng/ml basic fibroblast growth factor (R&D, Minneapolis, MN), 2 ng/ml platelet-derived growth factor (R&D, Minneapolis, MN), 1 ng/ml neurotrophin-3 (Peprotech, Rocky Hill, NJ) as previously described (See et al, 2007). Cultures were grown in the above medium until confluent.
After confluency was reached, oligodendrocyte precursor cells (OPCs) were isolated from primary culture by immunopanning. Coated Petri dishes were prepared as previously described (Grinspan et al, 1995). Type 1 astrocytes, neurons and microglial were removed during two sequential incubations on plates coated with RAN-2 antibody (Barres et al, 1992). OPCs were then collected by incubation on plates coated with A2B5 antibody (Grinspan et al, 2000). The purified OPCs were grown in growth media with growth factors (10ng/ml fibroblast growth factor, 2 ng/ml platelet-derived growth factor, 1 ng/ml neurotrophin-3) on lysine-coated 100 mm plates and on poly-D-lysine-coated coverslips.
OPCs were allowed to differentiate by placing them in differentiation medium containing 50% DMEM, 50% Ham's F12 with 50 μg/ml transferrin, 5 μg/ml putresine, 3 ng/ml progesterone, 2.6 ng/ml selenium, 12.5 μg/ml insulin, 0.4 μg/ml T4, 0.3% glucose, 2 mM glutamine and 10 ng/ml biotin as previously described (See et al, 2007). To induce oxidative stress, OPCs were treated for up to seventy-two hours with increasing concentrations of L-buthionine sulfoximine (BSO), an inhibitor of gamma glutamyl cysteine synthetase and ultimately glutathione reductase, or tert-butyl hydrogenperoxide (tBOOH), which leads to lipid peroxidation and the consumption of reduced glutathione and NADPH depletion.
To show that the oxidizing agents appropriately alter the levels of glutathione, we measured total glutathione, GSH and GSSG content in cells following 24 hours of treatment with BSO and tBOOH using a GSH assay kit (Cayman Chemical, Ann Arbor, MI). This assay uses glutathione reductase for the quantification of GSH. Both GSH and GSSG are measured and the assay reflects total glutathione.
SDS-Polyacrylamide Gel Electrophoresis and Western Blot Analysis
To determine protein expression of acetylated H3 and H4, Western blot analyses were performed on OPCs that had been exposed to BSO and tBOOH for 24 to 72 hours. Cells were harvested in ice-cold lysis buffer (containing 25 mM Tris, pH 7.6, 1 mM MgCl2, 1 mM EGTA, 1% Triton X-100, 1% SDS, 1 mM PMSF, 50 μg/ml antipain, 2 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A). The amount of protein was quantified by BCA assay (Pierce Chemical, Rockford, IL). Protein samples (25 μg) were denatured by boiling in sample buffer containing 2.8 M β-mercaptoethanol and separated on a 15% Tris-HCl gel (Bio-Rad Laboratories, Hercules, CA) by electrophoresis. The separated proteins were transferred electrophorectially to Immun-Blot PVDF Membranes (Bio-Rad Laboratories). Membranes were blocked in tris-buffered saline solution with 5% nonfat dried milk powder for 1 hour. Membranes were incubated with anti-acetylhistone 3 and anti-acetyl-histone 4 antibodies. Beta-actin was used as a loading control. For anti-acetyl-histone 3 antibody (Upstate, Lake Placid, NY) a 1:3000 dilution was used. The antibody is polyclonal and cross-reacts with human and mouse. For anti-acetylated H4 antibody (Upstate), a 1:5000 dilution was used. This antibody is polyclonal and cross-reacts with eukaryotes. Primary antibody incubation was followed by horse-radish peroxidase-conjugated goat-anti-rabbit IgG (Bio-Rad Laboratories), using a 1:3000 dilution. Immunoreactive protein was detected with ECL reagents (Amersham, Piscataway, NJ) according to the manufacturer's directions. Then the membranes were exposed to KODAK BioMax MR Film (Eastman Kodak Company, Rochester, NY) for analysis.
Real time PCR
OPCs were harvested after 24 or 72 hours of exposure to BSO or tBOOH. RNA was extracted using TRIzol (Molecular Research Center, Inc., Cincinnati, OH). Five μg of RNA per treatment group was used to generate complementary DNA (cDNA) using random hexamers. Specific primers were purchased from Applied Biosystems (Foster City, CA). Quantitative real time PCR using Taqman (Applied Biosystems) and equivalent dilutions of each cDNA sample was performed with ABI 7900 (Applied Biosystems). The comparative threshold cycle (Ct) method was used for quantification according to the protocol of the manufacturer. Measurement of Ct was performed at least three times per cycle. The relative abundance of the target was normalized to the relative abundance of 18S RNA in each sample. All samples were analyzed in triplicate.
Immunocytochemistry
To ensure purity of OPC culture and to determine differentiation of OPCs under experimental conditions, cells grown on lysine-coated coverslips were processed for detection of surface and internal antigens as previously described (Grinspan et al, 1990). Briefly, unfixed cells were incubated with primary antibody at room temperature for 25 minutes, washed and incubated in secondary antibody for 25 minutes. After fixation with ice-cold 95% ethanol/5% acetic acid (vol/vol) for 10 minutes, the coverslips were incubated with DAPI (4,6-diamidino-2-phenylidole, 10 mg/ml; Sigma, St. Louis, MO) for 5 minutes, mounted in Vectashield (Vector Laboratories, Burlingame, CA) and sealed in nail polish. Primary antibodies used for the recognition of surface antigens were A2B5 (hybridoma supernatant, undiluted; Eisenbarth et al, 1979), O4 (hybridoma supernatant, diluted 1:4: Sommer and Schachner, 1981) and galactocerebroside (GalC) (hybridoma supernatant, undiluted, Ranscht et al, 1982) followed by goat anti-mouse IgM (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). For the recognition of the internal antigen, glial fibrillary acidic protein (GFAP) antibody, we fixed with acid alcohol as above and incubated with anti-GFAP antibody (hybridoma supernatant, undiluted; gift of Dr. Virginia Lee, University of Pennsylvania) followed by goat anti-rat IgG(Jackson ImmunoResearch Laboratories, Inc). All secondary antibodies were diluted 1:100 in serum-free medium. Ten random fields per coverslip were scanned using both a 40× and a 63× oil immersion lens of a Leica DMR fluorescence microscope and the total number of cells, the number of cells labeled with each antibody and the total number of DAPI-positive cells per field were counted and compared. These determinations were performed on cultures from three separate preparations. One-way ANOVA was used to compare means of control, BSO and tBOOH treated OPCs.
Expression of Olig2 protein under conditions of oxidative stress was examined using Olig2 antibody (Chemicon-Millipore, Billerica, MA) at a 1:100 dilution with goat-anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc.) at 1:100 in serum-free medium. Cells expressing Olig2 protein were counted using a Leica DMR fluorescence microscope.
Measurement of Cell Death
In order to ensure that cell death was not responsible for changes in gene expression, cell death was measured by three different assays. Initiually, to analyze toxicity of the oxidizing agents, BSO and tBOOH, we examined cell death using propidium iodide incorporation assay. Cell death was examined by the incorporation of propidium iodide and assessed after 24 and 72 hours. At low concentrations, neither BSO nor tBOOH significantly increased cell death. We chose to treat OPCs with the highest doses of BSO (100uM) and tBOOH (5uM) that did not significantly increase cell death. We also used antibody to activated caspase 3 (Cell Signaling Technology, Beverly, MA) and TUNEL analysis on A2B5 positive OPCs as previously described (Grinspan et al, 1998). To estimate the number of activated caspase 3 or TUNEL positive OPCs, 10 random fields were examined per coverslip using the 40× lens. The number of activated caspase 3 or TUNEL positive cells was counted per field. These determinations were performed on cultures from 3 separate experiments. The average number of activated caspase 3 or TUNEL positive cells per field was used to calculate a mean and standard error after 24 and 72 hours of oxidant exposure.
Statistical Analyses
We analyzed real-time PCR and immunocytochemistry results with one-way analysis of variance (ANOVA) for repeated measurements of control, BSO and tBOOH exposed cell cultures. P value of <0.05 was defined as statistically significant.
Results
Oligodendrocyte Differentiation is Arrested Upon Exposure to Oxidative Stress
To show that both TBOOH and BSO significantly altered the redox ratio of cultured oligodendrocytes, we measured GSH and GSSG after 24 hours of treatment. There was a profound depletion of GSH in the OPCs, a slight increase in GSSG and a significant reduction in total glutathione following treatment with either agent (Table 1), indicating that both compounds are effective agents to model oxidative stress in oligodendrocyte lineage cells.
Table 1.
Measurement of glutathione levels in OPCs in culture 24 hours post treatment.
| Control | BSO | tBOOH | |
|---|---|---|---|
| GSH (nmol/mg protein) | 85.92 ± 4.99 | 49.54 ± 2.11* | 52.24 ± 1.89** |
| GSSG (nmol/mg protein) | 2.17 ± 0.28 | 2.92 ± 0.59* | 2.86 ± 0.52** |
| Redox ratio (GSH/GSSG) | 37.89 ± 2.14 | 22.33 ± 1.89* | 19.67 ± 1.89** |
p<0.05 BSO vs. controls
p<0.05 tBOOH vs. controls
To examine the effects of oxidative insult on the development and maturation of oligodendrocytes, purified OPC cultures were established from P0-1 rats and seeded on coverslips. A2B5 and GalC positivity were used to define developmental stages of the oligodendrocyte lineage as previously described (Robinson et al, 1996). After 24 (data not shown) and 72 hours of oxidant treatment, similar proportions of immature A2B5+ oligodendrocytes were present in all culture conditions while the proportion of mature, myelin-producing GalC+ oligodendrocytes was significantly reduced in the oxidant-exposed cultures compared to their timepoint controls (Figure 1). These data demonstrate that oxidant exposure leads to the arrest of oligodendrocyte differentiation at the precursor stage.
Figure 1.
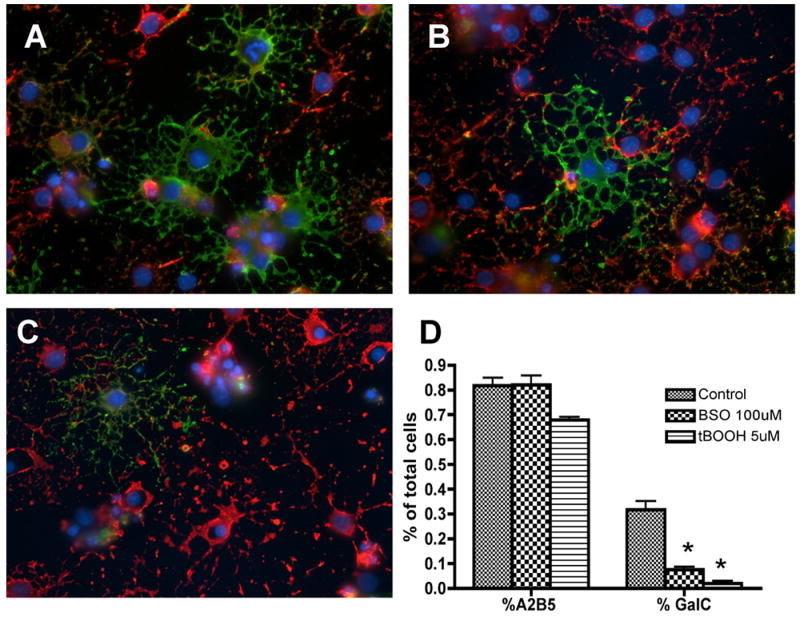
Oligodendrocyte differentiation after 72 hours of oxidant exposure. OPCs were double-labeled in vitro with oligodendroglial markers A2B5 (red) and GalC (green) after 72 hours of oxidant exposure. The number of mature GalC+ cells was significantly decreased in the BSO (B) and tBOOH (C) exposed cultures compared to control (A) cultures. The majority of cells exposed to oxidative stress were arrested in the immature A2B5+ cell stage. Quantitative analysis of A2B5+ and GalC+ cells is shown in (D). The proportion of positive cells per 40× field was determined by counting 10 fields per coverslip from three separate experiments following indirect immunofluorescence using antibody to A2B5 and GalC. Data represent the mean plus standard deviation. *P < 0.05 vs. control.
To rule out the possibility that TBOOH or BSO pushed the OPCs, which are still multipotential, to another neural lineage, we labeled cells following treatment with antibody to markers of astrocytes (GFAP) and neurons (Tuj1), counted the numbers of labeled cells versus DAPI+ nuclei and compared to controls. Fewer than 1% of the culture, treated or control, labeled with Tuj1. At 72 hours in defined medium, 5.1 ± 2.0% of the cells in the control cultures labeled with GFAP, 6.4 ± 3.0% of the cells in the BSO treated cultures and 6.2 ± 2.5% of the cells in the TBOOH treated cultures labeled with GFAP (n=3 separate preparations of cells). These cells clearly had the morphology of type 1 astrocytes (data not shown). This was not a significant increase. These data demonstrate that oxidant exposure does not inhibit differentiation by altering the neural lineage of the OPCs. The number of A2B5+ cells in the treated cultures is either the same (for BSO) as the controls or slightly lower (10% for TBOOH) suggesting that the cells that do not differentiate and remain precursors.
Oxidative Stress Alters Expression of Genes Regulating Oligodendrocyte Differentiation
To determine whether oxidative stress disrupts the normal step-wise progression of oligodendrocyte differentiation, we used quantitative real-time PCR to determine if expression of key genes that regulate normal oligodendrocyte differentiation was altered. Sox10 (Stolt et al, 2002), Shh (Nery et al, 2001; Pringle et al, 1996), HDAC3 (Shen et al, 2005), Olig1 (Ligon et al, 2006), Olig2 (Zhou et l, 2002), Id2 and Id4 (Samanta & Kessler, 2004, Marin-Husstege et al. 2006; Wang et al. 2001) are genes previously shown to be critical positive or negative regulators of oligodendrocyte differentiation. OPCs were exposed to oxidative agents BSO or tBOOH for 24 (data not shown) and 72 hours. Exposure to oxidative stress resulted in decreased expression of Sox10, Shh, and HDAC3, all genes important for promoting differentiation of oligodendrocytes (Figure 2). Inhibitors of Differentiation (Id) proteins sequester basic helix-loop-helix factors, such as Olig proteins, and act as dominant negative regulators of differentiation (Benezra et al, 1990). Exposure to oxidative stress resulted in significantly increased expression of Id2 and Id4, negative regulators of oligodendrocyte differentiation, and significantly decreased expression of Olig1 and Olig2.
Figure 2.
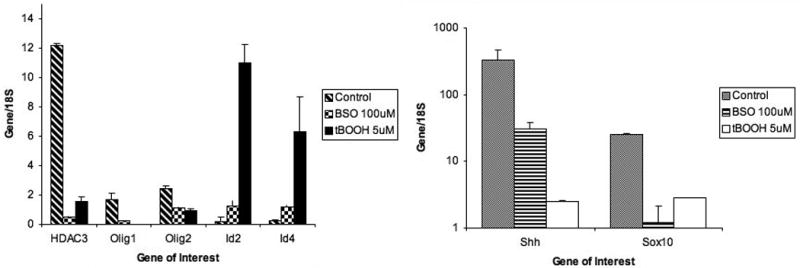
Expression of genes promoting or inhibiting differentiation of oligodendrocyte precursor cells. The graph shows significantly decreased expression of Shh, Sox10, HDAC3, Olig1 and Olig2 72 hours after exposure to oxidants compared to control and significantly increased expression of Id2 and Id4 after exposure to oxidants. cDNA expression from 3 separate experiments, each performed in triplicate, was averaged. The data are presented as the mean ± standard deviation. *P < 0.05 vs. control.
In addition to decreased Olig1 expression as seen by q-PCR, we saw a change in the expression of Olig2 upon exposure to oxidative stress by immunoctochemistry. Although active Olig2 is thought to be localized in the nucleus and moves to the cytoplasm when inactive (Samanta and Kessler, 2004), we observed an overall decrease in Olig2 expression in a significant number of treated cells by 6 hours after exposure to oxidative stress in either tBOOH or BSO (p< 0.014, Table 2 and Figure 3). Olig2 is necessary for differentiation and thus it's down regulation correlates with the decrease in other genes regulating maturation.
Table 2.
The number of cells strongly expressing Olig2 decreases 6 hours after treatment with reactive oxygen species.
| Treatment | % of cells with reduced Olig2 labeling |
|---|---|
| Control | 11.3 ± 1.8 |
| BSO | 31.3 ± 1.8 |
| tBOOH | 31.7 ± 3.8 |
Figure 3.
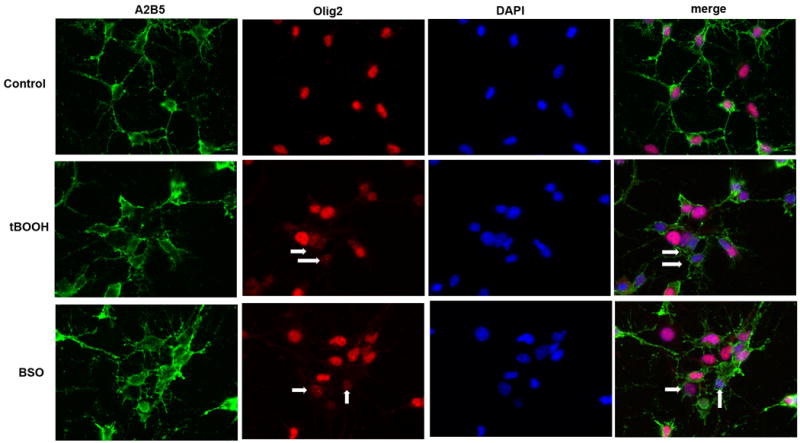
Expression of Olig2 protein after exposure to oxidants is decreased. OPCs were labeled with Olig2 (red), A2B5 (green), and DAPI (blue) after 6 hours of oxidant exposure. Treatment with both tBOOH and BSO significantly reduced the number of cells strongly expressing Olig2. Arrows mark sample A2B5+ cells with decreased Olig2 expression.
Arrested Maturation and Altered Gene Expression is Not Due to Cell Death
Oxidative stress leads to arrested maturation and altered expression of genes important for oligodendrocyte differentiation. The decrease in mature, myelin-producing GalC+ oligodendrocytes and expression of genes which regulate oligodendrocyte differentiation (Shh, Sox10, HDAC3, Olig1, Id2, Id4) is not caused by cell death as revealed by TUNEL and activated caspase 3 staining after 24 (data not shown) and 72 hours of exposure to oxidants BSO and tBOOH (Figure 4, A-F). Quantitative analysis of TUNEL+ and activated caspase 3+ nuclei is shown in Figure 4, G-H. There is a small but significant increase in the number of TUNEL positive nuclei in the tBOOH-exposed cultures. However, this increase does not appear to be large enough to account for the 10 to 20 fold decrease in key gene expression or decreased number of GalC+ oligodendrocytes. There was not a similar increased noted for activated caspase 3 treated cultures.
Figure 4.
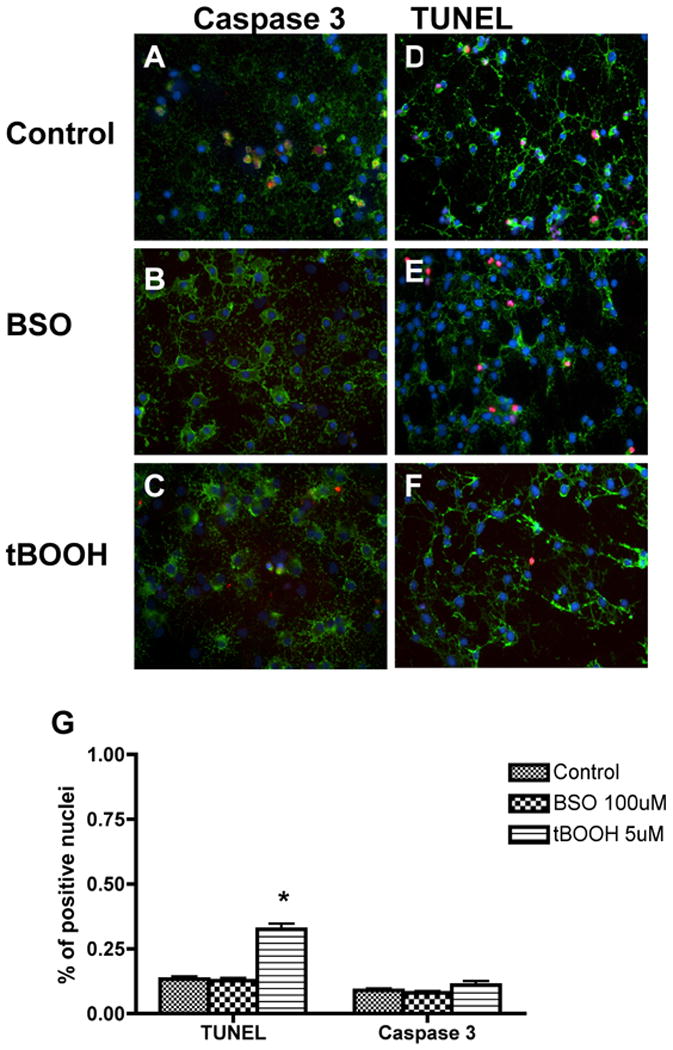
Cell death was quantitated using both the TUNEL assay and activated caspase 3 staining following 72 hours of exposure to oxidative agents. The density of activated caspase 3+ (A-C) or TUNEL+ (D-F) nuclei was unchanged between control and BSO-exposed culture conditions. There is a small but significant increase in the number of TUNEL+ positive nuclei in the tBOOH-exposed cultures compared to control, but this increase doesn't appear to be large enough to account for the 10 to 20 fold decrease in key gene expression or decreased number of GalC+ oligodendrocytes. Quantitative analysis of activated caspase 3+ and TUNEL+ (G) cells is also shown. The proportion of positive cells per 40× field was determined by counting 10 fields per coverslip in 3 separate experiments following indirect immunofluorescence using antibody to activated caspase 3 or TUNEL analysis. Data represent the mean plus standard deviation. *P <0.05 vs. control.
Global Histone Deacetylation is Prevented With Exposure to Oxidative Stress
Post-translational modifications of DNA and nucleosomal histones are known to play an important role in the differentiation of neural stem cells into astrocytes, neurons and oligodendrocytes (Feng et al, 2005; Hsieh et al, 2004; Roopra et al, 2000). Global histone acetylation increases the expression of genes associated with neurogenesis and astrogliogenesis during the early stages of brain development (Ballas et al, 2001; Hsieh et al, 2004; Song et al, 2004). In later stages of brain development, histone deacetylase activity increases allowing oligodendrocyte development and differentiation to begin (Marin-Husstage et al, 2002). Decreased histone deacetylase activity and persistent histone acetylation in oligodendrocyte progenitor cells has been shown to prevent differentiation to the mature phenotype (Liu et al, 2007; Shen et al, 2005).
Oxidative stress was shown to be associated with decreased levels of histone deacetylase activity by real-time PCR (Figure 2). Accordingly, exposure to BSO and tBOOH is associated with persistent global acetylation of histone proteins H3 and H4 compared to control as indicated by immunocytochemistry at 72 hours (Figure 5, A-F) (24 hour data not shown). Quantitative analysis of acetylated histone proteins H3 and H4 is shown in Figure 5G. Western blot analysis of acetylated histone H3 and H4 proteins reveals increased protein levels after 72 hours of exposure to oxidants in Figure 5H.
Figure 5.
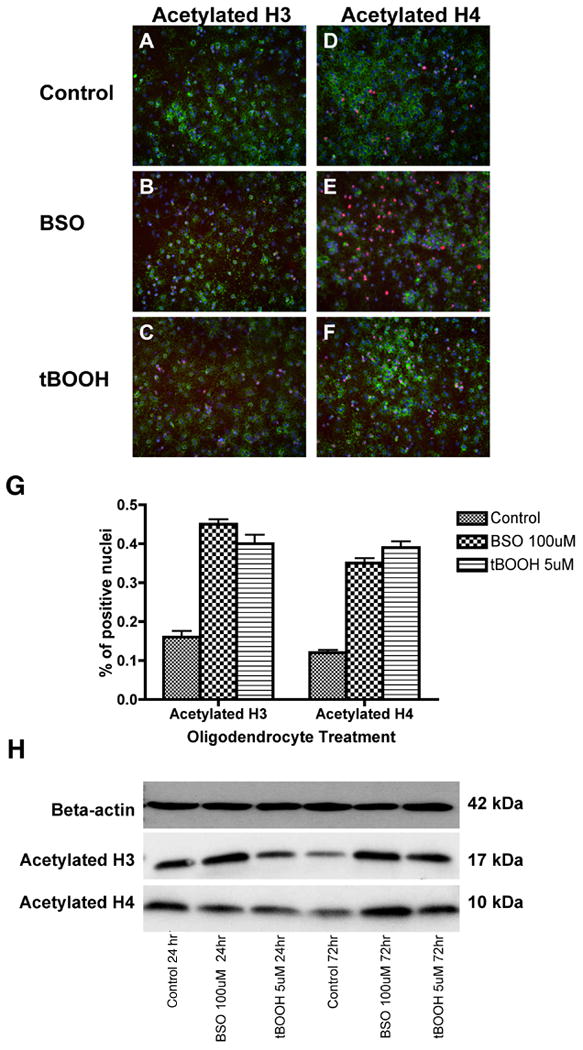
OPCs were stained in vitro with antibodies to acetylated histone H3 (A-C) and acetylated histone H4 (D-F) after 72 hours of exposure to oxidative agents. The density of acetylated histone H3 and H4 was significantly higher in BSO- and tBOOH-exposed cells. Quantitative analysis of acetylated histone H3+ and H4+ nuclei is also shown (G). The proportion of positive cells per 20× field was determined by counting 10 fields per coverslip in three separate experiments following indirect immunofluorescence using antibody to acetylated histone H3 or H4. Data represent the mean plus standard deviation. *P <0.05. Western blotting was performed on OPC lysate after 24 and 72 hours (H). Analysis reveals an increase in acetylated histone H3 and H4 protein after 72 hours of exposure to oxidative stress. Each lane contains 25 μg protein.
Discussion
In the present study, we examined how oxidative stress and reactive oxygen species (ROS) disrupts oligodendrocyte differentiation and leads to loss of mature myelinating oligodendrocytes. We have established an in vitro model of oxidative stress in oligodendrocytes and found arrested maturation of oligodendrocytes without an increase in cell death. In addition, oxidative stress was associated with alterations in expression of genes regulating oligodendrocyte differentiation and changes in global histone acetylation patterns. These results suggest that oxidative stress may directly interfere with the program of oligodendrocyte differentiation leading to the demyelination associated with neonatal brain injury.
Over the past two decades, oligodendrocytes have been the subject of research focusing on how injury to it's cellular lineage might result in periventricular white matter damage, the major form of brain injury and strong predictor of neurologic disability and cerebral palsy in survivors of preterm birth (Volpe, 2001b). Impaired myelination is the hallmark of PWMI, suggesting that oligodendrocytes, the myelinating cell of the CNS, is the major cell lineage damaged by the many clinical conditions associated with brain injury. The reduction of oligodendrocytes associated with PWMI has been shown by many investigators to be associated with pro-inflammatory cytokine expression (Baud et al, 1999; Damman et al, 1997; Kadhim et al, 2001, 2003; Yoon et al, 1997a,b). Other investigators have noted the vulnerability of the oligodendrocyte cell lineage to excitatory glutamate toxicity and oxidative stress (Kavahaugh et al, 2000; Tekkok et al, 2001).
Reactive oxygen species (ROS) production is a well established sequela of ischemia and reperfusion caused by sepsis, hypoxia and impaired cerebral autoregulation (Chan, 2001; Hagberg et al, 2002; Traystman et al, 1991). ROS are unstable molecules with unpaired electrons, capable of initiating cellular oxidative injury. Increased ROS production is linked to oxidation of proteins, DNA and lipids which cause direct cellular and tissue injury (Rahman, 2003). Additionally, when generated close to the cell membrane, ROS cause lipid peroxidation of membrane phospholipids. The premature neonatal brain is at an additional disadvantage because of its high concentration of unsaturated fatty acids (Halliwell, 1992). The peroxidative breakdown of unsaturated fatty acids impairs membrane function and inactivates cell surface receptors and enzymes. Several studies support the concept that oligodendrocytes display maturation-dependent vulnerability to oxidative damage caused by a developmental delay in the expression of antioxidant enzymes (superoxide dismutases-1 and -2, catalase, glutathione peroxidase), glutathione depletion or exogenous ROS (Back et al, 1998; Baud et al, 2004a,b; Fern et al, 2000; Folkerth et al, 2004). This intrinsic vulnerability of OPCs to oxidative stress from free radicals and excitotoxicity induced by sepsis and hypoxic-ischemic injury is considered to be central to the pathogenesis of PWMI in the preterm infant. Reactive oxygen species also play a destructive role in the adult in inflammatory demyelinating disorders such as multiple sclerosis and Guillain-Barre syndrome, possibly by overpowering antioxidant defense systems within the cells. This can cause death of mature oligodendrocytes by apoptosis and subsequent demyelination. Attack by macrophages may also occur (Smith et al, 1999).
Oxidant exposure to induce ROS in cell culture has been used extensively as a model to induce oxidative stress. Both L-buthionine sulfoximine (BSO) and tert-Butyl hydroperoxide (tBOOH) have been used widely in cell culture to study oxidative conditions. BSO is a selective inhibitor of gamma-glutamylcysteine synthetase. This enzyme catalyzes the first and rate-limiting step of glutathione synthesis, and its inhibition decreases cellular glutathione (Meister, 1992). tBOOH is a membrane-permeant oxidant compound that generates tert-butoxyl radicals resulting in lipid peroxidation and depletion of intracellular glutathione. BSO and tBOOH are easily added to cell culture media at concentrations which reliably deplete glutathione but do not affect cell viability (Back et al, 1998; Scotti et al, 2003; Zhao et al, 2005).
Our research supports the idea that oxidative stress decreases oligodendrocyte differentiation towards a mature, myelin-producing cell which was not caused by cell death as confirmed by TUNEL and activated caspase 3 immunocytochemistry. Other studies suggested cell death as the mechanism of decreased numbers of differentiated oligodendrocytes (Follett et al, 2000, 2004; Horiuchi et al, 2006; Pang et al, 2007). We suggest rather that differentiation does not proceed after oxidant exposure and OPCs are arrested in an immature, non-myelinating phenotype, identified by A2B5 positivity and GalC negativity. We did not determine whether oligodendrocyte differentiation would proceed normally if oxidant agents were removed from the cell media or if cell death would increase after 72 hours of exposure to oxidative agents.
To support our findings of decreased maturation, we found that oxidative stress downregulates the expression of genes known to play a major role in promoting oligodendrocyte differentiation at different stages of maturation. We have shown that oxidative stress decreases gene expression of Sonic Hedgehog (Shh), Sox10, histone deacetylase 3 (HDAC30, Olig1 and Olig2. Shh, a member of the hedgehog family of signaling molecules, can direct pluripotent neural stem cells to adopt an oligodendrocyte fate in both the spinal cord and forebrain (Nery et al, 2001; Orentas et al, 1999; Pringle et al, 1996). While Shh is normally thought of as a specification gene, it has been suggested that Shh expression prevents the inhibition of oligodendrogenesis by members of the TGFβ family which can force oligodendrocyte progenitors to commit to the astrocyte lineage (Miller 2002). Sox10, a transcription factor, is restricted in the CNS to myelin-forming oligodendrocytes and their progenitors. Sox10 knock out mice develop progenitor cells, but terminal differentiation to oligodendrocytes never occurs as determined by lack of expression of multiple myelin proteins (PLP, MBP, CNP) by maturing oligodendrocytes (Stolt et al, 2002). HDAC has been shown to be critical for differentiation in the developing brain by repressing genes that inhibit oligodendrocyte differentiation so that maturation and, therefore, CNS myelination can occur (Shen et a, 2005). Olig1 and Olig2 are basic helix-loop-helix transcription factors which are required for the generation of oligodendrocytes (Ligon et al, 2006). In Olig 1/2 double mutant mice, there is a complete failure of oligodendrocyte development in all areas of the brain along with an increase in astrocytogenesis (Zhou et al, 2002).
In addition to downregulation of factors which promote oligodendrocyte differentiation, oxidative stress increased the expression of Inhibitors of Differentiation (Id) Id2 and Id4. Members of the inhibitors of differentiation (ID) family of helix-loop-helix transcriptional inhibitors sequester basic helix-loop-helix factors, such as Olig genes, and act as dominant negative regulators of differentiation (Benezra et al, 1990; Wang et al, 2001). Using immunocytochemistry, we demonstrated a decrease in Olig2 expression upon exposure to oxidative stress. Previous observations in vitro indicate that Id2 and Id4 interact with Olig1 and Olig2 in the cytoplasm after exposure to inhibitors of differentiation (Samanta et al, 2004). Decreased expression of all of these factors contributes to decreased numbers of mature oligodendrocytes.
Oxidative stress decreased the activity of HDAC3 and coincided with prevention of global H3 and H4 histone deacetylation in OPCs. Prior research showed that persistent histone acetylation prevents the appearance of oligodendrocytes and favors the development of neural stem cells into neurons or astrocytes (Shen et al, 2005). It has been recently shown that histone deacetylation, carried out by histone deacetylase (HDAC), is important for developing oligodendrocytes by either repressing neuronal and astrocytic genes or repressing negative regulatory elements in oligodendrocyte gene promoters, thereby allowing oligodendrocyte differentiation to proceed (Marin-Husstege et al, 2002; Liu et al, 2007).
It was recently shown that oxidative stress and reactive oxygen species can alter nuclear chromatin remodeling leading to gene expression of pro-inflammatory mediators and pro-apoptotic and antiproliferative responses (Rahman et al, 1998; Guyton et al, 1996). Further evidence suggests that oxidative stress and pro-inflammatory mediators modify and increase histone acetylation by a mechanism dependent on the activation of the MAP kinase pathway (Miyata et al, 2001; Bohm et al, 1997). Several investigators reported that ROS increases histone acetylase (HAT) activity (Ito et al, 2001; Rahman et al, 2001). Decreased action of histone deacetylase also appears to play a role. Histone deacetylase enzyme activity has been shown to be decreased under conditions of oxidative stress in alveolar epithelial cells (Moodie et al, 2003). Because oligodendrocyte development from neural stem cells is dependent on decreased histone acetylase (HAT) activity and increased histone deacetylase (HDAC) activity, the imbalance towards HAT activity that occurs as a result of oxidative stress may favor the continued development of neurons and astrocytes at the expense of oligodendrocytes. Global histone acetylation is a characteristic of neurogenesis and astrocytogenesis. As suggested by other researchers, the lack of deacetylation likely represents a signal which contributed to the arrest of oligodendrocyte differentiation from the A2B5 precursor stage.
Defining the pathogenesis of PWMI in preterm infants and designing effective prevention and therapeutic strategies requires a more thorough understanding of cellular responses to inflammation, infection and oxidative stress. It is likely that multiple mechanisms contribute to the oligodendrocyte loss found in PWMI. Here we attempted to help elucidate how oxidative stress may lead to the disruption of oligodendrocyte differentiation, which may ultimately contribute to the diffuse hypomyelination found in PWMI. Our findings suggest that future research efforts should be directed towards further understanding the delicate signal transduction pathways that signal the beginning and proceedings of oligodendrocyte differentiation and how oxidative stress may lead to their disruption, with the ultimate goal of the development of antioxidant therapies to prevent or lessen the impact of PWMI.
Acknowledgments
This work was supported by NIH AG20898 (R.S.) and NMSS RG3662 (J.B.G.)
References
- Altman J, Bayer SA. The development of the rat spinal cord. In: Beck F, Hild W, van Limborgh J, Ortmann R, Pauly JE, Schiebler TH, editors. Advances in anatomy, embryology and cell biology. Vol. 85. Berlin: Springer Verlag; 1984. [DOI] [PubMed] [Google Scholar]
- Back SA, Gan XD, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo N, Borenstein N, Levine J, Volpe JJ, Kinney H. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Volpe JJ, Kinney HH. Immunocytochemical characterization of oligodendrocyte development in human cerebral white matter. Soc Neurosci Abstr. 1996;20:1722. [Google Scholar]
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, Federoff HJ, Rose DW, Rosenfeld MG, Brehm P, Mandel G. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart I, Coles HSR, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–47. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Baud O, Emilie D, Pelletier E, Lacaze-Masmontiel T, Zupan V, Fernandez H, Dehan M, Frydman R, Ville Y. Amniotic fluid concentrations of interleukin-1β, interleukin-6 and TNFα in chorioamnionitis before 32 weeks of gestation: histological associations and neonatal outcome. Br J Obstet Gynaecol. 1999;106:72–77. doi: 10.1111/j.1471-0528.1999.tb08088.x. [DOI] [PubMed] [Google Scholar]
- Baud O, Greene A, Li J, Wang H, Volpe JJ, Rosenberg PA. Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J Neurosci. 2004a;24:1531–1540. doi: 10.1523/JNEUROSCI.3989-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud O, Haynes RF, Wang H, Folkerth RD, Li J, Volpe JJ, Rosenberg PA. Developmental up-regulation of McSOD in rat oligodendrocytes confers protection against oxidative injury. European Journal of Neuroscience. 2004b;20:29–40. doi: 10.1111/j.0953-816X.2004.03451.x. [DOI] [PubMed] [Google Scholar]
- Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- Bohm L, Schneeweiss FA, Sharan RN, Feinendegen LE. Influence of histone acetylation on the modification of cytoplasmic and nuclear proteins by ADP-ribosylation in response to free radicals. Biochem Biophys Acta. 1997;1334:149–154. doi: 10.1016/s0304-4165(96)00085-2. [DOI] [PubMed] [Google Scholar]
- Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res. 2000;47:64–72. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Dammann O, Leviton A. Maternal intrauterine infection, cytokines and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8. doi: 10.1203/00006450-199707000-00001. [DOI] [PubMed] [Google Scholar]
- DiSalvo D. The correlation between placental pathology and intraventricular hemorrhage in the preterm infant. The Developmental Epidemiology Network. Pediatr Res. 1998;43:15–19. doi: 10.1203/00006450-199801000-00003. [DOI] [PubMed] [Google Scholar]
- Eisenbarth G, Walsh F, Nirenberg M. Monoclonal antibody to a plasma membrane antigen of neurons. Proc Natl Acad Sci USA. 1979;76:4913–4917. doi: 10.1073/pnas.76.10.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Chang H, Li E, Fan G. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J Neurosci Res. 2005;79:734–746. doi: 10.1002/jnr.20404. [DOI] [PubMed] [Google Scholar]
- Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, Volpe JJ, Jensen FE. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J Neurosci. 2004;24:4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkerth R, Haynes R, Borenstein NS, Belliveau RA, Trachtenberg F, Rosenberg PA, Volpe JJ, Kinney HC. Developmental lag in superoxide dismutases relative to other antioxidant enzymes in premyelinated human telencephalic white matter. J Neuropathol Exp Neurol. 2004;63:990–999. doi: 10.1093/jnen/63.9.990. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Kato T, Kakita H, Yamada Y, Hussein MH, Kato I, Suzuki S, Togari H. Hemodynamics of the cerebral arteries of infants with periventricular leukomalacia. Pediatrics. 2006;117:1–8. doi: 10.1542/peds.2004-1719. [DOI] [PubMed] [Google Scholar]
- Grinspan JB, Stern JL, Pustilnik SM, Pleasure D. Cerebral white matter contains PDGF-responsive precursors to O2A cells. J Neurosci. 1990;10:1861–1873. doi: 10.1523/JNEUROSCI.10-06-01866.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinspan JB, Franceschini B. Platelet-derived growth factor is a survival factor for PSA-NCAM+ oligodendrocyte pre-progenitor cells. J Neurosci Res. 1995;41:540–551. doi: 10.1002/jnr.490410414. [DOI] [PubMed] [Google Scholar]
- Grinspan JB, Coulalglou M, Beesley JS, Carpio DF, Scherer SS. Maturation-dependent apoptotic cell death of oligodendrocytes in myelin-deficient rats. J Neurosci Res. 1998;54:623–634. doi: 10.1002/(SICI)1097-4547(19981201)54:5<623::AID-JNR7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Grinspan JB, Edell E, Carpio DF, Beesley JS, Lavy L, Pleasure D, Golden JA. Stage-specific effects of bone morphogenetic proteins on the oligodendrocyte lineage. J Neurobiol. 2000;43:1–17. [PubMed] [Google Scholar]
- Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. J Biol Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Wennerholm UB, Savman K. Sequelae of chorioamnionitis. Curr Opin Infect Dis. 2002;15:301–306. doi: 10.1097/00001432-200206000-00014. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- Haynes RL, Baud O, Li J, Kinney HC, Volpe JJ, Folkerth DR. Oxidative and nitrative injury in periventricular leukomalacia: a review. Brain Pathol. 2005;15:225–233. doi: 10.1111/j.1750-3639.2005.tb00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Itoh A, Pleasure D, Itoh T. MEK-ERK signaling is involved in interferon-gamma-induced death of oligodendrocyte progenitor cells. J Biol Chem. 2006;281:20095–20106. doi: 10.1074/jbc.M603179200. [DOI] [PubMed] [Google Scholar]
- Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. PNAS. 2004;101:16659–64. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inder TE, Volpe JJ. Mechanisms of perinatal brain injury. Semin Neonatol. 2000;5:3–16. doi: 10.1053/siny.1999.0112. [DOI] [PubMed] [Google Scholar]
- Ito K, Lim G, Caramori G, Chung KF, Barnes PJ, Adcock IM. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression and inhibits glucocorticoid actions in alveolar macrophages. FASEB J. 2001;15:1110–1112. [PubMed] [Google Scholar]
- Kadhim H, Khalifa M, Deltenre P, Casimir G, Sebire G. Molecular mechanisms of cell death in periventricular leukomalacia. Neurology. 2006;67:293–299. doi: 10.1212/01.wnl.0000224754.63593.c4. [DOI] [PubMed] [Google Scholar]
- Kadhim H, Tabarki B, DePerez C, Sebire G. Cytokine immunoreactivity in cortical and subcortical neurons in perinventricular leukomalacia: are cytokines implicated in neuronal dysfunction in cerebral palsy? Acta Neuropathol. 2003;105:216–298. doi: 10.1007/s00401-002-0633-6. [DOI] [PubMed] [Google Scholar]
- Kadhim H, Tabarki B, Verellen G, DePerez C, Rona AM, Sebire G. Inflammatory cytokines in the pathogenesis of periventricular leukomalacia. Neurology. 2001;56:1278–1284. doi: 10.1212/wnl.56.10.1278. [DOI] [PubMed] [Google Scholar]
- Kavahaugh B, Beesley J, Itoh T, Itoh A, Grinspan J, Pleasure D. Neurotrophin-3 (NT-3) diminishes susceptibility of the oligodendroglial lineage to AMPA glutamate receptor-mediated excitotoxicity. J Neurosci Res. 2000;60:725–732. doi: 10.1002/1097-4547(20000615)60:6<725::AID-JNR4>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Kinney H, Back S. Human oligodendroglial development: relationship to periventricular leukomalacia. Semin Pediatr Neurol. 1998;5:180–189. doi: 10.1016/s1071-9091(98)80033-8. [DOI] [PubMed] [Google Scholar]
- Levison SW, Rothstein RP, Romanko MJ, Snyder MJ, Meyers RL, Vannucci SJ. Hypoxia/ischemia depletes the rat perinatal subventricular zone of oligodendrocyte progenitors and neural stem cells. Dev Neurosci. 2001;23:234–247. doi: 10.1159/000046149. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Fancy SPJ, Franklin RJM, Rowitch DH. Olig gene functions in CNS development and disease. Glia. 2006;54:1–10. doi: 10.1002/glia.20273. [DOI] [PubMed] [Google Scholar]
- Liu A, Han YR, Li J, Sun D, Ouyang M, Plummer MR, Casaccia-Bonnefil P. The glial or neuronal fate choice of oligodendrocyte progenitors is modulated by their ability to acquire an epigenetic memory. J Neurosci. 2007;27:7339–7343. doi: 10.1523/JNEUROSCI.1226-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabie PC, Mehler MF, Marmur R, Papavasiliou A, Song Q, Kessler JA. Bone morphogenic proteins induce astroglial differentiation of oligodendroglial-astroglial progenitor cells. J Neurosci. 1997;17:4112–4120. doi: 10.1523/JNEUROSCI.17-11-04112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Husstege M, Muggironi M, Liu A, Casaccia-Bonnefil P. Histone deacetylase activity is necessary for oligodendrocyte lineage progression. J Neurosci. 2002;22:10333–10345. doi: 10.1523/JNEUROSCI.22-23-10333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage: III. Gray matter lesions of the neocortex. J Neuropathol Exp Neurol. 1999;58:407–429. doi: 10.1097/00005072-199905000-00001. [DOI] [PubMed] [Google Scholar]
- Martin JA, Hamilton BE, Menacker F, Sutton PD, Mathews TJ. Preliminary births for 2004: Infant and maternal health Health E-stats. Hyattsville, MD: National Center for Health Statistics; 2005. [Google Scholar]
- Mehler MF, Mabie PC, Zhu G, Gokhan S, Kessler JA. Developmental changes in progenitor cell responsiveness to bone morphogenetic proteins differentially modulate progressive CNS lineage fate. Dev Neurosci. 2000;22:74–85. doi: 10.1159/000017429. [DOI] [PubMed] [Google Scholar]
- Meister A. Depletion of glutathione in normal and malignant human cells in vivo by L-buthionine sulfoximine: possible interaction with ascorbate. JNatl Cancer Inst. 1992;84:1601–1602. doi: 10.1093/jnci/84.20.1601-a. [DOI] [PubMed] [Google Scholar]
- Miller RH. Regulation of oligodendrocyte development in the vertebrate CNS. Progress in Neurobiology. 2002;67:451–467. doi: 10.1016/s0301-0082(02)00058-8. [DOI] [PubMed] [Google Scholar]
- Miyata Y, Towatari M, Maeda T, Ozawa Y, Saito H. Histone acetylation induces granulocyte colony-stimulating factor in a MAP kinase-dependent manner. Biochem Biophys Res Commun. 2001;283:655–600. doi: 10.1006/bbrc.2001.4840. [DOI] [PubMed] [Google Scholar]
- Moodie F, Anderson C, Marwick JA, Szulakowski P, Kilty I, MacNee W, Rahman I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-κB activation and proinflammatory cytokine release in alveolar epithelial cells. Am J Respir Crit Care Med. 2003;16:A160. doi: 10.1096/fj.04-1506fje. abstract. [DOI] [PubMed] [Google Scholar]
- Nery S, Wichterle H, Fishell G. Sonic hedgehog contributes to oligodendrocyte specification in the mammalian forebrain. Development. 2001;128:527–540. doi: 10.1242/dev.128.4.527. [DOI] [PubMed] [Google Scholar]
- Orentas DM, Hayes JE, Dyer KL, Miller RH. Sonic hedgehog signaling is required during the appearance of spinal cord oligodendrocyte precursors. Development. 1999;126:761–772. doi: 10.1242/dev.126.11.2419. [DOI] [PubMed] [Google Scholar]
- Pang Y, Zheng B, Fan LW, Rhodes PG, Cai Z. IGF-1 protects oligodendrocyte progenitors against TNFalpha-induced damage by activation of PI3K/Akt and interruption of the mitochondrial apoptotic pathway. Glia. 2007;55:1099–1107. doi: 10.1002/glia.20530. [DOI] [PubMed] [Google Scholar]
- Pringle NP, Yu WP, Guthrie S, Roelink H, Lumsden A, Peterson AC, Richardson WD. Determination of neuroepithelial cell fate: induction of the oligodendrocyte lineage by ventral midline cells and sonic hedgehog. Dev Biol. 1996;177:30–42. doi: 10.1006/dbio.1996.0142. [DOI] [PubMed] [Google Scholar]
- Rahman I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J Biochem Mol Biol. 2003;36:95–109. doi: 10.5483/bmbrep.2003.36.1.095. [DOI] [PubMed] [Google Scholar]
- Rahman I, Gilmour PS, Jimenez LA, MacNee W. Oxidative stress induces histone acetylation in alveolar epithelial cells. Am J Respir Crit Care Med. 2001;163:A549. abstract. [Google Scholar]
- Rahman I, MacNee W. Role of transcription factors in inflammatory lung diseases. Thorax. 1998;53:601–612. doi: 10.1136/thx.53.7.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranscht B, Clapshaw PA, Price J, Noble M, Seifert W. Development of oligodendrocytes and Schwann cells studied with a monoclonal antibody against galactocerebroside. Proc Natl Acad Sci USA. 1982;79:2709–2713. doi: 10.1073/pnas.79.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S, Miller RH. Environmental enhancement of growth factor-mediated oligodendrocyte precursor proliferation. Mol Cell Neurosci. 1996;8:38–52. doi: 10.1006/mcne.1996.0042. [DOI] [PubMed] [Google Scholar]
- Roopra A, Sharling L, Wood IC, Briggs T, Bachfischer U, Paquette AJ, Buckley NJ. Transcriptional repression by neuron-restrictive silencer factor is mediated via the Sin3-histone deacetylase complex. Mol Cell Biol. 2000;20:2147–2157. doi: 10.1128/mcb.20.6.2147-2157.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta J, Kessler JA. Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development. 2004;131:4131–4142. doi: 10.1242/dev.01273. [DOI] [PubMed] [Google Scholar]
- Scotti C, Iamele L, Alessandrini A, Vannini V, Cazzalini O, Lazze M, Melli R, Savio M, Pizzala R, Stivala LA, Biglieri S, Tomasi A, Bianchi L. Lack of molecular relationship between lipid peroxidation and mitochondrial DNA single strand breaks in isolated rat hepatocytes and mitochondria. Mitochondrion. 2003;2:361–373. doi: 10.1016/S1567-7249(03)00004-7. [DOI] [PubMed] [Google Scholar]
- See J, Mamontov P, Ahn K, Wine-Lee L, Crenshaw EB, 3rd, Grinspan JB. BMP signaling mutant mice exhibit glial cell maturation defects. Mol Cell Neurosci. 2007;35:171–182. doi: 10.1016/j.mcn.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. JCB. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Kapoor R, Felts PA. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9:69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer I, Schachner M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces. An immunocytochemical study in the central nervous system. Dev Biol. 1981;83:311–327. doi: 10.1016/0012-1606(81)90477-2. [DOI] [PubMed] [Google Scholar]
- Song MR, Ghosh A. FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat Neurosci. 2004;7:229–235. doi: 10.1038/nn1192. [DOI] [PubMed] [Google Scholar]
- Stolt CC, Rehberg S, Ader M, Lommes P, Riethmacher D, Schachner M, Bartsch U, Wegner M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes & Dev. 2002;16:165–170. doi: 10.1101/gad.215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkok S, Goldberg M. AMPA/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci. 2001;15:4237–4248. doi: 10.1523/JNEUROSCI.21-12-04237.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traysman RJ, Kirsch JR, Koehler RC. Oxygen radical mechanisms of brain injury following ischemia and reperfusion. Am J Physiol. 1991;71:1185–1195. doi: 10.1152/jappl.1991.71.4.1185. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001a;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Neurology of the newborn. Philadelphia: W.B. Saunders; 2001b. 2001. [Google Scholar]
- Wang S, Sdrulla A, Johnson JE, Yokota Y, Barres BA. A role for the helix-loop-helix protein Id2 in the control of oligodendrocyte development. Neuron. 2001;29:603–614. doi: 10.1016/s0896-6273(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Wilson-Costello D, Friedman H, Minich N, Fanaroff A, Hack M. Improved survival rates with increased neurodevelopmental disability for extremely low birth weight infants in the 1990s. Pediatrics. 2005;115:997–1003. doi: 10.1542/peds.2004-0221. [DOI] [PubMed] [Google Scholar]
- Yesilirmak DC, Kumral A, Baskin J, Ergur BU, Aykan S, Genc S, Genc K, Yilmaz O, Tugyan K, Giray O, Duman N, Ozkan H. Activated protein C reduces endotoxin-induced white matter injury in the developing rat brain. Brain Res. 2007;1164:14–23. doi: 10.1016/j.brainres.2007.04.083. [DOI] [PubMed] [Google Scholar]
- Yoon BH, Jun JK, Romero R, Park KH, Gomez R, Choi JH, Kim IO. Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1β, and tumor necrosis factor-α), neonatal brain white matter lesions, and cerebral palsy. Am J Obstet Gynecol. 1997a;177:19–26. doi: 10.1016/s0002-9378(97)70432-0. [DOI] [PubMed] [Google Scholar]
- Yoon BH, Kim C, Romero R, Jun JK, Park KH, Choi S, Chi J. Experimentally induced intrauterine infection causes fetal brain white matter lesions in rabbits. Am J Obstet Gynecol. 1997b;177:797–802. doi: 10.1016/s0002-9378(97)70271-0. [DOI] [PubMed] [Google Scholar]
- Zhao K, Luo G, Giannelli S, Szeto HH. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochemical Pharmacology. 2005;70:1796–1806. doi: 10.1016/j.bcp.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/s0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]