

Abstract
The cross-metathesis of terminal olefins using a novel ruthenium catalyst results in excellent selectivity for the Z-olefin homodimer. The reaction was found to tolerate a large number of functional groups, solvents, and temperatures while maintaining excellent Z-selectivity, even at high reaction conversions.
Olefin metathesis using a variety of transition metals has gained widespread popularity as a robust method for the formation of carbon-carbon bonds.1 Ruthenium-based catalysts, in particular, have been used in a wide variety of applications including biochemistry,2 materials chemistry,3 and synthetic organic chemistry.4 However, despite its widespread appeal, olefin metathesis is an equilibrium reaction and therefore, metathesis applications which require the formation of kinetic products are generally difficult if not altogether prohibited.1,5 Nevertheless, an increasingly sophisticated understanding of catalyst selectivity (both with ruthenium6 and Group VI metals7) has permitted the development of new catalysts which are capable of selectively forming kinetic products. Despite this progress, the cross-metathesis of terminal olefins to selectively form the Z-olefin product remained an elusive goal until the recent work of Hoveyda, Schrock, and coworkers.8 The Hoveyda-Schrock systems showed excellent selectivities and good turnover numbers (TONs). However, they concluded that obtaining similar selectivities using ruthenium-based catalysts would be challenging. Herein, we show that the cross-metathesis homocoupling of terminal olefins to selectively form Z-olefins is not only possible with ruthenium, but a viable alternative to catalysts based on Group VI metals.
We recently reported on the synthesis of a C-H activated ruthenium metathesis catalyst wherein the N-heterocyclic carbene (NHC) is chelated to the metal center through a Ru-C bond (Figure 1).9 Surprisingly, this catalyst represented the first example of a metathesis active complex which had undergone C-H activation.1e,10 Furthermore, despite 1's relatively poor activity in ring-opening metathesis polymerization (ROMP) and ring closing metathesis (RCM) reactions, it displayed remarkable Z-selectivity for the cross-metathesis product of allylbenzene (2) and cis-1,4-diacetoxy-2-butene (3) (Scheme 1). With this result in hand, we reasoned that 1 would excel in a catalytically less complex reaction, such as the homocoupling of terminal olefins (Scheme 2).
Figure 1.
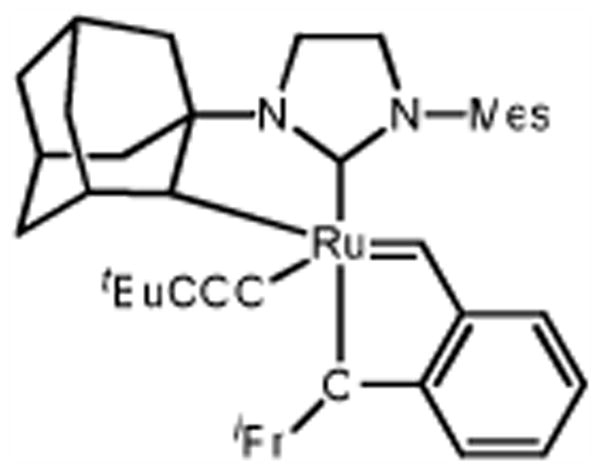
Previously Reported C-H Activated Catalyst 1.
Scheme 1.
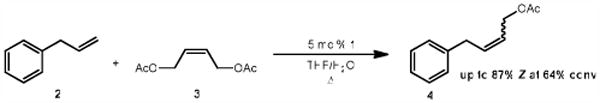
Previously reported Z-selectivity of 1.
Scheme 2.
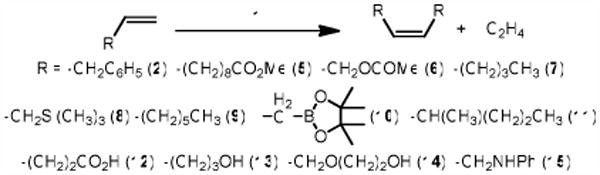
Homocoupling of Terminal Olefins with 1.
Due to the relatively large adamantane group on 1 and the associative initiation mechanism of complexes of this type, 1 requires fairly high temperatures in order to initiate (ca. 70 °C).11 Unfortunately, cross-metathesis reactions performed at this temperature and low olefin concentration gave relatively low conversion and showed significant amounts of catalyst decomposition. We suspected that the poor performance of 1 under these conditions was a result of the ethylene generated as a byproduct of the reaction, and indeed, exposure of 1 to an atmosphere of ethylene at RT resulted in complete decomposition within minutes.
While the decomposition of 1 in the presence of ethylene was disappointing, it is not uncommon among metathesis catalysts and can be mitigated by efficient removal of the gas from solution.12 Therefore, a series of cross-metathesis reactions were run under static vacuum, and under these conditions, 1 performed admirably (Table 1). For instance, 1 was stable at 70 °C in both THF and MeCN as long as oxygen was rigorously excluded, and was able to obtain high conversions and Z-selectivity for a variety of terminal olefin substrates. Some substrates showed a slight decrease in selectivity with increasing conversion, a result which is most likely caused by decomposition products of 1.14
Table 1.
Cross-metathesis of terminal olefins with 1 at 70 °C under static vacuum.a
| Substrate | Solvent | Time [h] | Conv.c [%] | Zc [%] |
|---|---|---|---|---|
| allyl benzene (2) | THF | 6 (10) | >95 (>95) | 83 (67) |
| methyl undecenoate (5)b | THF | 4 (6) | 78 (93) | 87 (85) |
| allyl acetate (6) | THF | 3 (6) | 53 (60) | 89 (83) |
| 1-hexene (7)b | THF | 6 (7.5) | 83 (87) | 80 (80) |
| allyl trimethylsilane (8) | THF | 6 (10) | 63 (72) | >95 (>95) |
| 1-octene (9) | THF | 3 (6) | 83 (97) | 80 (68) |
| allyl pinacol borane (10) | THF | 6 | 10 | >95 |
| 3-methyl-1-hexene (11) | THF | 12 | 0 | 0 |
| allyl benzene (2) | MeCN | 2.5 (21) | 12 (15) | >95 (>95) |
| methyl undecenoate (5) | MeCN | 2.5 (21) | 7 (11) | >95 (70) |
2 mol% catalyst in solvent (0.6 M in substrate) at 70 °C under static vacuum.
4 mol% catalyst.
Measured by 1H NMR Spectroscopy.
In contrast to the Group VI metal systems, olefin migration instead of metathesis was observed in some substrate (10, see Figure S1).13 Attempts to prevent olefin migration via the use of additives such as benzoquinone or mild acid met only with catalyst decomposition.14 This type of reactivity, although usually undesirable, can be valuable in certain situations.15 Regardless, olefin migration can be eliminated via careful optimization of reaction conditions (vide infra). Finally, substrates with even a small amount of substitution (11) were disappointingly resistant to homodimerization, even at temperatures exceeding 100 °C.
Although 1 is clearly functional at high temperature, the presence of deleterious side reactions encouraged us to search for conditions in which 1 would initiate at lower temperatures. Extensive optimization revealed that 1 could affect the homodimerization of terminal olefins at 35 °C with high olefin concentration (ca. 3 M). This result is not surprising, considering that the initiation of 1 should depend on olefin concentration. Nevertheless, we did not anticipate that the activity and selectivity of 1 would be superior at 35 °C. Furthermore, reactions performed at lower temperature and higher concentration had the additional advantage of not requiring any special technique to remove ethylene.16
For most substrates, reactions with 1 at 35 °C showed similar selectivity to reactions performed at 70 °C, but improved activity (Table 2). Isolated yields of the homodimerization products were also good. Gratifyingly, in the case of 10, no detectable amount of olefin migration was observed and excellent Z-selectivity was maintained up to very high conversion. Emboldened by this success, we attempted to dimerize several more advanced substrates (11-14). Unfortunately, in the case of hindered (11) or acidic substrates (12), no activity was observed. On the other hand, 1 was able to dimerize alcoholic substrates (13, 14) with excellent conversion and good selectivity. This latter result is particularly important since it is the first example of Z-selective cross-metathesis with alcohol substrates
Table 2.
Cross-metathesis of terminal olefins with 1 at 35 °C.a
| Substrate | Time [h] | Conv.b [%] | Zb [%] | Yieldc [%] |
|---|---|---|---|---|
| allyl benzene (2) | 1 | >95 | 92 | 81 |
| methyl undecenoate (5) | 5.5 | >95 | 73 | >95 |
| allyl acetate (6) | 4 | >95 | 89 | 62 |
| 1-hexene (7)d | 3 | 73 | 69 | 21 |
| allyl trimethylsilane (8) | 3 | >95 | >95 | 54 |
| 1-octene (9) | 4 | >95 | 83 | 79 |
| allyl pinacol borane (10) | 4 | >95 | >95 | 74 |
| 3-methyl-1-hexene (11) | 24 | 0 | - | - |
| pentenoic acid (12) | 24 | 0 | - | - |
| 4-penten-1-ol (13) | 1 | >95 | 72 | 72 |
| 2-(allyloxy)ethanol (14) | 1 | 87 | 66 | 73 |
| N-allylaniline (15) | 2 | 70 | 71 | 67 |
2 mol% catalyst in THF (3.33 M in substrate) at 35 °C.
Measured by 1H NMR Spectroscopy.
Isolated yield.
Run in sealed container.
Given that 1 is not only stable to water and other protic media, but shows increased activity, we deemed it appropriate to examine a wide variety of different solvents for the homodimerization of 5 at RT (Table 3).9 Several polar and nonpolar solvents were tested and the majority were conducive to the transformation. Coordinating solvents (e.g. MeCN) resulted in slower reactions but were able to achieve roughly equivalent TONs to reactions run in noncoordinating solvents. Protic solvents such as MeOH and EtOH yielded highly Z-olefin enriched product while hexafluoroisopropanol resulted in immediate catalyst decomposition.17 The fact that high Z-selectivity is maintained in protic solvents further demonstrates the functional group compatibility of 1. Nevertheless, mildly acidic substrates and solvents appear to result in catalyst decomposition.
Table 3.
Solvent screen for cross-metathesis of 5 with 1 at RT.a
| Substrate | Solvent | Time [h] | Conv.b [%] | Zb [%] |
|---|---|---|---|---|
| MeCN | 3 (28) | 19 (76) | 94 (91) | |
| MeOH | 3 (28) | 49 (87) | 88 (75) | |
| EtOH | 3 (28) | 50 (86) | 89 (76) | |
| C6H6 | 3 (21) | 13 (77) | >95 (84) | |
| methyl undecenoate (5) | Et2O | 3 (7) | 50 (85) | 93 (73) |
| DMF | 3 (21) | 44 (77) | 92 (87) | |
| CH2Cl2 | 3 (21) | 35 (81) | 93 (85) | |
| (CF3)2CHOH | 3 (28) | 0 (0) | - | |
| diglyme | 3 (28) | 31 (81) | 95 (80) |
2 mol% catalyst in solvent (2.25 M in substrate) at 25 °C.
Measured by 1H NMR Spectroscopy.
In conclusion, we have demonstrated the first example of Z-selective homodimerization of terminal olefins using a ruthenium-based catalyst. Optimization of reaction conditions revealed that 1 was effective for a multitude of substrates at different temperatures and in a variety of solvents. The selectivity and activity (TONs from 20-50) of 1 were comparable to previously reported molybdenum and tungsten catalysts. Notably, 1 was able to dimerize several challenging substrates, including alcohols, with excellent conversion and good selectivity for the Z-olefin. However, despite the recent success of ruthenium and Group VI systems, new catalysts, which undergo more turnovers and function under practical experimental conditions, are clearly needed to tackle more advanced olefin substrates and metathesis reactions.
Supplementary Material
Acknowledgments
This work was financially supported by the NIH (NIH 5R01GM031332-27), the NSF (CHE-1048404), Mitsui Chemicals, Inc. (K.E.), and the NDSEG (fellowship to B.K.K.). Instrumentation facilities on which this work was carried out were supported by NIH RR027690. Materia, Inc. is thanked for its donation of metathesis catalysts.
Footnotes
Supporting Information: Supporting Information containing experimental details and NMR spectra is available free of charge via the internet at http://pubs.acs.org
References
- 1.(a) Fürstner A. Angew Chem Int Ed. 2000;39:3013. [Google Scholar]; (b) Trnka TM, Grubbs RH. Acc Chem Res. 2001;34:18. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]; (c) Schrock RR. Chem Rev. 2002;102:145. doi: 10.1021/cr0103726. [DOI] [PubMed] [Google Scholar]; (d) Schrock RR, Hoveyda AH. Angew Chem Int Ed. 2003;42:4592. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; (e) Vougioukalakis G, Grubbs RH. Chem Rev. 2009;110:1746. doi: 10.1021/cr9002424. [DOI] [PubMed] [Google Scholar]; (f) Samojłowicz C, Bieniek M, Grela K. Chem Rev. 2009;109:3708. doi: 10.1021/cr800524f. [DOI] [PubMed] [Google Scholar]
- 2.(a) Lin YA, Chalker JM, Davis BG. J Am Chem Soc. 2010;132:16805. doi: 10.1021/ja104994d. [DOI] [PubMed] [Google Scholar]; (b) Matson JB, Grubbs RH. J Am Chem Soc. 2008;130:6731. doi: 10.1021/ja802010d. [DOI] [PubMed] [Google Scholar]
- 3.(a) Leitgeb A, Wappel J, Slugovc C. Polymer. 2010;51:2927. [Google Scholar]; (b) Liu X, Basu A. J Organomet Chem. 2006;691:5148. [Google Scholar]; (c) Xia Y, Verduzco R, Grubbs RH, Kornfield JA. J Am Chem Soc. 2008;130:1735. doi: 10.1021/ja077192j. [DOI] [PubMed] [Google Scholar]
- 4.For a discussion on the use of metathesis catalysts in natural product synthesis see: Cossy J, Arseniyadis S, Meyer C. Metathesis in Natural Product Synthesis : Strategies, Substrates, and Catalysts. 1st. Wiley-VCH; Weinheim, Germany: 2010.
- 5.Grubbs RH. Handbook of Metathesis. Wiley-VCH; Weinheim: 2003. [Google Scholar]
- 6.(a) Stewart IC, Keitz BK, Kuhn KM, Thomas RM, Grubbs RH. J Am Chem Soc. 2010;132:8534. doi: 10.1021/ja1029045. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) van der Eide EF, Piers WE. Nature Chemistry. 2010;2:571. doi: 10.1038/nchem.653. [DOI] [PubMed] [Google Scholar]
- 7.(a) Schrock RR, Hoveyda AH. Angew Chem Int Ed. 2003;42:4592. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; (b) Cortez GA, Baxter CA, Schrock RR, Hoveyda AH. Org Lett. 2007;9:2871. doi: 10.1021/ol071008h. [DOI] [PubMed] [Google Scholar]; (c) Meek SJ, Malcolmson SJ, Li B, Schrock RR, Hoveyda AH. J Am Chem Soc. 2009;131:16407. doi: 10.1021/ja907805f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Jiang AJ, Zhao Y, Schrock RR, Hoveyda AH. J Am Chem Soc. 2009;131:16630. doi: 10.1021/ja908098t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Marinescu SC, Schrock RR, Müller P, Takase MK, Hoveyda AH. Organometallics. 2011;30:1780. doi: 10.1021/om200150c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Meek SJ, O'Brien RV, Llaveria J, Schrock RR, Hoveyda AH. Nature. 2011;471:461. doi: 10.1038/nature09957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Endo K, Grubbs RH. J Am Chem Soc. 2011 doi: 10.1021/ja202818v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Trnka TM, Morgan JP, Sanford MS, Wilhelm TE, Scholl M, Choi TL, Ding S, Day MW, Grubbs RH. J Am Chem Soc. 2003;125:2546. doi: 10.1021/ja021146w. [DOI] [PubMed] [Google Scholar]; (b) Leitao EM, Dubberley SR, Piers WE, Wu Q, McDonald R. Chem Eur J. 2008;14:11565. doi: 10.1002/chem.200801584. [DOI] [PubMed] [Google Scholar]
- 11.(a) Hejl A. PhD Dissertation. California Institute of Technology; 2007. [Google Scholar]; (b) Vorfalt T, Wannowius KJ, Plenio H. Agnew Chem Int Ed. 2010;1:5533. doi: 10.1002/anie.201000581. [DOI] [PubMed] [Google Scholar]
- 12.(a) Hong SH, Wenzel AG, Salguero TT, Day MW, Grubbs RH. J Am Chem Soc. 2007;129:7961. doi: 10.1021/ja0713577. [DOI] [PubMed] [Google Scholar]; (b) Schrock RR, Jiang AJ, Marinescu SC, Simpson JH, Müller P. Organometallics. 2010;29:6816. [Google Scholar]
- 13.A small amount (ca. 15%) of migration was also observed for 8 although in this case, it did not appear to inhibit the cross-metathesis reaction.
- 14.Hong SH, Sanders DP, Lee CW, Grubbs RH. J Am Chem Soc. 2005;127:17160. doi: 10.1021/ja052939w. [DOI] [PubMed] [Google Scholar]
- 15.(a) Schmidt B. Eur J Org Chem. 2003;34:816–819. [Google Scholar]; (b) Alcaide B, Almendros P, Luna A. Chem Rev. 2009;109:3817. doi: 10.1021/cr9001512. [DOI] [PubMed] [Google Scholar]; (c) Donohoe TJ, O'Riordan TJC, Rosa CP. Angew Chem Int Ed. 2009;48:1014. doi: 10.1002/anie.200804617. [DOI] [PubMed] [Google Scholar]; (d) Gauthier D, Lindhardt AT, Olsen EPK, Overgaard J, Skrydstrup T. J Am Chem Soc. 2010;132:7998. doi: 10.1021/ja9108424. [DOI] [PubMed] [Google Scholar]
- 16.A vent to an inert atmosphere was sufficient. See the Supporting Information.
- 17.1 was only sparingly soluble in MeOH.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.