

Abstract
The factor inhibiting HIF (FIH) is a proximate oxygen sensor for human cells, hydroxylating Asn803 within the α subunit of the hypoxia inducible factor (HIF). FIH is an α-ketoglutatrate (αKG) dependent, non-heme Fe(II) dioxygenase, in which Fe(II) is coordinated by a (His2Asp) facial triad, αKG, and H2O. Hydrogen bonding between the facial triad, the HIF-Asn803 sidechain, and various second-sphere residues suggests a functional role for the second coordination sphere in tuning the chemistry of the Fe(II) center. Point mutants of FIH were prepared to test the functional role of the αKG-centered (Asn205, Asn294) or HIF-Asn803 centered (Arg238, Gln239) second-sphere residues. The second sphere was tested for local effects on priming Fe(II) to react with O2, oxidative decarboxylation, and substrate positioning. Steady-sate kinetics were used to test for overall catalytic effects, autohydroxylation rates were used to test for priming and positioning, and electronic spectroscopy was used to assess the primary coordination sphere and the electrophilicity of αKG. Asn205→Ala and Asn294→Ala exhibited diminished rates of steady-state turnover, while minimally affecting autohydroxylation, consistent with impaired oxidative decarboxylation. Blue shifted MLCT transitions for (Fe+αKG)FIH indicated that these point mutations destabilized the π* orbitals of αKG, further supporting a slowed rate of oxidative decarboxylation. The Arg238→Met mutant exhibited steady-state rates too low to measure and diminished product yields, suggesting impaired substrate positioning or priming; Arg238→Met was capable of O2-activation for the autohydroxylation reaction. The Gln239→Asn mutant exhibited significantly slowed steady-state kinetics and diminished product yields, suggesting impaired substrate positioning or priming. As HIF binding to Gln239→Asn stimulated autohydroxylation, it is more likely that this point mutant simply mis-positions the HIF-Asn803 sidechain. The present work combines kinetics and spectroscopy to show that these second sphere hydrogen bonds play roles in promoting oxidative decarboxylation, priming Fe(II) to bind O2, and positioning HIF-Asn803.
A small number of Fe(II), α-ketoglutarate (αKG)-dependent dioxygenases directly control cellular oxygen sensing in humans.(1–3) These enzymes hydroxylate specific residues within the α-subunit of the hypoxia inducible factor (HIF), thereby affecting the transcriptional level of hundreds of genes.(4) The best characterized of these enzymes is ‘factor inhibiting HIF-1’ (FIH), which hydroxylates the β-carbon of HIF-Asn803;(5) HIF-Asn803 lies within the C-terminal transactivation domain (CTAD) of HIFα. Hydroxylating HIF-Asn803 turns off HIF-dependent gene expression and is a key step in down-regulating angiogenesis. Understanding the chemical basis of how FIH reacts with HIFα and O2 is crucial to understanding tissue O2 homeostasis, and holds potential for treating disease states such as cancer or stroke.(6–8)
According to the consensus chemical mechanism (Scheme 1), O2 binds to (FeII+αKG)FIH only after HIFα binds.(9, 10) In this mechanism, the active site Fe(II) of FIH is proposed to change its coordination geometry upon binding HIFα, thereby creating a binding site for O2. A close correlation between pO2 and HIF hydroxylation would then result, as the rate would be proportional to pO2. X-ray crystallography of FIH in various substrate-bound states revealed a structural linkage between HIF-binding and O2 activation through changes in multiple hydrogen-bonding interactions.(11–13) Controlling oxygenation chemistry through varied hydrogen bonding to atoms coordinated to the metal has been found in other enzymes and models,(14–16) suggesting that the structural changes in FIH might also have functional significance.
Scheme 1.

Consensus chemical mechanism
The chemistry within the FIH active site occurs at the Fe(II) cofactor, making the extended coordination environment of this metal the focus of our investigation. FIH provides a primary coordination sphere of His199, Asp201, and His279, forming a facial triad,(17) with other coordination sites occupied by H2O or αKG (Figure 1). The secondary coordination sphere is comprised of those residues that form hydrogen bonds to the ligands. Spectroscopic and structural data from FIH and related enzymes suggest that coordination changes at Fe are crucial to turnover.(9, 18, 19) Based on evidence from related enzymes, these key coordination geometries are proposed to be a six-coordinate (6C) center for the (FeII+αKG)FIH, and a five-coordinate (5C) center for (FeII+αKG+CTAD)FIH.
Figure 1.
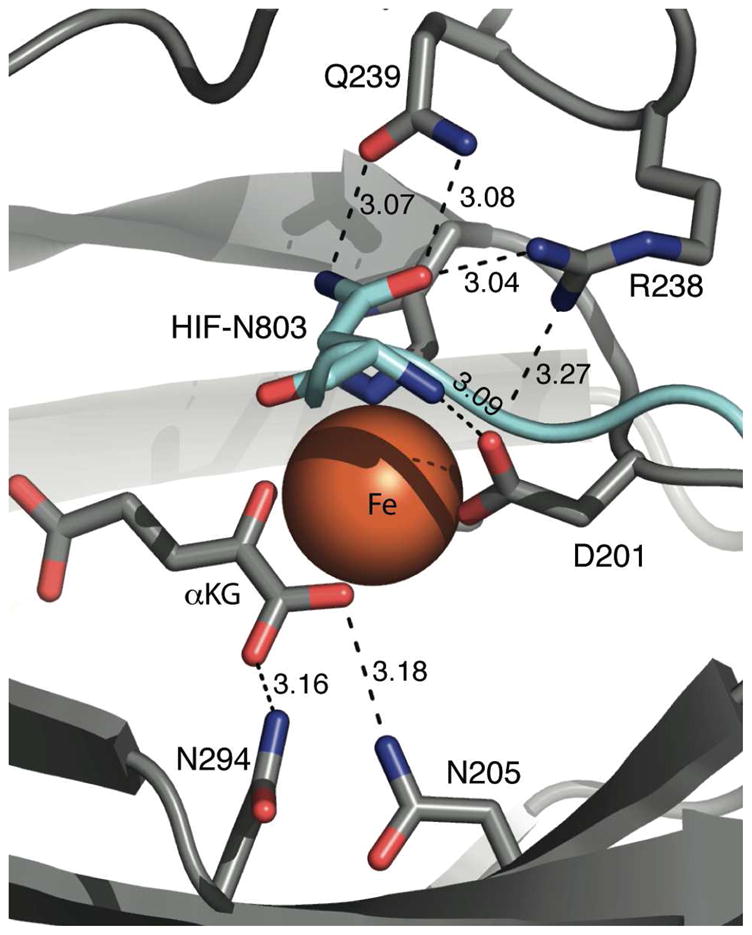
Hydrogen bonding network in the active site of wild type FIH (PDB 1H2K). FIH (gray) and CTAD (cyan) shown as strands; hydrogen bond distances (Å), FIH residues, and HIF-Asn803 are noted.
Hydrogen bonds between surrounding residues, substrates, and iron ligands may comprise a functionally significant second-coordination sphere for FIH, both for priming as well as for oxidative decarboxylation. X-ray crystal structures of FIH imply that the single H2O-ligand is lost from the 6C (FeII+αKG)FIH upon binding CTAD, which would prime the Fe(II) to react with O2.(11–13) Concomitant alterations in nearby hydrogen bonds between Arg238, Asp201, and the H2O ligand suggest that these second-sphere interactions may be necessary for priming. We prepared and characterized the isosteric mutant Arg238→Met in order to determine how loss of this hydrogen bond linkage affected priming of the Fe(II) to react with O2.
Decarboxylating α-ketoacids generally requires stabilization of negative charge within the transition state.(18–20) Notably, a Lys or Arg is positioned as a hydrogen bond donor to the C1-carboxylate of αKG in most structures of the αKG dioxygenase superfamily.(9) In the case of FIH, Asn205 and Asn294 appear to fulfill this role via hydrogen-bond donation to the C-1 carboxylate. We prepared and characterized the single-point Asn → Ala mutants to test how loss of each hydrogen bond affected O2-activation in normal turnover as well as autohydroxylation.
Subsequent to O2-activation, CTAD hydroxylation likely proceeds via a hydrogen transfer/rebound steps involving a putative [FeO]2+ oxidant observed in related enzymes.(21, 22) Precise substrate positioning is necessary to ensure that the putative [FeO]2+ oxidant attacks the proper C-H bond. The side chain of the HIFα-Asn803 residue is located directly above the open coordination site of the Fe(II) in FIH, and forms a hydrogen bond pair to FIH-Gln239. This interaction may be crucial for substrate positioning, such that the [FeO]2+ oxidant reacts selectively with the β-carbon of HIF-Asn803. Although Gln239 is not strictly a second-sphere residue in FIH, the Gln239→Asn mutant was included in the present study as a way to investigate the role of CTAD positioning on catalysis.
Testing the functional role of the second-sphere is crucial to understand the intricate relationship between the overall structure and function of FIH. This manuscript reports the activity, metal binding, and catalytic precision of second-sphere point mutants of FIH. Our results indicate that several second-sphere residues are key to tuning the O2-reactivity of FIH.
Materials and methods
Protein expression and purification
FIH and its mutants were expressed from E. coli with an N-terminal His6 tag and purified as previously described.(23) Following purification via Ni-NTA column hromatography, the His6 tag was removed by thrombin digestion. Exogenous metal was removed by prolonged incubation with EDTA, and then FIH was further purified by size-exclusion chromatography to yield the FIH dimer.
Differential Scanning Calorimetry
DSC experiments were performed using a MicroCal VP-DSC microcalorimeter.(24) 50 mM Hepes buffer pH 7.50 was used and 50 μM samples were heated in the calorimeter over a 25–75 °C range at a scan rate of 60°C/hour. A buffer scan was subtracted from each dataset to correct for base-line drift. Data analysis was performed using Origin Microcal software,(25) the results of which are provided in the supplementary material.
Metal binding
The experimental protocol was modified from Marletta et al.(26) Citrate was used as a chelator to buffer the concentration of free Co(II) ion, [Co(H2O)6]2+, for which stability constants are reported.(27) Co(II) binding to FIH was monitored by fluorescence quenching in a deoxygenated solution at room temperature. FIH was present in a fluorescence cuvette as a 200 μL solution of 1.00 mM sodium citrate, 20 μM FIH, 100 μM α-ketoglutarate, and 50 mM HEPES pH 7.50. Small volumes of CoCl2 were added from a solution of 1.00 mM CoCl2 and 1.00 mM sodium citrate, 50 mM HEPES pH 7.50. Following addition of Co(II), the cuvette was gently rocked by hand and the fluorescence measured over several minutes until a steady reading was obtained. Samples were excited at 280 nm and the fluorescence intensity was measured at 340 nm. Data fitting and results are provided in the supplementary material.
EPR
X-band EPR spectra were recorded on a Bruker Elexsys E-500 ESR Spectrometer equipped with a Bruker ER 4118CF-O LHe/LN2 cryostat. EPR samples were prepared by reconstituting enzymes with CuSO4 in a FIH:Cu2+ ratio of 1:0.9; the CuSO4 solution was slowly added to the enzyme solution to prevent precipitation. Similarly, FIH(Cu+αKG) was prepared by adding αKG slowly as the final step. Samples totaling 300 μL of 1 mM enzyme, 0.9 mM CuSO4 and 1 mM αKG in 50 mM Hepes, pH 7.50, were frozen in quartz tubes with liquid nitrogen. The spectra were obtained by averaging 4 scans at 9.438 GHz frequency, 20 mW power, 20 G modulation amplitude, 100 GHz modulation frequency and a 327 ms time constant. The microwave power was variatied to ensure the samples were not saturated under reported conditions.
UV-Vis spectroscopy
Enzyme stocks, FeSO4, and αKG were made anaerobic under an Argon flush. 50 mM HEPES pH 7.50 was also made anaerobic by a repeating cycle of vacuum and nitrogen flush. (FeII+αKG)FIH spectra were obtained on anaerobic samples containing FIH (250 μM), FeSO4 (230 μM) and αKG (500 μM) in buffer. Apo FIH spectra were recorded for similar samples by omitting αKG and FeSO4, and used as background spectra.
Activity assays
Initial rate measurements were performed in 50 mM HEPES pH 7.50 and incubated at 37 °C in 50 μL reaction volume. Reaction buffer included 2.00 mM ascorbate, 100 μM DTT, 5 u/μL catalase, 600 μM αKG, 25 μM FeSO4, and 0–600 μM CTAD. The reaction was initiated by adding the enzyme (0.5–5 μM), and at certain time points 5 μL aliquots were taken and quenched in 45 μL 0.1% formic acid. For each reaction 10 time points were collected and quenched, then analyzed by LC-ESI-MS. Samples were first loaded onto a C8 column for desalting, and CTAD and hydroxylated CTAD (CTADOH) were detected by ESI-MS to determine the mole fraction of peptide that had been converted to product, χCTAD-OH. Product concentrations were calculated as [CTADOH] = χ(CTAD-OH) × [CTAD]0, and used to determined initial rates, which were then used for Michaelis-Menten fits.
Autohydroxylation
Autohydroxylation was measured as described previously, with minor changes.(28) FIH (100 μM), FeSO4 (500 μM), and αKG (500 μM) were anaerobically incubated in 50 mM HEPES pH 7.50 for 20 min. Autohydroxylation was initiated by adding an equal volume of buffer that had been equilibrated under air, and the reaction monitored at 583 nm. Competitive autohydroxylation assays included 50 μM CTAD that was pre-incubated with the FIH.
Coupling
A Hamilton PRP-X300 anion exclusion column and UV detection at 210 nm was used to separate and detect succinate yield from quenched steady-state reactions of FIH. The concentration of hydroxylated CTAD, CTADOH, was determined as per the activity assays. The coupling ratio was defined as C = [succinate]/[CTADOH].
Results
Inspection of the X-ray crystal structure of FIH revealed hydrogen-bonding networks surrounding the Fe, which were centered on either αKG or CTAD. Asn205 and Asn294 residues donate hydrogen-bonds to the C-1 carboxylate of αKG, suggesting that these second-sphere residues may stabilize charge buildup during decarboxylation. Arg238 and Gln239 make hydrogen bonds with CTAD, suggesting that these residues may position the HIF-Asn803, or may play a role in priming the Fe for oxygenation chemistry (Figure 1). The role of these hydrogen bonds was tested by functional assays and electronic spectroscopy of point mutants.
The effect of hydrogen bonding network on turnover was investigated by steady-state kinetics (Table 1). Saturating conditions were determined for ascorbate, αKG and Fe(II) and then initial rates of WT-FIH and its mutants were acquired by varying the CTAD concentration. DTT and catalase were used in the steady-state assays to prevent ferroxidase chemistry by Fe(II) in solution. WT-FIH showed kcat of 31 min−1 with a kcat/KM(CTAD) of 0.41 μM−1 min−1, similar to previously reported values (Figure 2).(29–31) Each of the mutants had a modest effect on KM(CTAD), as both kcat and kcat/KM were diminished by a similar factor for each mutant: N205A (3-fold), N294A (15-fold), and Q239N (150-fold) (Figure 2 and 3). R238M exhibited very low activity, which we could only detect through endpoint assays.
Table 1.
Kinetic parameters for WT-FIH and its mutants for αKG and the synthetic peptide CTAD (39 residues).a,b
| kcat (min−1)a | KM(αKG) (μM)b | KM(CTAD) (μM)a | kcat/KM(CTAD) (μM−1 min−1)a | |
|---|---|---|---|---|
| WT-FIH | 31 ± 4 | 22 ± 6 | 77 ± 22 | 0.41 ± 0.1 |
| N205A | 10.2 ± 0.5 | 7.5 ± 2 | 43 ± 8 | 0.24 ± 0.07 |
| N294A | 2.2 ± 0.1 | 14 ± 2 | 137 ± 26 | 0.016 ± 0.004 |
| Q239N | 0.123 ± 0.002 | 24 ± 4 | 38 ± 2 | 0.0032 ± 0.0002 |
| R238Mc | - | - | - | - |
Assays in which CTAD was the varied substrate were performed with FIH and mutants (0.5–5 μM), ascorbate (2 mM), DTT (100 μM), catalase (5 u/mL), αKG (500 μM), FeSO4 (25–50 μM), CTAD (0–600 μM) in 50 mM HEPES pH 7.50 at 37 °C. CTAD corresponds to a Cys→Ala point mutant of HIF-1α788–826
Assays in which αKG was the varied substrate were as above, with these exceptions: CTAD (200 μM), and αKG varied (3 – 160 μM).
R238M was too slow to assay initial rates.
Figure 2.
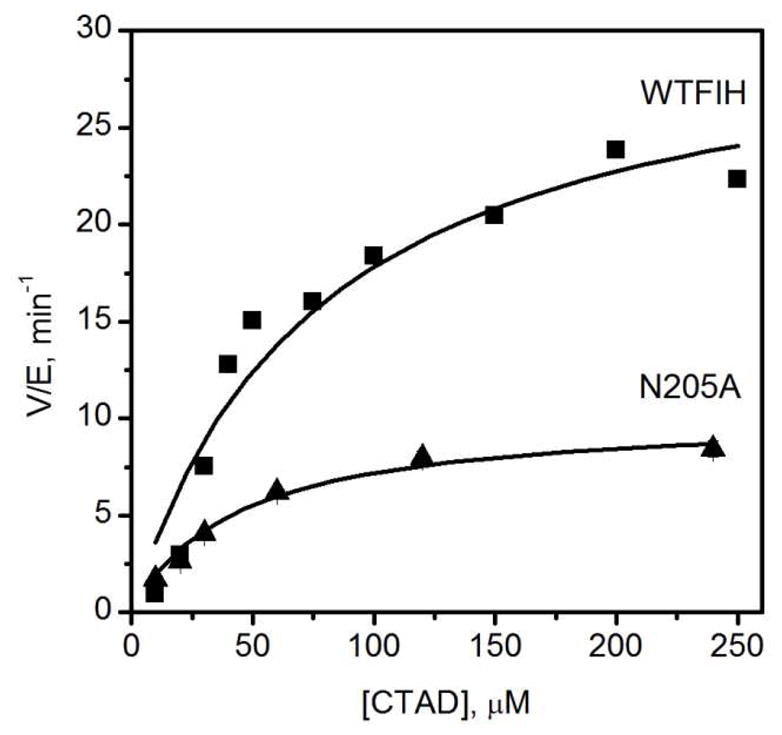
Steady state kinetics of WT-FIH and N205A. WT-FIH (0.5 μM), N205A (1 μM), ascorbate (2 mM), DTT (100 μM), catalase (5 u/mL), αKG (500 μM), FeSO4 (25 μM), CTAD (0–250 μM) in 50 mM HEPES pH 7.50.
Figure 3.
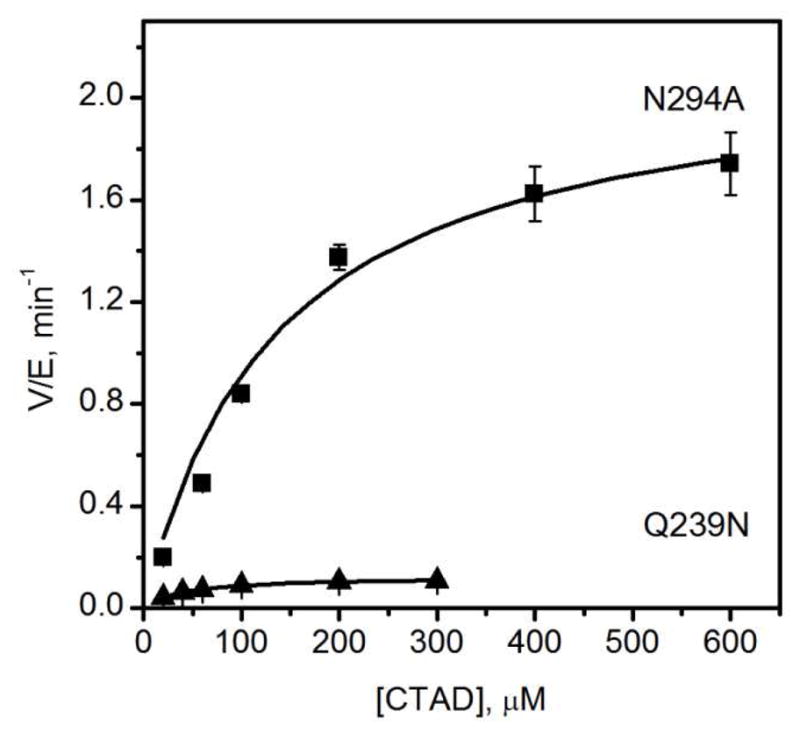
Steady-state kinetics of N294A and Q239N. N294A (5 μM), Q239N (5 μM), ascorbate (2 mM), DTT (100 μM), catalase (5 u/mL), αKG (500 μM), FeSO4 (50 μM), CTAD (0–600 μM) in 50 mM HEPES pH 7.50.
Endpoint assays showed that CTAD-centered mutants were impaired in terms of product yield, relative to WT-FIH and the αKG-centered mutants (Table 2). WT-FIH hydroxylated 94% of CTAD during prolonged incubations; the αKG-centered mutants, N205A and N294A, were nearly as thorough as WT-FIH, converting over 79% of CTAD to product. In contrast, both CTAD-centered mutants were significantly compromised in their ability to hydroxylate CTAD, as Q239N and R238M converted less than 25% of CTAD to hydroxylated product.
Table 2.
Coupling and yield of hydroxylated CTAD by FIH variants.
| Ca | % CTADOHb | |
|---|---|---|
| WT-FIH | 0.98 ± 0.03 | 94% |
| N205A | 1.08 ± 0.04 | 90 |
| N294A | 1.3 ± 0.1 | 79 |
| Q239N | 3.1 ± 0.4 | 23 |
| R238M | n.d. | 5 |
C = [succinate]/[CTADOH] as determined under steady-state turnover conditions; see text for details. Not determined (n.d.).
Qualitative endpoints: FIH (100 μM) FeSO4 (500 μM), αKG (500 μM), CTAD (50 μM) 50 mM HEPES, pH 7.50.
The ratio of succinate production to CTAD hydroxylation was measured, to check for coupling between O2-activation and hydroxylation (Table 2). As O2-activation produces succinate, but might not lead to hydroxylated CTAD, uncoupling between these two enzymatic steps would lead to elevated coupling ratios, C = [succinate]/[CTADOH]. WT-FIH was tightly coupled,(43) with C equal to unity within experimental uncertainty (C = 0.98 ± 0.03). The Q205A and Q294A point mutants exhibited slightly elevated C values, however Q239N exhibited a coupling ratio was appreciably greater than unity (C = 3.1 ± 0.4).
FIH will slowly activate O2 in the absence of CTAD, autohydroxylating Trp296 to form an Fe(III)-O-Trp296 chromophore with λmax = 583 nm.(23, 28) While the rate-limiting step in autohydroxylation is not known, priming of the (FeII+αKG)FIH is a likely requirement to permit O2-activation. WT-FIH and the point mutants were tested for autohydroxylation rates in the absence of CTAD in order to determine how the priming of Fe(II) changed upon mutation. The autohydroxylation rates for the point mutants were only moderately altered from WT-FIH (Table 3), suggesting that the second coordination sphere had minor effects on steps directly involved in autohydroxylation. It is notable that both αKG-centered mutants exhibited 2-fold increases in the autohydroxylation rates relative to that for WT-FIH.
Table 3.
Autohydroxylation rates for FIH variants.
| auto-hydroxylation initial rates (μM/min)
|
||
|---|---|---|
| 0 CTAD | 50 μM CTAD | |
| WT FIH | 0.0442 ± 0.0007 | 0.0265 ± 0.0008 |
| N205A | 0.100 ± 0.002 | 0.078 ± 0.001 |
| N294A | 0.0816 ± 0.0008 | 0.068 ± 0.002 |
| Q239N | 0.034 ± 0.001 | 0.0564 ± 0.0008 |
| R238M | 0.0262 ± 0.0008 | 0.0183 ± 0.0013 |
100 μM FIH, 500 μM FeSO4, 500 μM αKG, 0 or 50 μM CTAD, 50 mM HEPES, pH 7.50.
We felt that a competition assay, in which autohydroxylation was monitored in the presence of CTAD, would be an interesting way to further test the effect of second-sphere mutations on positioning. In the competition assay, HIF-Asn803 and Trp296 were both present as hydroxylation targets (Table 3). The rate of autohydroxylation for WT-FIH, and most of the point mutants, decreased in the presence of CTAD. The lone exception to this was the CTAD-centered mutant Q239N, which underwent autohydroxylation 60% faster in the presence of CTAD than in its absence. This increased autohydroxylation rate for Q239N strongly suggested that CTAD binding primed Fe(II) to react with O2, but that HIF-Asn803 was improperly oriented to receive the oxidant in this point mutant.
The ability of WT-FIH and its mutants to stabilize negative charge on C-1 of αKG was analyzed spectroscopically by the energy of the metal to ligand charge transfer (MLCT) transition. UV-Vis absorption spectra of (FeII+αKG)FIH form of each enzyme was measured, with the spectrum of the apo enzyme subtracted as a background. Each of the (FeII+αKG)FIH samples exhibited an MLCT peak near 500 nm (Figure 4). This absorption band has been attributed to three overlapping transitions between populated Fe T2g(π) orbitals and empty π* orbitals delocalized over the C1 carboxylate and C2 keto group of αKG.(18) WT-FIH displayed an MLCT maximum at 500 nm, similar to that of other αKG hydroxylases such as TauD (530 nm)(32) and CS2 (476 nm).(33) In αKG-centered mutants this MLCT was blue shifted (Table 4), with the MLCT peaks for N205A and N294A appearing at 485 and 490 nm respectively. This blue shift indicated that the αKG π* orbitals in these point mutants were destabilized from WT-FIH, consistent with the hydrogen-bonds donated by Asn205 and Asn294 pulling electron density away from the keto-group of αKG.
Figure 4.
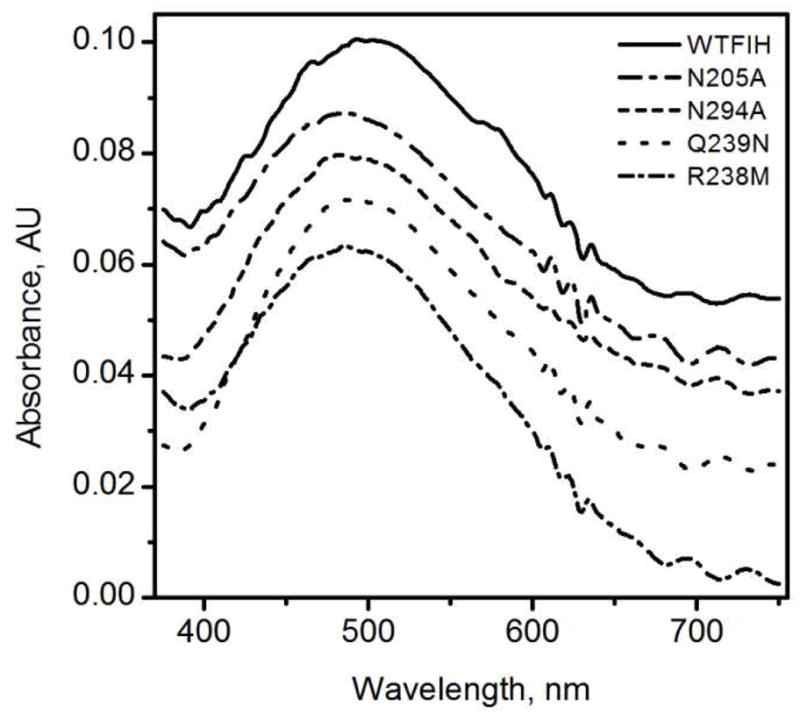
UV-Vis spectra of (FeII+αKG)FIH under anaerobic conditions after subtraction of (FeII)FIH spectra. FIH & mutants (250 μM), FeSO4 (230 μM), αKG (250 μM) in 50 mM HEPES pH 7.50.
Table 4.
MLCT transitions in FIH variants.
| λmax (nm) | |
|---|---|
| WT FIH | 500 |
| N205A | 485 |
| N294A | 490 |
| Q239N | 495 |
| R238M | 488 |
FIH (250 μM), FeSO4 (230 μM), αKG (250 μM) in 50 mM HEPES pH 7.50.
Substrate centered mutations also showed blue-shifted MLCT bands (Table 4). Charge transfer complexes of Q239N and R238M appeared at 495 and 488 nm respectively. The slight shift in the MLCT energy for Q239N is consistent with the short distance between residue 239 and αKG, suggesting only minor structural perturbations. The larger shift observed for R238M is similar in magnitude to that observed for N294A, and may be tentatively attributed to polarization effects from the loss of the positive charge at residue 238.
Cu(II) was used as a spectroscopic probe for the electronic fine structure in FIH. EPR spectra of both (Cu)FIH and (Cu+αKG)FIH were measured for WT-FIH and each point mutant (Table 5). The spectra of the point mutants were similar to that of WT-FIH, with some heterogeneity evident in the (Cu)FIH samples that diminished significantly in the (Cu+αKG)FIH samples. As FIH only provided three protein derived ligands, heterogeneity in the (Cu)FIH samples likely arose due to the presence of two or three solvent-derived ligands with variable bond lengths to the Cu(II). The WT (Cu)FIH sample exhibited an axial spectrum with apparent geff and A|| values (g⊥ = 2.06; g|| = 2.3; A|| = 148.1 G) appropriate for a “Type 2” Cu(II) site with mixed N and O-donor ligands (Figure 5).(34) Spectra of the WT (Cu+αKG)FIH sample exhibited increased anisotropy in geff (g⊥ = 2.06, g|| = 2.35) and reduced hyperfine coupling (A|| = 136.4 G) as compared to (Cu)FIH (Figure 6). The small A|| is similar to that reported for other αKG hydroxylases,(35–37) where it was attributed to an O-rich anionic ligand set and distorted planarity.(37) The spectra for the (Cu+αKG)FIH point mutants were nearly identical to those of WT-FIH, showing that the point mutations did not alter the metal coordination geometry in the (Cu+αKG)FIH enzyme form.
Table 5.
EPR spectral parameters for (Cu)FIH and (Cu+αKG)FIH.
| EPR parameters
|
|||
|---|---|---|---|
| g|| | g⊥ | A|| (G) | |
| (Cu)FIH | 2.30 | 2.06 | 146.2 |
| (Cu)N205A | 2.31 | 2.06 | 131.0 |
| (Cu)N294A | 2.31 | 2.06 | 134.6 |
| (Cu)Q239N | 2.29 | 2.06 | 135.0 |
| (Cu)R238M | 2.31 | 2.06 | 144.8 |
| (Cu+αKG)FIH | 2.35 | 2.06 | 136.4 |
| (Cu+αKG)N205A | 2.35 | 2.06 | 136.4 |
| (Cu+αKG)N294A | 2.35 | 2.07 | 136.4 |
| (Cu+αKG)Q239N | 2.35 | 2.07 | 136.8 |
| (Cu+αKG)R238M | 2.35 | 2.07 | 136.1 |
Parameters observed using Spincount(44)
Figure 5.
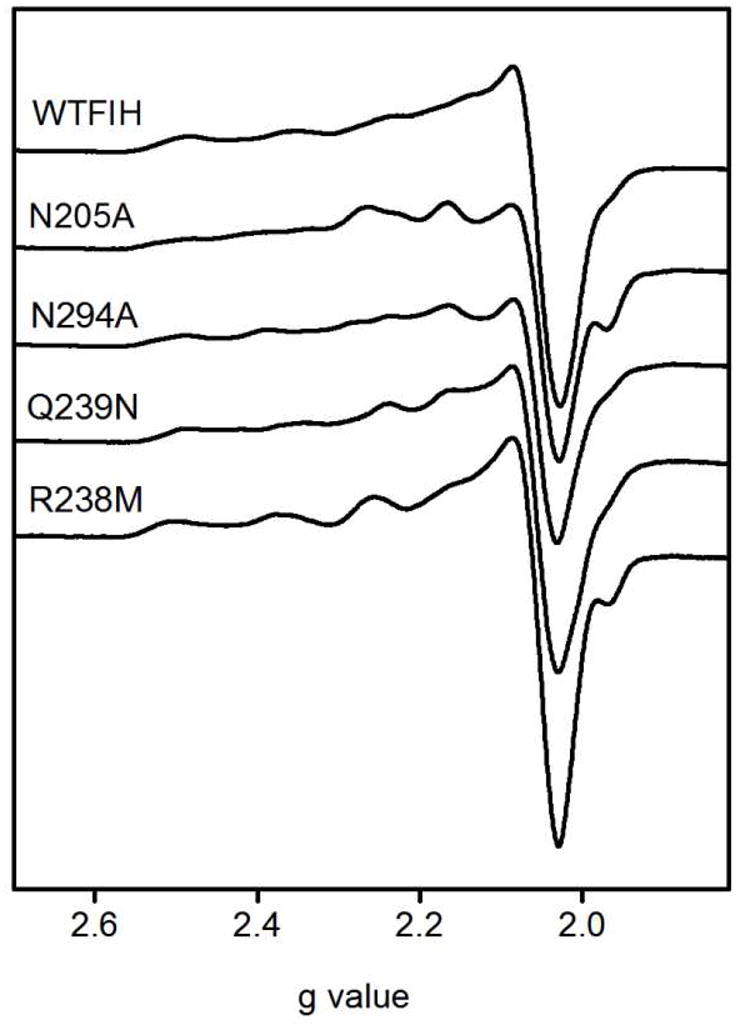
X-band EPR spectra of (CuII)FIH variants. FIH and point mutants (1 mM), CuSO4 (0.9 mM) in 50 mM HEPES pH 7.50. 9.438 GHz, 20 mW power, 20 G modulation amplitude, 100 GHz modulation frequency, 327 ms time constant, 77 K.
Figure 6.

X-band EPR spectra of (CuII+αKG)FIH variants. FIH and mutants (1 mM), CuSO4 (0.9 mM), αKG (1 mM) in 50 mM HEPES pH 7.50. 9.438 GHz frequency, 20 mW power, 20G modulation amplitude, 100 GHz modulation frequency, 327 ms time constant at 77 K.
Global enzyme stability was not altered by point mutation, as shown by the irreversible melting temperatures (TM(app)) determined by DSC (Table S1). The TM(app) of wild type FIH was 54.5 °C, whereas the TM(app) for the mutants were, in fact, slightly higher. Substrate centered mutants R238M and Q239N had slightly higher TM(app) than wild type, 56.2 and 56.7 °C respectively. The TM(app) of αKG centered mutants N205A and N294A were 58.6 and 58.9 °C respectively, again indicating that the second sphere mutations did not alter overall protein stability.
The metal binding of FIH was slightly affected by point mutation, as shown by the Co(II) titrations (Table S2). The equilibrium dissociation constant for (Co2++αKG)FIH was 1.38(6) × 10−7, indicating a relatively strong affinity for Co2+. Each of the point mutants exhibited a similar dissociation constant, with KD ranging from 1.0 – 1.9 × 10−7. This indicated that the primary coordination sphere of the active site was not altered by mutagenesis in the secondary sphere.
Discussion
Hydrogen bonding networks have been shown to be crucial to the function of enzymes reacting with O2-derived species, such as lipoxygenase and SOD,(15, 16) as well as for O2-activating models.(14) In the case of FIH, hydrogen bonding was observed between the facial triad ligands of Fe(II), the αKG, and various second-sphere residues, suggesting a functional role in tuning the chemistry of the Fe(II) center. In addition Gln239 hydrogen bonds to the CTAD substrate, and it’s role in substrate positioning has been included. The present work combines kinetics and spectroscopy to show that these second sphere hydrogen bonds play roles in promoting oxidative decarboxylation, priming Fe(II) to bind O2, and positioning HIF-Asn803.
The steady-state rate constants, kcat and kcat/KM(CTAD), were diminished for each point mutant of FIH (Table 1), indicating a reduction in the rate of one or more steps during turnover. These rate constants can be interpreted within the context of an ordered sequential mechanism, which is the consensus chemical mechanism for αKG hydroxylases.(9, 10) A minimal kinetic model (Scheme 2) for FIH in the presence of saturating αKG involves separate microscopic steps for binding S = CTAD (k1) and O2 (k2), with an irreversible chemical step (k3) followed by product release (k4).
Scheme 2.
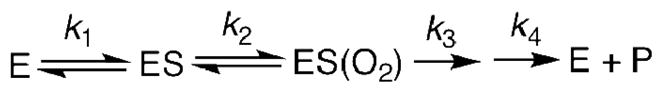
The steady-state rate constants are composites of the above microscopic steps. All steps after substrate binding contribute to kcat, while kcat/KM(CTAD) is a function of all steps up through the first irreversible step. The reduced values to both kcat and kcat/KM(CTAD) for each point mutant suggests that the second coordination sphere plays a significant role in determining k3, the common microscopic step. This first irreversible step may be oxidative decarboxylation of αKG to form the [FeO]2+ intermediate, as seen for TauD,(38, 39) or a another step forming an Fe(II) center, as implied for PHD.(40) In the absence of additional mechanistic probes such as kinetic isotope effects, we turned to the use of electronic spectroscopy and inactivation kinetics to test the effect of the second sphere residues.
Point mutations to second sphere residues were non-perturbing to the protein structure and primary coordination of the metal center in mutated FIH, as shown by the thermodynamic stability and electronic spectroscopy of each mutant. EPR spectra of the point mutants showed that the metal center in (Cu+αKG)FIH retained a virtually identical ground-state electronic environment to WT-FIH (Figure 6). The MLCT transitions for (FeII+αKG)FIH variants revealed that the αKG was a bidentate ligand to Fe(II), while also providing a view of the changing Lewis acidity of the C2-position of αKG (Figure 4). As nucleophilic attack of superoxide at C2 is thought to be a key step in O2-activation by αKG-dependent oxygenases,(41) the Lewis acidity of the keto group is essential for oxidative decarboxylation. The blue shifts in the MLCT energies for point mutants indicated that the αKG π* orbitals were destabilized in the N294A and N205A mutants, suggesting that the hydrogen bonds found in WT-FIH serve to pull electron density away from the C2 position of αKG.
Oxidative decarboxylation is promoted by stabilizing negative charge buildup in the transition state, as shown for enzymes(20) and model complexes.(41) Structural analysis and the MLCT shifts in FIH indicate that both Asn205 and Asn294 pull electron density away from αKG, although the Fe(II) likely plays the predominant role in activating αKG for decarboxylation. Asn294 plays a more significant role than Asn205 in stabilizing charge, as shown by the relative values for kcat: WT > N205A > N294A (Table 1). This may be due to Asn294 being the sole residue pulling electron density away from the distal O of the C1 carboxylate of αKG, while both Asn205 and Fe(II) pull electron density from the proximal O of the C1 carboxylate (Figure 1).
The significant decrease in the steady-state rate constants for CTAD-centered mutants may arise from changes in Fe(II) priming or CTAD positioning. The consensus chemical mechanism suggests that mutations affecting priming should principally reduce kcat/KM(CTAD), as steps up through O2-activation determine this rate constant. Conversely, mutations affecting positioning should reduce kcat, as steps following O2-binding determine this rate constant. Although the structure of FIH with CTAD bound suggests that there may be hydrogen bonds with specific roles, such as those between Gln239 and HIF-Asn803 (positioning), and Arg238 and Asp201 (priming), the kinetics data is less clear as each point mutation reduced kcat as well as kcat/KM(CTAD) when compared to WT FIH. This may arise from an effect on a chemical step common to both rate constants, such as O2-activation, or from the extended hydrogen bond network linking Asp201 to HIF-Asn803 thorugh Arg238 and Gln239 (Figure 1). It appears that positioning the sidechain of HIF-Asn803 is integral to the priming of Fe(II), and may even be a design feature to ensure tight coupling between O2-activation and substrate hydroxylation.
While priming could not be separated from substrate positioning by steady-state kinetics, the two roles could be distinguished by the coupling ratio, and by analysis of hydroxylation products, as shown for TauD.(42) A functional view of priming is the abilty of FIH to activate O2 only once CTAD binds, leading to close coupling between the production of succinate and CTADOH. In the case of WT-FIH, breakdown in priming leads solely to autohydroxylation as FIH does not release reactive oxygen species.(23, 28, 43) This was supported by our observation of a coupling value of unity (C = 0.98 ± 0.03) for WT-FIH, and a near-unity coupling ratio for N205A and N294A.
Positioning HIF-Asn803 in the proper registry within the active site is crucial to ensure that the [FeO]2+ oxidant attacks the correct residue and forms the CTADOH product. The Q239N mutant exhibited notable effects on the coupling ratio, indicating that Gln239 was crucial for local positioning (Table 2). Q239N was the only mutant in which the coupling ratio differed significantly from unity (3.1 ± 0.4), and the only mutant that autohydroxylated faster in the presence of CTAD. These observations indicate that, while Q239N was able to activate O2, the HIF-Asn803 sidechain was improperly positioned to form normal product.
FIH serves a crucial role in sensing hypoxia within human cells. As an O2-sensing enzyme, it must maintain tight coupling between O2-activation and substrate hydroxylation, which it achieves by hydrogen bonding between ligands to the Fe and the second coordination sphere. These second sphere hydrogen bonds play roles in promoting O2-activation as well as maintaining the structural registry between CTAD and Fe. Hydrogen bonds from Asn205 and Asn294 pulled electron density aweay from αKG, as shown by the LMCT transitions; removal of eityher hydrogen bond in the Asn→Ala point mutants slighly accelerated autohydroxylation, but made normal turnover much slower in the case of Asn294→Ala. It is likely that autohydroxylation rates reflect the intrinsic reactivity of αKG, whereas the rate of normal turnover is more dependent on precise positioning of CTAD. For this reason, the rate constants for normal turnover are greatly reduced in Gln239→Asn and Arg238→Met, both CTAD-centered mutants. By virtue of their respective locations relative to Asp201 and HIF-Asn803, it is likely that Arg238 is necessary for priming of the Fe(II) in response to CTAD binding, whereas Gln239 is more involved in positioning HIF-Asn803. The second sphere interactions play a significant role in O2-sensing function of FIH by tuning the chemistry at the Fe(II) center.
Supplementary Material
Acknowledgments
We thank the NIH for funding (R01-GM077413); the NSF for support of the EPR facility (CHE-0443180 NSF-CRIF); and Prof. Stephen Eyles for technical assistance.
Abbreviations
- αKG
alpha-ketoglutarate
- CTAD
C-terminal transactivation domain of HIFα
- DSC
differential scanning calorimetry
- EPR
electron paramagnetic resonance
- FIH
factor inhibiting HIF-1
- HIF
hypoxia inducible factor
- LC-ESI-MS
liquid chromatography – electrospray ionization mass spectrometry
- MLCT
metal-to-ligand charge transfer
- NTA
nitrilotriacetate
- SOD
superoxide dismutase
Footnotes
Supported by the NIH (R01-GM077413)
Supporting Information Available: Tabulated results for thermal melting (DSC), and Co(II) binding titrations for FIH variants. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 2.Ozer A, Bruick RK. Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007;3:144–153. doi: 10.1038/nchembio863. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology. 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 4.Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, Ratcliffe PJ. Genome-wide Association of Hypoxia-inducible Factor (HIF)-1 alpha and HIF-2 alpha DNA Binding with Expression Profiling of Hypoxia-inducible Transcripts. J Biol Chem. 2009;284:16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McNeill LA, Hewitson KS, Claridge TD, Seibel JF, Horsfall LE, Schofield CJ. Hypoxia-inducible factor asparaginyl hydroxylase (FIH-1) catalyses hydroxylation at the beta-carbon of asparagine-803. Biochem J. 2002;367:571–575. doi: 10.1042/BJ20021162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanauske-Abel HM, Gunzler V. A stereochemical concept for the catalytic mechanism of prolylhydroxylase: Applicability to classification and design of inhibitors. J Theor Biol. 1982;94:421–455. doi: 10.1016/0022-5193(82)90320-4. [DOI] [PubMed] [Google Scholar]
- 7.Hewitson KS, Schofield CJ. The HIF pathway as a therapeutic target. Drug Discovery Today. 2004;9:704–711. doi: 10.1016/S1359-6446(04)03202-7. [DOI] [PubMed] [Google Scholar]
- 8.Nagel S, Talbot NP, Mecinovic J, Smith TG, Buchan AM, Schofield CJ. Therapeutic Manipulation of the HIF Hydroxylases. Antioxid Redox Signal. 2010;12:481–501. doi: 10.1089/ars.2009.2711. [DOI] [PubMed] [Google Scholar]
- 9.Hausinger RP. FeII/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 10.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem Rev. 2000;100:235–350. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 11.Dann CE, Bruick RK, Deisenhofer J. Structure of factor-inhibiting hypoxia-inducible factor 1: An asparaginyl hydroxylase involved in the hypoxic response pathway. Proc Natl Acad Sci USA. 2002;99:15351–15356. doi: 10.1073/pnas.202614999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1 alpha. J Biol Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 13.Lee C, Kim SJ, Jeong DG, Lee SM, Ryu SE. Structure of human FIH-1 reveals a unique active site pocket and interaction sites for HIF-1 and von Hippel-Lindau. J Biol Chem. 2003;278:7558–7563. doi: 10.1074/jbc.M210385200. [DOI] [PubMed] [Google Scholar]
- 14.Shook RL, Borovik AS. The effects of hydrogen bonds on metal-mediated O2 activation and related processes. Chem Commun. 2008:6095–6107. doi: 10.1039/b810957e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomchick DR, Phan P, Cymborowski M, Minor W, Holman TR. Structural and functional characterization of second-coordination sphere mutants of soybean lipoxpgenase-1. Biochemistry. 2001;40:7509–7517. doi: 10.1021/bi002893d. [DOI] [PubMed] [Google Scholar]
- 16.Miller AF. Redox tuning over almost 1 V in a structurally conserved active site: Lessons from Fe-containing superoxide dismutase. Acc Chem Res. 2008;41:501–510. doi: 10.1021/ar700237u. [DOI] [PubMed] [Google Scholar]
- 17.Hegg EL, Que L. The 2-His-1-carboxylate facial triad - An emerging structural motif in mononuclear non-heme iron(II) enzymes. Eur J Biochem. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 18.Pavel EG, Zhou J, Busby RW, Gunsior M, Townsend CA, Solomon EI. Circular dichroism and magnetic circular dichroism spectroscopic studies of the non-heme ferrous active site in clavaminate synthase and its interaction with alpha-ketoglutarate cosubstrate. J Am Chem Soc. 1998;120:743–753. [Google Scholar]
- 19.Zhou J, Kelly WL, Bachmann BO, Gunsior M, Townsend CA, Solomon EI. Spectroscopic studies of substrate interactions with clavaminate synthase 2, a multifunctional alpha-KG-dependent non-heme iron enzyme: Correlation with mechanisms and reactivities. J Am Chem Soc. 2001;123:7388–7398. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 20.Walsh CT. Enzymatic Reaction Mechanisms. W. H. Freeman & Co; San Francisco: 1979. [Google Scholar]
- 21.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an alpha-ketoglutarate-dependent dioxygenase: a high-spin FeIV complex in taurine/alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 22.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. Direct detection of oxygen intermediates in the non-heme Fe enzyme taurine/alpha-ketoglutarate dioxygenase. J Am Chem Soc. 2004;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 23.Chen YH, Comeaux LM, Herbst RW, Saban E, Kennedy DC, Maroney MJ, Knapp MJ. Coordination changes and auto-hydroxylation of FIH-1: uncoupled O2-activation in a human hypoxia sensor. J Inorg Biochem. 2008;102:2120–2129. doi: 10.1016/j.jinorgbio.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plotnikov VV, Brandts JM, Lin LN, Brandts JF. A new ultrasensitive scanning calorimeter. Anal Biochem. 1997;250:237–244. doi: 10.1006/abio.1997.2236. [DOI] [PubMed] [Google Scholar]
- 25.Origin. OriginLab Corporation; Northampton, MA 01060: [Google Scholar]
- 26.Mills SA, Marletta MA. Metal binding characteristics and role of iron oxidation in the ferric uptake regulator from Escherichia coli. Biochemistry. 2005;44:13553–13559. doi: 10.1021/bi0507579. [DOI] [PubMed] [Google Scholar]
- 27.Martell AE, Smith RM. Critical Stability Constants. Plenum Press; New York, New York: 1974. [Google Scholar]
- 28.Chen YH, Comeaux LM, Eyles SJ, Knapp MJ. Auto-hydroxylation of FIH-1: an Fe(II), alpha-ketoglutarate-dependent human hypoxia sensor. Chem Commun. 2008:4768–4770. doi: 10.1039/b809099h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ehrismann D, Flashman E, Genn DN, Mathioudakis N, Hewitson KS, Ratcliffe PJ, Schofield CJ. Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem J. 2007;401:227–234. doi: 10.1042/BJ20061151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koivunen P, Hirsila M, Gunzler V, Kivirikko KI, Myllyharju J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem. 2004;279:9899–9904. doi: 10.1074/jbc.M312254200. [DOI] [PubMed] [Google Scholar]
- 31.Linke S, Stojkoski C, Kewley RJ, Booker GW, Whitelaw ML, Peet DJ. Substrate requirements of the oxygen-sensing asparaginyl hydroxylase factor-inhibiting hypoxia-inducible factor. J Biol Chem. 2004;279:14391–14397. doi: 10.1074/jbc.M313614200. [DOI] [PubMed] [Google Scholar]
- 32.Ryle MJ, Padmakumar R, Hausinger RP. Stopped-Flow Kinetic Analysis of Escherichia coli Taurine/α-Ketoglutarate Dioxygenase: Interactions with α-Ketoglutarate, Taurine, and Oxygen. Biochemistry. 1999;38:15278–15286. doi: 10.1021/bi9912746. [DOI] [PubMed] [Google Scholar]
- 33.Zhou J, Gunsior M, Bachmann BO, Townsend CA, Solomon EI. Substrate Binding to the α-Ketoglutarate-Dependent Non-Heme Iron Enzyme Clavaminate Synthase 2: Coupling Mechanism of Oxidative Decarboxylation and Hydroxylation. J Am Chem Soc. 1998;120:13539–13540. [Google Scholar]
- 34.Peisach J, Blumberg WE. Structural Implications Derived from Analysis of Electron-Paramagnetic Resonance-Spectra of Natural and Artificial Copper Proteins. Arch Biochem Biophys. 1974;165:691–708. doi: 10.1016/0003-9861(74)90298-7. [DOI] [PubMed] [Google Scholar]
- 35.Bleijlevens B, Shivarattan T, Sedgwick B, Rigby SEJ, Matthews SJ. Replacement of non-heme Fe(II) with Cu(II) in the alpha-ketoglutarate dependent DNA repair enzyme AlkB: Spectroscopic characterization of the active site. Journal of Inorganic Biochemistry. 2007;101:1043–1048. doi: 10.1016/j.jinorgbio.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 36.Hegg EL, Whiting AK, Saari RE, McCracken J, Hausinger RP, Que L. Herbicide-degrading alpha-keto acid-dependent enzyme TfdA: Metal coordination environment and mechanistic insights. Biochemistry. 1999;38:16714–16726. doi: 10.1021/bi991796l. [DOI] [PubMed] [Google Scholar]
- 37.Whiting AK, Que L, Saari RE, Hausinger RP, Fredrick MA, McCracken J. Metal coordination environment of a Cu(II)-substituted alpha-keto acid-dependent dioxygenase that degrades the herbicide 2,4-D. J Am Chem Soc. 1997;119:3413–3414. [Google Scholar]
- 38.Grzyska PK, Ryle MJ, Monterosso GR, Liu J, Ballou DP, Hausinger RP. Steady-state and transient kinetic analyses of taurine/alpha-ketoglutarate dioxygenase: Effects of oxygen concentration, alternative sulfonates, and active-site variants on the fe(IV)-oxo intermediate. Biochemistry. 2005;44:3845–3855. doi: 10.1021/bi048746n. [DOI] [PubMed] [Google Scholar]
- 39.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr Evidence for hydrogen abstraction from C1 of taurine by the high-spin Fe(IV) intermediate detected during oxygen activation by taurine:alpha-ketoglutarate dioxygenase (TauD) J Am Chem Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 40.Flashman E, Hoffart LM, Hamed RB, Bollinger JM, Krebs C, Schofield CJ. Evidence for the slow reaction of hypoxia-inducible factor prolyl hydroxylase 2 with oxygen. FEBS J. 2010;277:4089–4099. doi: 10.1111/j.1742-4658.2010.07804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mehn MP, Fujisawa K, Hegg EL, Que L. Oxygen activation by nonheme iron(II) complexes: alpha-keto carboxylate versus carboxylate. J Am Chem Soc. 2003;125:7828–7842. doi: 10.1021/ja028867f. [DOI] [PubMed] [Google Scholar]
- 42.McCusker KP, Klinman JP. An Active-Site Phenylalanine Directs Substrate Binding and C-H Cleavage in the alpha-Ketoglutarate-Dependent Dioxygenase TauD. J Am Chem Soc. 2010;132:5114–5120. doi: 10.1021/ja909416z. [DOI] [PubMed] [Google Scholar]
- 43.Saban E, Flagg SC, Knapp MJ. J Inorg Biochem. 2011 doi: 10.1016/j.jinorgbio.2011.01.007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hendrich MP. Spincount software. Carnegie Mellon Univ; [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.