

Abstract
The glutamate transporter-1 [GLT-1 (excitatory amino acid transporter 2)] subtype of glutamate transporter ensures crisp excitatory signaling and limits excitotoxicity in the CNS. Astrocytic expression of GLT-1 is regulated during development, by neuronal activity, and in neurodegenerative diseases. Although neurons activate astrocytic expression of GLT-1, the mechanisms involved have not been identified. In the present study, astrocytes from transgenic mice that express enhanced green fluorescent protein (eGFP) under the control of a bacterial artificial chromosome (BAC) containing a very large region of DNA surrounding the GLT-1 gene (BAC GLT-1 eGFP mice) were used to assess the role of nuclear factor-κB (NF-κB) in neuron-dependent activation of the GLT-1 promoter. We provide evidence that neurons activate NF-κB signaling in astrocytes. Transduction of astrocytes from the BAC GLT-1 eGFP mice with dominant-negative inhibitors of NF-κB signaling completely blocked neuron-dependent activation of a NF-κB reporter construct and attenuated induction of eGFP. Exogenous expression of p65 and/or p50 NF-κB subunits induced expression of eGFP or GLT-1 and increased GLT-1-mediated transport activity. Using wild-type and mutant GLT-1 promoter reporter constructs, we found that NF-κB sites at −583 or −251 relative to the transcription start site were required for neuron-dependent reporter activation. Electrophoretic mobility shift and supershift assays reveal that p65 and p50 interact with these same sites ex vivo. Finally, chromatin immunoprecipitation showed that p65 and p50 interact with these sites in adult cortex, but not in kidney (a tissue that expresses no detectable GLT-1). Together, these studies strongly suggest that NF-κB contributes to neuron-dependent regulation of astrocytic GLT-1 transcription.
Introduction
In the CNS, glutamate is cleared by a family of Na+-dependent transporters, including GLAST (glutamate-aspartate transporter), GLT-1 (glutamate transporter-1), EAAC1 [also called excitatory amino acid transporter 1 (EAAT1), EAAT2, and EAAT3, respectively], EAAT4, and EAAT5 (for review, see Danbolt, 2001; Beart and O'Shea R, 2007). Genetic, pharmacological, and electrophysiologic studies indicate that clearance of synaptic glutamate in the mammalian forebrain is primarily dependent upon GLT-1 (for review, see Robinson, 1998). Reduced expression of GLT-1 is observed in postmortem specimens from humans with several neurodegenerative diseases and/or rodent models of these diseases, including amyotrophic lateral sclerosis, epilepsy, Alzheimer's disease, and Huntington's disease (Rothstein et al., 1995; Li et al., 1997; Tanaka et al., 1997; Rao et al., 1998; Mathern et al., 1999; Kawahara et al., 2005; Shin et al., 2005; Yamada et al., 2006). In fact, recent studies support exploration of transcriptional activation of GLT-1 as a therapeutic approach for some of these disorders (Rothstein et al., 2005; Ganel et al., 2006; Chu et al., 2007; Rawls et al., 2007; Weller et al., 2008).
GLT-1 expression is dynamically regulated in vivo and in vitro. Expression of GLT-1 increases during synaptogenesis in vivo (Sutherland et al., 1996; Furuta et al., 1997). Whisker stimulation in rodents, used to increase sensory activation, results in increased expression of astrocytic GLT-1 in the corresponding cortical column of barrel cortex (Genoud et al., 2006). Deafferentation of various projection neurons is associated with a decrease in GLT-1 protein and mRNA in target areas (Ginsberg et al., 1995, 1996; Levy et al., 1995; Hein et al., 2001). These studies indicate that astroglial expression of GLT-1 is dependent upon the presence of neurons and is responsive to neuronal activation in vivo. Direct evidence for this functional interaction comes from in vitro studies. Astrocytes in culture express very low levels of GLT-1; however, coculturing with neurons induces expression of GLT-1 in astrocytes (Gegelashvili et al., 1997; Swanson et al., 1997; Perego et al., 2000; Zelenaia et al., 2000; Poitry-Yamate et al., 2002). This effect is partially mimicked by neuron-conditioned medium or by separating neurons from astrocytes with a semipermeable membrane (Schlag et al., 1998). In microfluidic chambers, axons (presynaptic termini) cause a clear induction of GLT-1 that is almost exclusively restricted to astrocytes that contact these processes. Furthermore, neuronal membranes also induce astrocytic GLT-1 expression (Yang et al., 2009). Together, these studies show that astrocytic expression of GLT-1 depends on both neuron-secreted molecules and contact with neurons. Very little is known about the transcriptional mechanisms that contribute to neuron-dependent induction of astrocytic GLT-1 expression.
Some studies have linked the nuclear transcription factor nuclear factor-κB (NF-κB) to the regulation of GLT-1 that is observed with epidermal growth factor (EGF) or ceftriaxone (Sitcheran et al., 2005; Lee et al., 2008); however, it is not clear whether neuron-dependent activation of astrocytic GLT-1 expression is dependent upon NF-κB signaling in astrocytes, nor is it known whether NF-κB interacts with GLT-1 promoter regions in vivo. In the present study, we provide evidence that NF-κB contributes to neuron-dependent induction of GLT-1.
Materials and Methods
Animals.
Generation of the bacterial artificial chromosome (BAC) GLT1 enhanced green fluorescent protein (eGFP) transgenic mice was described previously (Regan et al., 2007). These animals were rederived, and a small colony was maintained in the animal facility of Children's Hospital of Philadelphia. Mice were housed in humidity-, temperature-, and light-controlled environment, and provided with food and water ad libitum in accordance with the principles and procedures of the National Institutes of Health Guidelines for Care and Use of Laboratory Animals. Pregnant Sprague Dawley rats were obtained from Charles River Laboratories; pregnant C57BL/6J mice were obtained from Jackson Laboratories. The studies involving animals were reviewed and approved by the institutional animal care and use committee of the Children's Hospital of Philadelphia.
Species comparisons of the GLT-1 gene.
Genomic sequences from mouse, rat, and human for GLT-1 were aligned using Dcode database (www.dcode.org). Standard parameters (70% homology for at least 100 nt) were used to identify evolutionary conserved regions, and putative transcription factor binding sites within these regions were identified.
Primary cultures and experimental timelines.
Primary cultures of astrocytes were prepared from postnatal day 1 (P1) to P2 BAC GLT-1 eGFP promoter reporter mice (both female and males were used) as reported earlier with minor modifications (Susarla et al., 2004; Li et al., 2006). After dissection and removal of the meninges and blood vessels, cortices were incubated with trypsin-EDTA (0.05%, 0.2 mm) for 20 min at 37°C. Tissue was triturated and suspended in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone), 10% Hams F-12, and 0.24% penicillin/streptomycin (10,000 U/ml penicillin, 10,000 mg/ml streptomycin). Cells (14 ml) were plated at a density of 2.5 × 105 cells/ml (3 × 104 cells/cm2) in 75 cm2 flasks and maintained in a 5% CO2 incubator at 37°C. The growth medium was completely exchanged with fresh medium twice a week until cells were ∼90% confluent (9–10 d). To eliminate the oligodendrocyte precursors (A2B5-positive cells) that express GLT-1 (Zelenaia et al., 2000), cultures were washed with HBSS, and then incubated in DMEM (5 ml/75 cm2 flask) with A2B5 hybridoma supernatant (diluted 1:50; from Dr. Judy Grinspan, Children's Hospital of Philadelphia, Philadelphia, PA) and Low Tox-M rabbit complement (diluted 1:20; Cederlane) for 45 min at 37°C and 5% CO2 with occasional shaking (Li et al., 2006). The flasks were struck firmly against a hard surface to remove loosely adherent cells. After three washes with HBSS, the cells were maintained for 2 d in growth medium and then replated onto 12 well plates for glutamate uptake assays and onto 6 well plates for all other experiments at a density of 1 × 106 cells/ml (1 and 2 ml/well, respectively). Under these conditions, astrocyte cultures contain no neurons and <1% microglia/oligodendroglia (Zelenaia et al., 2000; Saura, 2007).
After 3–5 d, astrocytes were transduced with lentiviral vectors or by transfection. After an additional 3–5 d, cell suspensions, containing neurons and astrocytes, were prepared from the cortices of embryonic (day 17–18) Sprague Dawley rats or from wild-type mice (González et al., 2007a,b). Cells were layered over the astrocyte cultures at a density of 4 × 105 cells/ml; control astrocytes underwent a media change. Both sets of cultures (astrocytes or astrocytes with layered cell suspension) were fed with a one-fourth medium exchange every 3–4 d. After 7–10 d, the cultures were similar to classic mixed cultures of neurons and astrocytes with an intact monolayer of polygonal and stellate-shaped astrocytes under a moderate to high density of phase-bright, process-bearing neurons. As a similar induction of eGFP expression was observed with cell suspensions from either wild-type mice or rats, we used cell suspensions from rats because these cells were being produced for other ongoing experiments. This allowed us to reduce the overall numbers of animals needed.
In some experiments, astrocytes were treated with EGF (30 ng/ml; BD Biosciences). In these experiments, the complete medium supplemented with vehicle or EGF was exchanged twice a week. In some experiments, NF-κB-SN50 inhibitory peptide or the control peptide SN50M (SM; Calbiochem) was added to the medium 3 d after transfection. After an additional 2 d, cell suspensions were introduced. Except where noted, cells were harvested as described for analysis of luciferase activity or using RIPA buffer (Sheldon et al., 2008) after 10 d.
Constructs.
The wild-type 958bp GLT-1 promoter fragment containing from −921 to +37 bp of the human gene has been previously described (Yang et al., 2009). To mutate the putative NF-κB binding sequence at −272, the wild-type sequence of 5′-GCAAATCCCCA-3′ was changed to 5′-CGTACTTACGA-3′ by PCR-based mutagenesis using a site-directed mutagenesis kit (Stratagene). The putative NF-κB binding sequence at −583 5′-GGGGCATCCCGC-3′ was changed to 5′-CATCTGGATTCA-3′, and the sequence at −251 5′-GGGGCTAAACC-3′ was changed to 5′-TGGTCTAAACC-3′. A GLT-1 promoter fragment with all three sites mutated was prepared using sequential mutagenesis. These constructs were cloned between XhoI and HindIII restriction sites within the multiple cloning site of pGL4.14 luciferase reporter vector (Promega). The NF-κB luciferase reporter vector (Clontech) containing four repeats of a NF-κB consensus sequence, dominant-negative constructs of IκBα (IκBα super-repressor-SR and IκBα-ΔN; from Dr. Dean Ballard, Vanderbilt University School of Medicine, Nashville, TN), NF-κB p50 (Invitrogen), NF-κB p65 (from Dr. Dean Ballard) or Discosoma species Red (dsRED) fluorescent protein were subcloned into the lentiviral transfer plasmid (pTY-CMV). The sequences of all constructs were verified before use.
Lentiviral vector production/astrocyte infection.
Concentrated stocks of pseudotyped lentiviral vectors were produced with slight modification of recently published procedures (Watson and Wolfe, 2003; Susarla et al., 2004; Li et al., 2006). HEK-293T cells were grown in poly-d-lysine-coated (Sigma) 15 cm diameter plates and were triple-transfected with a packaging plasmid, pCMVΔr8.2 (16.8 μg), an envelop plasmid, JS-86 (5.6 μg), and a transfer plasmid (pTY-CMV) containing the transgene of interest (22.5 μg) using a Ca2+-phosphate transfection kit (Clontech). After ∼8–12 h, the cells were rinsed and fresh medium (12 ml) was added. On each of the following 2 d, the virus-containing medium was collected from three plates and filtered through 0.45 μm filters and concentrated by centrifugation at 50,000 × g for 2 h at 4°C. The resulting pellet was suspended in 1 ml of ice-cold growth medium and stored in aliquots at −80°C (never stored for >1 month). Concentrations of virus were determined using HIV-1 P24 ELISA assay kit (PerkinElmer).
To infect astrocytes, virus was thawed on ice, mixed with growth medium, and added to astrocyte cultures (200 ng of p24/ml). After an overnight incubation, cells were rinsed once with DMEM, and then fresh medium was added. In initial experiments, transgene expression was optimized to achieve transduction of a high percentage of cells using a lentivirus that expresses eGFP. At this concentration of virus, we observed no evidence of toxicity and 80–90% transduction efficiency. Fluorescence intensity was stable from 3 d after infection for at least 20 d. There was no effect on morphology and no cell death.
Transient transfection and luciferase assay.
Astrocytes in 6 well plates were transfected with a mixture of promoter reporter construct (1 μg) and pSV40-β-galactosidase (1 μg; Promega) using FuGENE HD transfection reagent (Roche) according to manufacturer's instructions. In parallel, the percentage of cells transfected was examined using dsRED; under these conditions between 20 and 25% express detectable fluorescence. Ten days after transfection, cells were harvested; luciferase and β-galactosidase activities were analyzed according the manufacturer's instructions (Luciferase Reporter Gene Assay and β-Galactosidase Enzyme Assay kits from Promega). Luciferase activity was normalized to β-galactosidase activity and/or total protein, where appropriate. The data from each experiment were expressed relative to mock transfection (empty vector).
Western blot analysis.
After analysis of total protein using bicinchoninic acid (kit from Pierce), samples were resolved by SDS-PAGE using minigels (Bio-Rad) and transferred to Immobilon membrane (Millipore) using a Bio-Rad transblot apparatus. After incubation for 1 h at room temperature in blocking solution (5% nonfat dry milk in 20 mm Tris, 0.9% NaCl, 0.1% Tween 20, pH 7.5), membranes were then probed with C-terminal-directed rabbit anti-GLT-1 (1:5000) (Rothstein et al., 1994), rabbit anti-actin (1:5000; Sigma), rabbit anti-GFP (1:5000; Sigma), rabbit anti-IκBα (1:5000; Cell Signaling Technology), or, in some cases, combinations of these antibodies. After three washes in blocking solution containing 1% nonfat milk, immunoblots were incubated in the same solution with anti-rabbit IRDye 680 antibody (1:10,000; LI-COR Biosciences) and visualized using an Odyssey Infrared Imaging system (LI-COR Biosciences). In initial experiments, protein linearity of both the eGFP and GLT-1 signals was confirmed (data not shown). As was previously reported (Haugeto et al., 1996), GLT-1 immunoreactivity was observed as both monomers and multimers. These bands were quantified separately, and there was no evidence that either responded differently to any of the treatments. Thus, data are presented as the sum of the amount of immunoreactivity found in the monomer and the multimers. The effects of treatments on actin immunoreactivity were examined in every experiment, and there was no evidence for effects of either lentiviral vector or other treatments on actin levels. Therefore, all data were normalized to actin as a loading control, and the amount of immunoreactivity was expressed relative to the corresponding control.
Electrophoretic mobility shift assay.
For electrophoretic mobility shift assays (EMSAs), nuclear proteins were extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce) from freshly dissected cortex of adult mice. Oligonucleotides (45 mer) corresponding to wild-type sequences surrounding the three putative NF-κB binding sites at −583, −272, and −251, and mutant variants (same as used for GLT-1 promoter reporter, see above) were synthesized by integrated DNA technology. Control oligonucleotides (22 mer) containing a mixture of known NF-κB binding sites (consensus sequence: GGG RNN YYC C, where r = purine and Y = pyrimidine) were used as a positive control (Parry and Mackman, 1994; May and Ghosh, 1997). EMSA was performed with procedures described in LightShift Chemiluminescent EMSA kit (Pierce). In each binding reaction, 1 μl of the following were incubated with 2 μl of nuclear extract in total volume of 20 μl at room temperature for 25 min: biotin-labeled oligo (20 fmol/ml); poly (dI-dC) (1 mg/ml); 50% glycerol, MgCl2 (100 mm); and 1% NP-40. In a subset of experiments, 200-fold excess unlabeled oligonucleotide (4 pmol) was included to test specificity. For supershift assays, 10 μg of rabbit anti-p65 or anti-p50 (Rockland) was added to the binding reaction that was incubated for an additional 2 h. Protein–DNA complexes were resolved on a 6% DNA retarding gel (polyacrylamide), transferred to nylon hybridization membrane (Hybond-N+; GE Healthcare), cross-linked with UV light, and finally biotin-labeled DNA was detected by chemiluminescence (chemiluminescent nucleic acid detection module kit; Pierce).
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) analysis was performed using EZ-CHIP Chromatin Immunoprecipitation Kit (Millipore) following the manufacturer's instruction with minor modifications. Briefly, tissues from cortex and kidney were harvested from adult wild-type mice and cross-chopped with two razor blades. After incubation with 1.5% formaldehyde for 15 min to cross-link protein–DNA complexes, excess glycine was used to quench the formaldehyde. After washing twice with 1× PBS, tissue was triturated repeatedly with a glass Pasteur pipette. The resulting suspension was centrifuged at 500 × g for 10 min, and the pellet resuspended in SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris, pH 8.1). After sonication (to achieve a smear of DNA 100–400 nucleotides in length, data not shown), 100 μl of supernatant was mixed with 900 μl of ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, 167 mm NaCl, pH 8.1). After preclearing, 1% of this reaction was saved for direct PCR amplification (input). The remainder of the reaction was incubated with rabbit anti-NF-κB p65 antibody, rabbit anti-NF-κB p50 antibody, or control rabbit IgG (Invitrogen). After isolation and washing of antibody containing complexes, DNA was extracted for PCR analysis. PCRs were performed using the following primers: 583 site 5′ GTGAGTGTGAGCTGAAGCGGG 3′ (−662 to −641 relative to the transcription start site) and 5′ GTTTCCCCTGAAGCCCGCGTG 3′ (−524 to −503); 251/272 site 5′ GCGGGTGATGTCAGCTCTC 3′ (−315 to −296) and 5′ GTGAGCGTGCGTGCGCGTGTG 3′ (−167 to −146). Products were resolved on 1% agarose gel with 100 bp DNA ladder (New England Biolabs) and visualized under UV light using a ChemiImager 4400 imaging system. Band intensities were analyzed densitometrically using the ImageJ 1.41o program. The band intensities were normalized to input (see above).
Glutamate uptake assay.
Ten days after lentiviral transduction of astrocytes, cells were rinsed twice with either Na+- or choline-containing buffer, and transport activity was measured in 12 well plates as described earlier (Sheldon et al., 2008). Na+-dependent glutamate (0.5 μm) transport activity was calculated as a difference in the amount of radioactivity accumulated in the presence or absence of Na+ by substituting equimolar concentrations of choline for Na+ in the assay. Cells were solubilized in 1 ml of 0.1 N NaOH; aliquots of this suspension were used for measurement of radioactivity and for analysis of protein (Lowry et al., 1951). Dihydrokainate (DHK)-sensitive sodium-dependent transport was determined by measuring glutamate uptake in the presence or absence of 300 μm DHK (Tocris Bioscience), a selective inhibitor of GLT-1-mediated glutamate uptake (for recent discussion, see Dabir et al., 2006). DHK-sensitive transport was normalized to the levels observed in astrocytes transduced with p65 and p50 because in some experiments there was no DHK-sensitive transport in control astrocytes, and normalization to 0 is not possible.
Statistical analysis.
Results are expressed as the mean ± SEM of independent experiments. Except where noted, one-way ANOVA followed by Bonferroni post hoc analysis was performed for all statistical comparisons using Instat 3 (GraphPad Software). A p value <0.05 was considered significant.
Results
Effects of neurons and EGF on NF-κB signaling in astrocytes
NF-κB has been implicated in EGF- or ceftriaxone-induced expression of GLT-1 (Sitcheran et al., 2005; Lee et al., 2008), but it is not known whether NF-κB contributes to neuron-dependent induction of GLT-1. Efforts in this area were significantly hampered by the fact that neurons in culture also express GLT-1 (Brooks-Kayal et al., 1998; Mennerick et al., 1998). In the present study, we took advantage of a recently generated transgenic BAC-GLT-1 promoter reporter mouse line. The BAC used for the generation of these mice carries 192 kb of chromosomal material surrounding the GLT-1 gene; it contains the entire coding region plus 45 kb upstream of the first exon and 24 kb downstream of the last exon. In these mice, expression of eGFP faithfully replicates in vivo expression of GLT-1, and GLT-1 is not expressed from the transgene, so there is no increase in GLT-1 expression (Regan et al., 2007). Layering cell suspensions from embryonic brain tissue from wild-type animals over these astrocytes results in induction of eGFP expression; the levels of eGFP can be used as a “surrogate” measure for activation of the GLT-1 promoter (Yang et al., 2009).
As we recently reported (Yang et al., 2009), no eGFP fluorescence was observed in astrocytes grown from BAC GLT-1 eGFP mice. Overlaying a cell suspension from embryonic cortex of wild-type rodent (rat) resulted in a continuous monolayer of green fluorescence (from the eGFP) and increased steady-state levels of eGFP expression (Fig. 1A). Before overlaying the cell suspension, astrocytes were polygonal. After overlaying the suspension, eGFP was observed in stellate-shaped cells, indicating that the astrocyte had undergone a change in morphology (data not shown). We also examined the effects of cell suspensions from embryonic cortex of wild-type mice; these suspensions also induced eGFP expression (Fig. 1A). We previously found that TTX abolishes induction of eGFP using organotypic cultures of spinal cord (Yang et al., 2009). To determine whether the effects observed in the current study can attributed to the neurons in the cell suspension (as opposed to other cell types), we added either tetrodotoxin (TTX) or NMDA to the cultures shortly after overlaying the mixed cell suspension; both of these treatments abolished induction of eGFP expression (Fig. 1A). Finally, we grew astrocytes from wild-type rats and replated them over astrocytes from the BAC GLT-1 eGFP mice, and found no evidence for induction of eGFP expression (Fig. 1A). Together with the fact that axons/presynaptic termini extending through a microfluidic chamber are sufficient to induce eGFP expression in astrocytes from these mice (Yang et al., 2009), these data provide strong evidence that induction of eGFP observed under these conditions is neuron dependent.
Figure 1.

Effects of neurons on NF-κB reporter activity and effects of dominant-negative NF-κB variants on induction of eGFP/GLT-1. A, Cortical astrocyte cultures from BAC GLT-1 eGFP transgenic mice were overlaid with a cell suspension from embryonic brain tissue obtained from wild-type animals (rats or mice) or a cell suspension containing astrocytes cultured from wild-type rat. In a subset of cultures, TTX (50 μm) or NMDA (1 mm) was added after 1 h. Cells were harvested after 10 d, and eGFP protein levels were analyzed. Data are representative of three independent experiments. B, Astrocyte cultures were infected with lentivirus engineered to express a NF-κB luciferase reporter and either dsRED (negative control) or dominant-negative constructs of IκBα (ΔN or SR). After 5 d, a fresh cell suspension containing neurons and astrocytes from wild-type E17 brain was overlaid on the top of infected astrocytes, and luciferase activity was measured after 10 d. Top, Summary of luciferase activity normalized to total protein content and expressed as a percentage of that measured in control astrocytes without neurons. Bottom, Western blot used to confirm expression of IκBα dominant-negative constructs in transduced astrocytes. Fifteen micrograms of cell lysate was loaded in each lane. Note that deletion of 36 aa in IκBα-ΔN construct results in faster migration of immunoreactive bands than in IκBα-SR. No other immunoreactive bands were observed in these blots. C, Top, Representative Western blot for eGFP protein. In all three experiments, eGFP was never detected in the absence of neurons and was observed in all experiments with neurons. Bottom, Graphic summary of data. D, GLT-1 protein levels were also analyzed in these same specimens. Top, A representative Western blot; bottom, summary of independent experiments. Data are the mean ± SEM of three independent experiments. *p < 0.05, ***p < 0.001 compared with corresponding astrocyte control; ###p < 0.001 compared with astrocytes infected with vector control plus neurons.
To determine whether NF-κB is activated in astrocytes under these conditions, astrocyte-enriched cultures prepared from the BAC GLT-1 eGFP mice were transduced using lentivirus with a NF-κB reporter construct that contains multiple copies of NF-κB consensus sequence. Overlaying a cell suspension from embryonic cortex of wild-type animals increased NF-κB promoter reporter activity (Fig. 1B). Since the reporter is only expressed in astrocytes, this shows that neuron-dependent increases in eGFP expression are associated with increased NF-κB signaling in astrocytes. To determine whether this activation of NF-κB can be attributed to neurons, we examined the effects of adding TTX or NMDA shortly after layering the cell suspension. We found that the neuron-dependent increase in NF-κB promoter reporter activity was reduced to 22% of control in TTX-treated samples and abolished (0% of control) in NMDA-treated samples (data are the mean of two independent experiments with values that were within 6% of the mean).
To determine whether activation of NF-κB in astrocytes is required for neuronal activation of GLT-1 transcription, astrocyte cultures derived from the BAC GLT-1 eGFP mice were first transduced using lentivirus with one of two different dominant-negative inhibitors of NF-κB signaling or control vector. The first, IκBα-SR, carries a mutation in both serine residues required for proteasomal degradation of IκBα. The second, IκBα-ΔN, lacks the first 36 aa of IκBα. Both of these constructs selectively block NF-κB signaling (Brockman et al., 1995). Expression of the dominant-negative variants of IκBα was verified by Western blotting (Fig. 1B, bottom). The effects of the dominant-negative variants on neuronal activation of the NF-κB reporter construct were also examined as a positive control. Both constructs completely blocked neuronal activation of the reporter construct (Fig. 1B). In these same samples, the dominant-negative constructs significantly reduced neuron-dependent induction of eGFP by 51 or 55%, but eGFP levels were significantly elevated compared with astrocytes alone (Fig. 1C). These data indicate that NF-κB contributes to promoter activation, but other signals also contribute to induction of eGFP. GLT-1 was also examined in these samples. As was observed with eGFP, the dominant-negative inhibitors of NF-κB signaling partially blocked neuronal induction of GLT-1 (Fig. 1D). Together these studies show that specifically blocking NF-κB signaling in astrocytes reduces induction of GLT-1.
Previously, we and others showed that EGF mimics the effects of neurons on GLT-1 protein and mRNA levels (Zelenaia et al., 2000; Sitcheran et al., 2005). These effects were blocked by a nonspecific inhibitor that blocks NF-κB signaling by blocking proteasomal function. To further test the hypothesis that EGF induces GLT-1 through an NF-κB-dependent mechanism, the effects of EGF on activation of the NF-κB reporter construct were examined in astrocytes. EGF increased luciferase activity in astrocytes transduced with NF-κB reporter construct using lentivirus (Fig. 2A). EGF did not activate this reporter construct in astrocytes transduced with either the IκBα-SR or the IκBα-ΔN dominant-negative inhibitors of NF-κB. In these same samples, EGF increased GLT-1 protein levels, but these dominant-negative variants did not completely block the EGF-induced increase in GLT-1 protein (Fig. 2B). These data are in agreement with studies showing that the IκBα-SR construct blocks the EGF-induced increase in GLT-1 mRNA in a human astroglioma cell line (Sitcheran et al., 2005).
Figure 2.
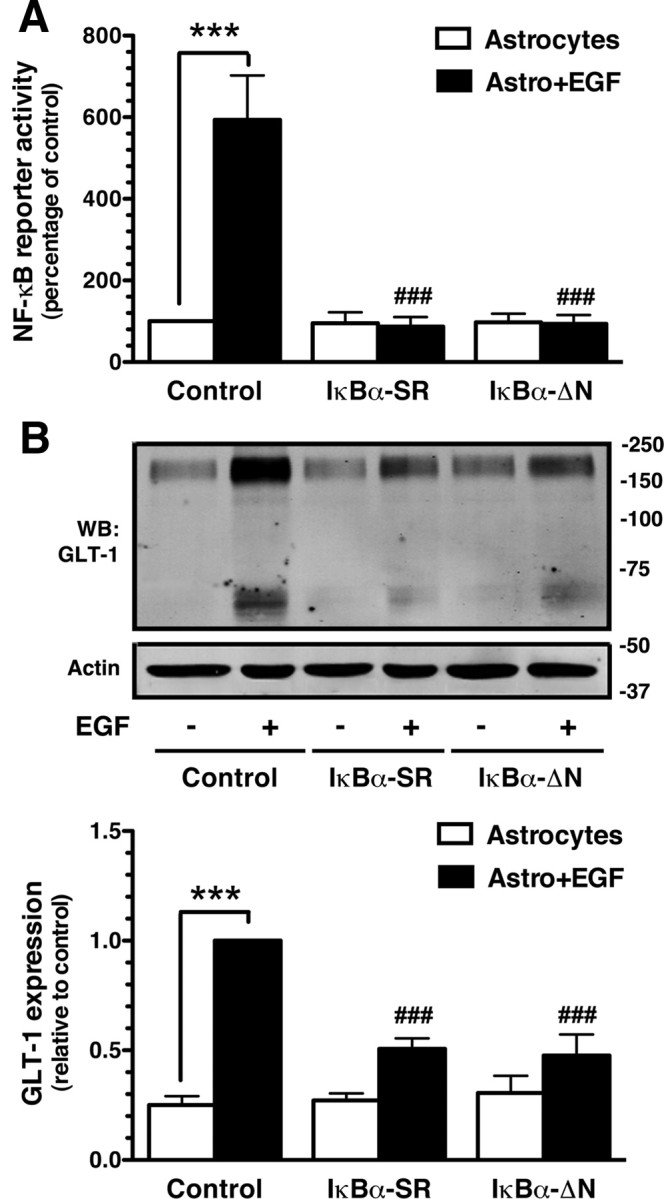
Effects of EGF on NF-κB reporter activity, and effects of dominant-negative NF-κB variants on EGF-dependent induction of GLT-1. Cortical astrocyte cultures were infected with lentiviral particles carrying NF-κB luciferase reporter and either dsRED or dominant-negative constructs of IκBα (ΔN or SR). Five days postinfection, cultures were treated with EGF (30 ng/ml) or vehicle for 7 d. A, NF-κB luciferase reporter activity. Luciferase activity was normalized to protein and expressed as a percentage of that measured in control astrocytes without neurons. B, GLT-1 protein levels were analyzed in the same samples. Fifteen micrograms of cell lysate was loaded in each lane. Bottom: Summary of quantification of GLT-1 protein levels normalized to actin and expressed relative to the levels observed in control astrocytes treated with EGF. Data are the mean ± SEM of four independent experiments. ***p < 0.001 compared with corresponding astrocyte control; ###p < 0.001 compared with astrocytes infected with vector control and treated with EGF.
Effects of neurons and EGF on regulation of 958 bp GLT-1 promoter reporter activity
Neuron-dependent activation of several constructs containing up to ∼2.5 kb of the proximal GLT-1 promoter were recently examined by us; we found that a 958 bp fragment was maximally activated (Yang et al., 2009). Within this region, there are several evolutionarily conserved putative transcription factor binding sites, including three potential NF-κB binding sites (Fig. 3). This 958 bp GLT-1 promoter reporter and variants harboring mutations of the putative NF-κB binding sites were engineered to control luciferase expression. These constructs were transfected into astrocytes. As might be expected from data showing neurons and other stimuli increase GLT-1 protein or mRNA relatively slowly (Schlag et al., 1998), neurons caused a slow, time-dependent increase in luciferase activity (Fig. 4A). This effect was abolished by mutation of the putative NF-κB sites at −583 or −251, but was not affected by mutation of the site at −272 (Fig. 4B). These observations show that neuronal activation requires two of the three putative NF-κB binding sites in the region of the GLT-1 promoter.
Figure 3.

Schematic representation of the GLT-1 gene intron–exon structure and its promoter. Exons are indicated as bold rectangular boxes. Location of various putative transcription factor binding sites within 958 bp GLT-1 promoter fragment are depicted. The size of the first intron is indicated. Sequences of the three putative NF-κB binding sites that are conserved in human, rat, and mouse are provided.
Figure 4.
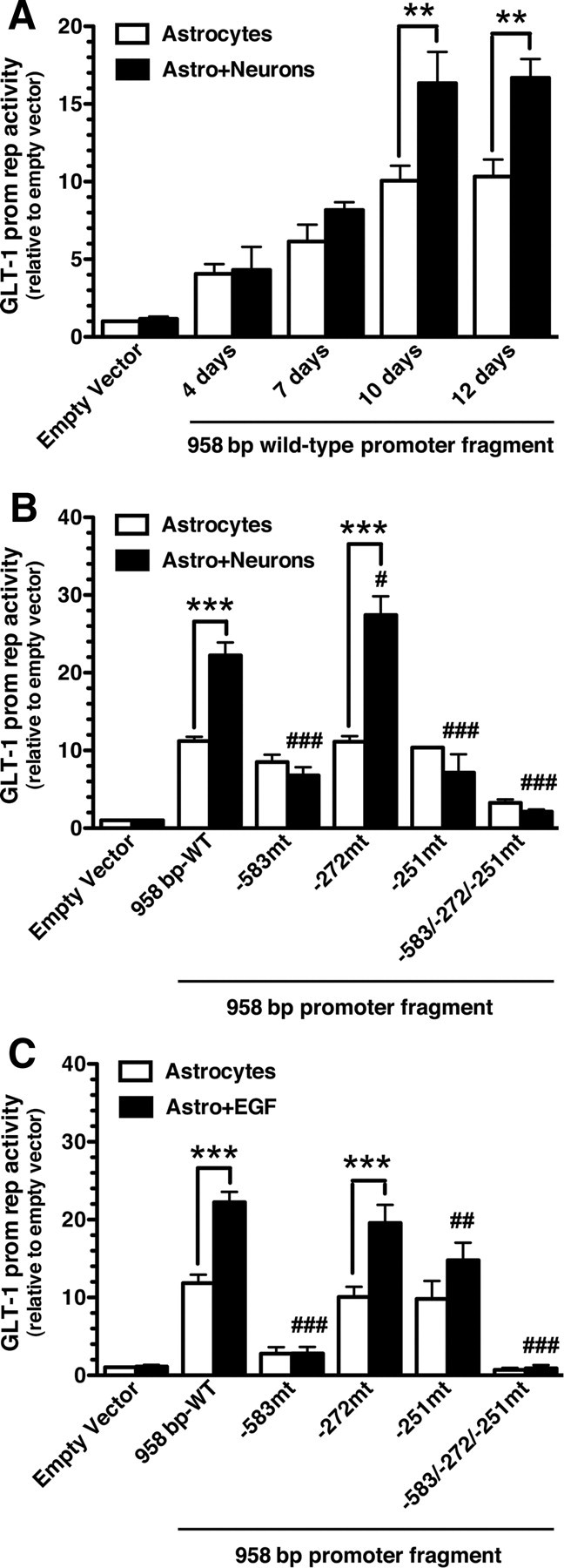
Neuron- and EGF-dependent activation of the GLT-1 promoter reporter. A, Cortical astrocyte cultures from BAC GLT1 eGFP mice were transfected (FuGENE) with a 958 bp wild-type GLT-1 promoter fragment. After 5 d, a fresh cell suspension containing neurons and astrocytes from embryonic brain of wild-type rodent was layered onto the transfected astrocytes, and luciferase activity was measured at different time points. Luciferase activity was normalized to β-galactosidase activity and expressed relative to that measured in astrocytes transfected with empty vector. B, C, Astrocytes were transfected (FuGENE) with either the wild-type 958 bp promoter fragment or with a promoter fragment harboring mutations of the various NF-κB binding sites. Five days post-transfection, cultures were overlaid with a cell suspension containing neurons and astrocytes (B) or treated with EGF (30 ng/ml) (C). Luciferase activity was measured after 10 d, normalized to β-galactosidase activity, and expressed relative to that measured in astrocytes transfected with empty vector. Data are the mean ± SEM of five independent experiments. **p < 0.01, ***p < 0.001 compared with corresponding astrocyte control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with astrocytes transfected with wild-type promoter fragment plus neurons or EGF.
As previously reported (Sitcheran et al., 2005), mutation of the site at −583 completely blocked EGF-dependent activation of this GLT-1 promoter reporter in primary astrocytes (Fig. 4C). We extended these analyses to other putative sites and found that mutation of the site at −251 also blocked EGF-dependent promoter activation. The NF-κB site at −272 has been implicated in ceftriaxone-dependent promoter activation (Lee et al., 2008). Interestingly mutation of this site had no effect on neuron- or EGF-dependent promoter activation (Fig. 4B,C). Mutation of all three putative NF-κB binding sites abolished neuron- or EGF-dependent activation of this reporter construct.
Effects of peptide-mediated inhibition of p50 nuclear translocation
NF-κB consists of homodimers and heterodimers of five structurally related proteins, p50, p52, p65 (RelA), RelB, and c-Rel. Although all of these subunits are observed in brain, it is widely accepted that p50/p65 heterodimers represent the most abundant form of active NF-κB in adult brain (for review, see Karin and Lin, 2002; Li and Verma, 2002; Meffert and Baltimore, 2005; Kaltschmidt and Kaltschmidt, 2009). To determine whether p50 contributes to neuronal induction of GLT-1, the effects of SN50, a cell-permeable peptide that specifically blocks the nuclear import of p50, were examined (Lin et al., 1995; Gutierrez et al., 2008). SN50 completely blocked neuronal activation of the 958 GLT-1 promoter reporter fragment; control peptide SM was without effect (Fig. 5A). These same samples were probed for expression of GLT-1; SN50 markedly attenuated neuronal induction of GLT-1 (Fig. 5B), providing pharmacological evidence that p50 contributes to promoter activation. Combined with the data presented in Figure 1, these data strongly suggest that NF-κB contributes to neuronal induction of GLT-1 and that NF-κB-independent effects may depend on elements that are outside the promoter reporter used in the current study.
Figure 5.
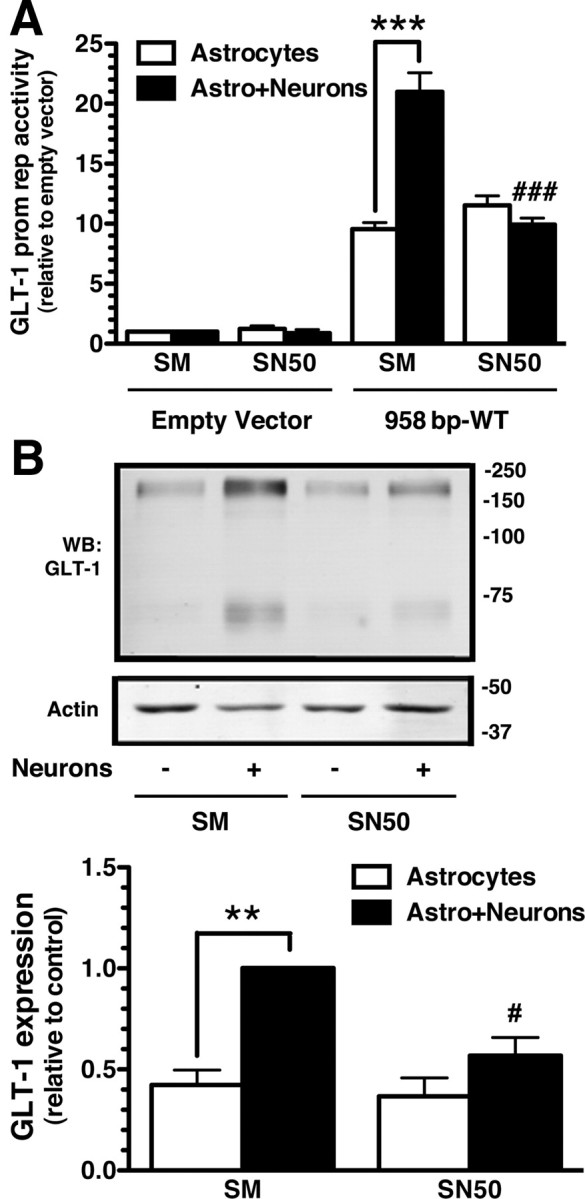
Effects of NF-κB inhibitor on neuron-dependent promoter activation or GLT-1 expression. A, Cortical astrocytes were transfected (FuGENE) with a 958 bp wild-type GLT-1 promoter fragment and treated with NF-κB-SN50 inhibitor peptide or SM inactive control peptide (20 μm). Five days post-transfection, cultures were overlaid with a cell suspension containing neurons and astrocytes from wild-type E17 brain. Cells were harvested, and luciferase activity was measured after 10 d. Luciferase activity was normalized to β-galactosidase activity and expressed relative to that measured in astrocytes transfected with empty vector. B, GLT-1 protein levels were analyzed in the same samples. Ten micrograms of cell lysate was loaded in each lane. GLT-1 protein levels were normalized to actin and expressed relative to the levels observed in astrocytes (with neurons) treated with control peptide. Data are the mean ± SEM of three independent experiments. **p < 0.01, ***p < 0.001 compared with corresponding astrocyte control; #p < 0.05, ###p < 0.001 compared with astrocytes with neurons and treated with control peptide.
Effects of NF-κB subunits on regulation of 958 bp EAAT2 promoter
Exogenous expression of wild-type NF-κB subunits is sufficient to activate transcription of NF-κB-responsive genes (for review, see Perkins, 2007). Therefore, the effects of exogenous expression of p50, p65, and p50 with p65 were examined in astrocytes prepared from BAC GLT-1 eGFP mice. Transfection of astrocytes with p50, p65, or p50 with p65 increased eGFP by >10-fold and GLT-1 by ∼2-fold (Fig. 6A,B). The transfection mixture included the GLT-1 promoter reporter constructs (Fig. 6C). As was observed with either EGF or neurons, mutation of the sites at −583 or −251 abolished p65-dependent promoter activation, and mutation of the site at −272 had no effect. In contrast, mutation of any of these three sites, including −272, abolished promoter activation induced by p50 or p50 with p65. These studies suggest that p65-dependent activation of the promoter does not depend on the NF-κB binding site located 272 nt upstream of the transcription start site, whereas p50-dependent activation is dependent on all three sites.
Figure 6.

Effects of exogenous expression of NF-κB subunits on eGFP expression, GLT-1 expression, and activation of the GLT-1 promoter reporter. A, B, Cortical astrocytes were transfected (FuGENE) with p50, p65, p50 with p65, or dsRED plus the GLT-1 promoter reporter. After 10 d, the levels of eGFP (A) or GLT-1 (B) were analyzed by Western blot. Fifteen micrograms of cell lysate was loaded in each lane. Bottom, Summary of quantification of eGFP and GLT-1 protein levels normalized to actin and expressed relative to the levels observed in p65, p50, or p65 with p50 transfected astrocytes, respectively. C, Luciferase activity was measured, normalized to protein, and expressed relative to that observed in astrocytes transfected with empty vector. Data are the mean ± SEM of three to five independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with corresponding dsRED transfected control; ###p < 0.001 compared with astrocytes transfected with wild-type promoter fragment and either of the NF-κB subunits.
Interactions of p50 or p65 with the putative NF-κB binding sites ex vivo
EMSAs and supershift assays were used to test for interactions of p50 or p65 with biotin-labeled, double-stranded oligonucleotides corresponding to the sequence surrounding the sites at −583 and −251. The effects of incubation of nuclear extracts from adult cortex were examined as were incubation with anti-p50 or p65 antibodies. Because of the proximity of the regions at −272 and −251, the oligonucleotide used surrounds both sites. An oligonucleotide containing a known p50/p65 interaction domain was included as a positive control. Incubation of the control oligonucleotide or the oligonucleotides corresponding to the −583 or −272/−251 GLT-1 promoter regions with nuclear extracts from cortex slowed migration of the oligonucleotides (Fig. 7, labeled shift). These shifts were not observed when the incubation included 200-fold excess unlabeled oligonucleotide or when the putative NF-κB binding sites were mutated, strongly suggesting that this shift is specific and dependent upon these specific sites. As was observed earlier (Sitcheran et al., 2005), incubation with antibodies specific for p50 or p65 caused an additional shift in the migration of the oligonucleotide that included the site at −583 (labeled supershift). Using the oligonucleotide that included both the −272 and −251 sites, incubation with nuclear extracts caused a shift that was abolished when both sites were mutated. Anti-p65 or anti-p50 antibodies caused supershifts of this oligonucleotide. Interestingly, mutation of the site at −251 abolished the supershift observed with anti-p65 antibody. This suggests that p65 does not interact with the site at −272 but does interact with the site at −251. We also found that mutation of the site at −272 had no effect on the supershift induced by either the anti-p50 or the anti-p65 antibodies, providing complementary evidence that p65 selectively interacts with the −251 site. Notice that mutation of the site at −251 did not abolish the supershift observed with anti-p50 antibody; this provides strong evidence that p50 interacts with the site at −272. These data are consistent with the effects of mutation of specific sites in the promoter region mentioned above (Fig. 6C) that show that p65-dependent activation of the promoter does not depend on the site at −272. Since p65 is not thought to function as a homomultimer (Meffert et al., 2003), the simplest explanation of these data is that the sites at −583 or −251 bind p50/p65 heterodimers ex vivo and that the site at −272 binds either p50 homomers or heteromers of p50 with subunits other than p65.
Figure 7.

Analysis of p50 or p65 binding ex vivo. EMSAs were performed using oligonucleotide probes designed surrounding the −583 or −272/−251 NF-κB binding sites from the GLT-1 promoter with nuclear extracts prepared from adult mouse cortex. Analyses of binding were also performed using oligonucleotides containing mutations in the putative NF-κB binding sites (see Materials and Methods). For the −272/−251 sites, binding to oligonucleotides with both sites mutated or with just the −251 or −272 site mutated was examined. An oligonucleotide containing known κB consensus binding sequences (Control Oligo) was included as a positive control; these oligonucleotides migrate out of the bottom of the gel because they are approximately half the length of the other nucleotides used in these studies. Shifted and supershifted oligonucleotide complexes are indicated with single and double arrows, respectively. All of the samples from the analyses of the −272/−251 oligonucleotide binding were prepared at the same time, but the control oligo was analyzed on a different gel because of the number of samples. These results are representative of between three or four independent experiments.
Interactions of p50 or p65 with the putative NF-κB binding sites in vivo
To test for interactions of p50 and/or p65 with the native GLT-1 promoter, nuclear DNA and interacting proteins were cross-linked in tissues from wild-type mice. After immunoprecipitation of p50 or p65 the DNA–protein complexes were un-cross-linked, and PCR with appropriate primers was used to test for the presence of the sites at −583 or −272/−251. As a positive control, PCRs were also conducted with a small volume of the precleared supernatant before immunoprecipitation (input). Using cortical tissue, PCR with primers surrounding either the −583 or the −272/−251 regions of the promoter generated fragments of the appropriate length (Fig. 8A). PCRs with IgG control immunoprecipitations support the specificity of these pull downs. For these experiments, DNA was sheared by sonication to a range of 100–400 bp. Therefore, it is theoretically possible that both the −583 and −272/−251 regions are present within a single complex. To test this possibility, the primers surrounding both sites were used for PCR amplification of the immunoprecipitated products (Fig. 8B). No signal was observed in the p50 or p65 immunoprecipitates. Intact (unsheared) genomic material was used as a positive control for this PCR. In fact, very low signal was observed in the sheared DNA, as might be expected given the small sizes of genomic DNA used in these studies. These data provide strong evidence that p50 and p65 interact with the −583 region of the promoter and also independently interact with the −272/−251 region of the promoter in vivo.
Figure 8.
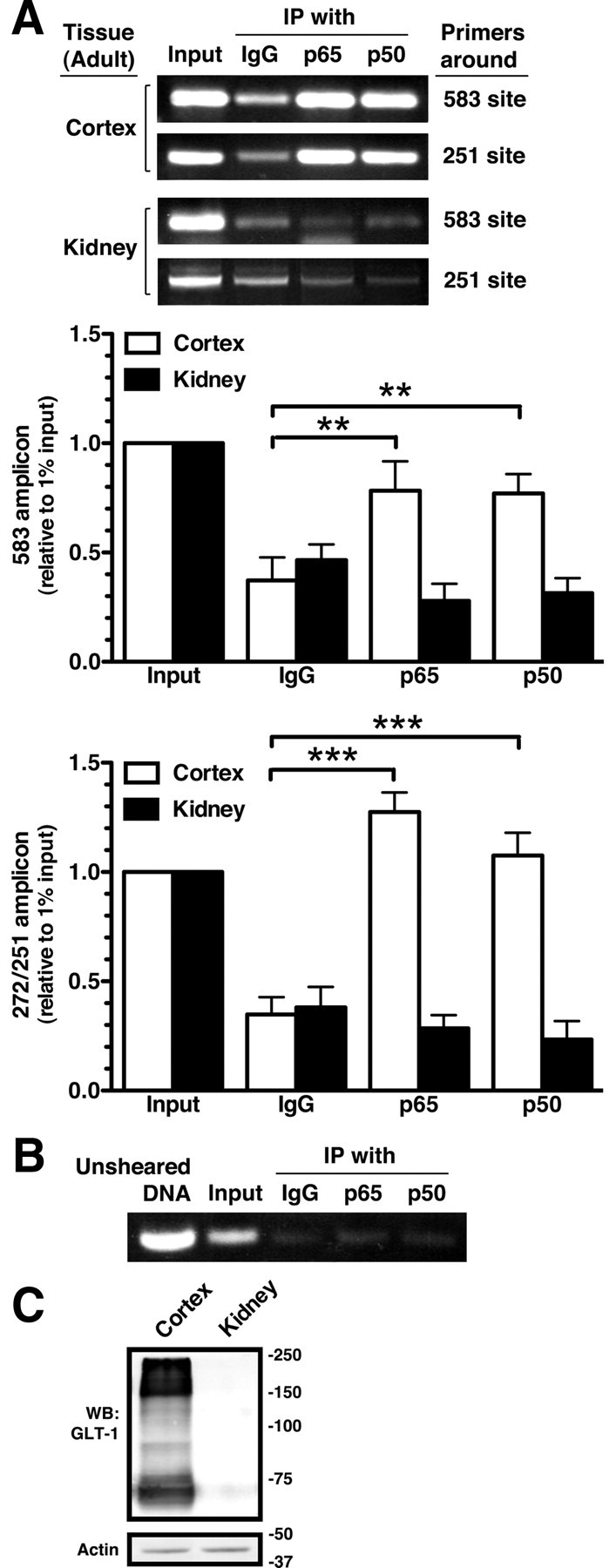
Analysis of interaction of p65 and p50 with GLT-1 promoter in vivo by ChIP. A, Nuclear extracts from adult mouse cortex or kidney were treated with formaldehyde to cross-link DNA binding proteins to chromatin and then subjected to ChIP with control IgG, anti-p65, or anti-p50 antibodies. Eluted DNA fragments were subjected to PCR amplification with primers surrounding the −583 and −272/251 putative NF-κB binding sites. One percent of the sheared chromatin was used as a positive control (input) for the PCR. Bottom, Summary of quantification of amplicons generated by PCR analysis using primers surrounding the −583 or the −272/−251 sites. Intensities of amplified fragments were normalized and expressed relative to that observed in 1% input. B, Immunoprecipitated DNAs from the same cortical samples were amplified using primers that span both NF-κB binding sites (−583 and −272/−251). Unsheared DNA served as a positive control for this primer pair. As expected based on the average size of the DNA after shearing, the signal from the input is much lower than that observed in the other reactions. C, GLT-1 protein levels were analyzed in the same samples. Fifteen micrograms of cell lysate was loaded in each lane. These results are representative of at least three independent experiments. **p < 0.01, ***p < 0.001 compared with the amplified fragment generated by normal IgG.
These ChIP studies were extended to test for an interaction of p50 and/or p65 with these regions of the GLT-1 promoter in kidney, a tissue that expresses little or no GLT-1 protein. As was previously reported (Shayakul et al., 1997), GLT-1 was not detected in kidney by Western blot (Fig. 8C). No specific interaction between either p50 or p65 and sequences corresponding to the GLT-1 promoter were observed in immunoprecipitates from kidney DNA (Fig. 8A). These data are consistent with the hypothesis that interaction of p50 and/or p65 with these promoter regions contributes to induction of GLT-1.
Effects of p50 and p65 on Na+-dependent l-[3H]glutamate transport
The studies above implicate both p50 and p65 in the regulation of GLT-1 expression in vitro and in vivo. To determine whether the effects of p50 and p65 on eGFP and GLT-1 levels are associated with an increase in GLT-1-mediated uptake, we measured total Na+-dependent uptake and dihydrokainate-sensitive transport in astrocytes infected with lentiviral vectors engineered to express p50 and p65 (100 ng of p24 of each virus/ml). We found that transduction with p50 and p65 had a small and nearly significant effect on total Na+-dependent transport (0.66 ± 0.09 nmol/mg/min in control-dsRED-transduced astrocytes, 0.80 ± 0.15 nmol/mg/min in astrocytes transduced with p65 and p50; p = 0.07 by paired t test; n = 4). As previously reported, we observed essentially no inhibition with DHK in control (mean 7.8 ± 2.78% inhibition; not significant). Lentiviral transduction of astrocytes with p65 and p50 caused a significant (4.4-fold) increase in DHK-sensitive transport compared with control (0.23 ± 0.14 expressed as a fraction of that observed in astrocytes transduced with p65 and p50; n = 4; p = 0.005 by one-sample t test). These studies strongly suggest that exogenous expression of p50 with p65 is sufficient to increase GLT-1-mediated activity.
Discussion
Genetic deletion or knock-down studies demonstrate that GLT-1 has a critical role in normal brain function (Rothstein et al., 1996; Tanaka et al., 1997). Over a decade ago, it was shown that astrocytes in culture express little (or no) GLT-1 protein, but that coculturing with neurons induces astrocytic expression (Gegelashvili et al., 1997; Swanson et al., 1997; Schlag et al., 1998). NF-κB signaling was linked to EGF- or dibutyryl cAMP-dependent regulation of GLT-1 by showing that inhibitors of proteasomal function block induction of GLT-1 in astrocyte cultures; this approach has obvious limitations related to the broad effects of inhibition of proteasomal function (Zelenaia et al., 2000). Sitcheran et al. (2005) demonstrated that IκBα-SR, a dominant-negative construct of NF-κB signaling, blocks EGF-dependent activation of GLT-1 expression in a human astroglioma. Using either pharmacological approaches or this dominant-negative construct, NF-κB signaling has been linked to ceftriaxone-, brain-derived neurotrophic factor-, or β-amyloid-dependent upregulation of GLT-1 (Rodriguez-Kern et al., 2003; Lee et al., 2008). NF-κB signaling has also been linked to amitriptyline-dependent upregulation of GLT-1 in vivo (Tai et al., 2008).
Despite these advances, it is not known whether NF-κB signaling might contribute to neuron-dependent induction of GLT-1. Using a relatively simple strategy, we provide several lines of evidence that activation of NF-κB signaling in astrocytes contributes to this neuron-dependent transcriptional activation of GLT-1 through an interaction of NF-κB subunits with specific sites in the GLT-1 promoter. First, when an NF-κB reporter construct is specifically introduced into astrocytes, overlaying neurons increases reporter activity. Second, specific expression of dominant-negative inhibitors of NF-κB signaling in astrocytes attenuates neuron-dependent induction of eGFP. Third, exogenous expression of p65 and/or p50 increases eGFP expression in astrocytes prepared from the BAC GLT-1 eGFP transgenic mice and also increases DHK-sensitive (GLT-1-mediated) transport activity in astrocytes. Forth, mutation of putative NF-κB binding sites in a GLT-1 promoter abolishes neuron-dependent activation of a GLT-1 promoter reporter. Finally, binding of p50 and p65 to these specific sites was detected both in vitro and in vivo. Together, these studies strongly suggest that the effects of NF-κB are dependent upon a direct interaction with elements in the GLT-1 promoter rather indirect through transcriptional regulation of other trans factors. Consistent with earlier findings that link glutamatergic signaling to induction of GLT-1 (Gegelashvili et al., 1996; Yang et al., 2009), glutamate activates nuclear translocation of p50 and p65 in primary astrocytes (Caccamo et al., 2005). The fact that induction of eGFP expression is not completely blocked by dominant-negative inhibitors that completely block NF-κB signaling strongly suggests that other signals also contribute to neuronal induction of GLT-1, but this was not explored.
As was observed previously (Sitcheran et al., 2005), we found that EGF-dependent induction of GLT-1 was blocked by dominant-negative inhibitors of NF-κB signaling. Also in agreement with this study, we found that mutation of the putative NF-κB binding site at −583 blocked EGF-dependent activation of a GLT-1 promoter reporter. We report that mutation of the NF-κB binding site at −251 also abolishes EGF-dependent activation of a GLT-1 promoter reporter. This same group showed that ceftriaxone-dependent induction of GLT-1 was dependent upon activation of NF-κB signaling and promoter reporter activation was blocked by mutation of the NF-κB binding site at −272 (Lee et al., 2008). We found that neuron-dependent activation of the promoter reporter was abolished by mutation of sites at either −583 or −251 but not −272, similar to that observed with EGF. Interestingly, we found that p50- but not p65-dependent activation of the promoter was abolished by mutation of the −272 NF-κB binding site. We also found evidence that p50 interacts with the site at −272, but that p65 does not. This contradicts an earlier study suggesting that p65 can interact with the site at −272 (Lee et al., 2008). The reason for this discrepancy is not clear at this time, but it is possible that the differences are related to the different model systems used for these analyses. Regardless, these data suggest that different NF-κB binding sites in the GLT-1 promoter are activated by different signals, ceftriaxone activating one of these signaling pathways and neurons/EGF activating a distinct pathway. This would not be unprecedented, as different stimuli have been shown to activate translocation of different NF-κB dimers in the same cellular system (Tong et al., 2004). It should also be noted that tumor necrosis factor-α downregulates GLT-1 expression by a pathway dependent upon n-myc and NF-κB activation using a distinct signaling pathway (Sitcheran et al., 2005).
Until recently essentially nothing was known about the specific transcriptional mechanisms that contribute to neuron-dependent induction of GLT-1. We recently demonstrated that mutation of a site ∼680 nt upstream of the transcription start site also abolished neuron-dependent activation of the 958 bp GLT-1 promoter fragment (Yang et al., 2009). In this study, κB motif binding phosphoprotein (KBBP) was found to interact with the site ∼680 nt upstream of the transcription start site, and exogenous expression of KBBP in astrocytes increased GLT-1 expression. As was observed with inhibition of NF-κB signaling, impairing KBBP function (with shRNA interference) attenuated, but did not abolish induction of GLT-1 (or eGFP). In this same study, we also showed that mutation of the −583 site abolished neuron-dependent activation of this promoter reporter (Fig. 3D, mutant 3). Regrettably, in this earlier study, we included text indicating that mutation of NF-κB binding sites had no dramatic influence on neuron-stimulated promoter activation. Despite this error in the text of the earlier study, we conclude that mutation of any one of three different sites within this region is sufficient to abolish neuron-dependent activation. In future experiments, it will be interesting to study the interdependence of these protein interactions with the GLT-1 promoter.
NF-κB signaling was originally linked to a variety of pathologic conditions including inflammation, infection, injury, and stress (Nonaka et al., 1999; Schwaninger et al., 1999; Yu et al., 1999; Mattson and Camandola, 2001). In fact, there is evidence that NF-κB signaling contributes to development and learning/memory (for review, see Kaltschmidt and Kaltschmidt, 2009). Although frequently thought of as a transient transcriptional activator, transgenic lines of mice engineered to express β-galactosidase under the control of a NF-κB promoter demonstrates constitutive activation in the brain, particularly neurons (for review, see Kaltschmidt and Kaltschmidt, 2009). All of the NF-κB family members have been observed in the CNS, but the most commonly active complexes are p50/p50 homodimers and p50/p65 heterodimers (Naumann and Scheidereit, 1994; Meffert et al., 2003). In the present study, both p50 and p65 have been linked to GLT-1 promoter regions required for neuron-induced transcription. Mice genetically deleted of either p50 or p65 have been described. Knockout of p65 results in death during embryogenesis (Beg et al., 1995; Meffert et al., 2003); therefore, it is not possible to determine whether they express a similar phenotype to that observed in GLT-1 knock-out mice, including seizures and susceptibility to ischemic insults (Tanaka et al., 1997). Mice deleted of p50 have not been examined for GLT-1 expression, but the fact that these animals survive and appear relatively normal in adulthood (Denis-Donini et al., 2008), suggests that subunits other than p50 can support expression of GLT-1.
In summary, both in vitro and in vivo studies strongly suggest that astrocytic GLT-1 expression is dynamically regulated by neurons/neuronal activity (see Introduction). Furthermore, decreases in GLT-1 expression are observed in several neurodegenerative disorders (for review, see Sheldon and Robinson, 2007). The identification of transcriptional events involved in regulation of GLT-1 expression may lead to the development of therapeutic strategies to limit that loss of GLT-1 and neurodegeneration.
Footnotes
This work was supported by NIH Grant NS36465 to M.B.R. and J.D.R. We thank Dr. Dean Ballard (Vanderbilt University School of Medicine) for the dominant-negative constructs of IκBα; Olga Zelanaia and Janice Watch for subcloning some of these cDNAs into the lentiviral backbone; and Dr. Judy Grinspan (Children's Hospital of Philadelphia) for providing the A2B5 hybridoma supernatant. We thank members of the Robinson laboratory for their advice and suggestion with this project.
References
- Beart PM, O'Shea RD. Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150:5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Munir M, Jin H, Robinson MB. The glutamate transporter, GLT-1, is expressed in cultured hippocampal neurons. Neurochem Int. 1998;33:95–100. doi: 10.1016/s0197-0186(98)00018-7. [DOI] [PubMed] [Google Scholar]
- Caccamo D, Campisi A, Marini H, Adamo EB, Li Volti G, Squadrito F, Ientile R. Glutamate promotes NF-kappaB pathway in primary astrocytes: protective effects of IRFI 016, a synthetic vitamin E analogue. Exp Neurol. 2005;193:377–383. doi: 10.1016/j.expneurol.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Chu K, Lee ST, Sinn DI, Ko SY, Kim EH, Kim JM, Kim SJ, Park DK, Jung KH, Song EC, Lee SK, Kim M, Roh JK. Pharmacological induction of ischemic tolerance by glutamate transporter-1 (EAAT2) upregulation. Stroke. 2007;38:177–182. doi: 10.1161/01.STR.0000252091.36912.65. [DOI] [PubMed] [Google Scholar]
- Dabir DV, Robinson MB, Swanson E, Zhang B, Trojanowski JQ, Lee VM, Forman MS. Impaired glutamate transport in a mouse model of tau pathology in astrocytes. J Neurosci. 2006;26:644–654. doi: 10.1523/JNEUROSCI.3861-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Denis-Donini S, Dellarole A, Crociara P, Francese MT, Bortolotto V, Quadrato G, Canonico PL, Orsetti M, Ghi P, Memo M, Bonini SA, Ferrari-Toninelli G, Grilli M. Impaired adult neurogenesis associated with short-term memory defects in NF-κB p50-deficient mice. J Neurosci. 2008;28:3911–3919. doi: 10.1523/JNEUROSCI.0148-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci. 1997;17:8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganel R, Ho T, Maragakis NJ, Jackson M, Steiner JP, Rothstein JD. Selective up-regulation of the glial Na+-dependent glutamate transporter GLT1 by a neuroimmunophilin ligand results in neuroprotection. Neurobiol Dis. 2006;21:556–567. doi: 10.1016/j.nbd.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Civenni G, Racagni G, Danbolt NC, Schousboe I, Schousboe A. Glutamate receptor agonists up-regulate glutamate transporter GLAST in astrocytes. Neuroreport. 1996;8:261–265. doi: 10.1097/00001756-199612200-00052. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem. 1997;69:2612–2615. doi: 10.1046/j.1471-4159.1997.69062612.x. [DOI] [PubMed] [Google Scholar]
- Genoud C, Quairiaux C, Steiner P, Hirling H, Welker E, Knott GW. Plasticity of astrocytic coverage and glutamate transporter expression in adult mouse cortex. PLoS Biol. 2006;4:e343. doi: 10.1371/journal.pbio.0040343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Martin LJ, Rothstein JD. Regional deafferentation down-regulates subtypes of glutamate transporter proteins. J Neurochem. 1995;65:2800–2803. doi: 10.1046/j.1471-4159.1995.65062800.x. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Rothstein JD, Price DL, Martin LJ. Fimbria-fornix transections selectively down-regulate subtypes of glutamate transporter and glutamate receptor proteins in septum and hippocampus. J Neurochem. 1996;67:1208–1216. doi: 10.1046/j.1471-4159.1996.67031208.x. [DOI] [PubMed] [Google Scholar]
- González MI, Krizman-Genda E, Robinson MB. Caveolin-1 regulates the delivery and endocytosis of the glutamate transporter, excitatory amino acid carrier 1. J Biol Chem. 2007a;282:29855–29865. doi: 10.1074/jbc.M704738200. [DOI] [PubMed] [Google Scholar]
- González MI, Susarla BT, Fournier KM, Sheldon AL, Robinson MB. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J Neurochem. 2007b;103:1917–1931. doi: 10.1111/j.1471-4159.2007.04881.x. [DOI] [PubMed] [Google Scholar]
- Gutierrez H, O'Keeffe GW, Gavaldà N, Gallagher D, Davies AM. Nuclear factor κB signaling either stimulates or inhibits neurite growth depending on the phosphorylation status of p65/RelA. J Neurosci. 2008;28:8246–8256. doi: 10.1523/JNEUROSCI.1941-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honoré T, Nielsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- Hein C, Horváth E, Kugler P. Glutamate transporter expression in astrocytes of the rat dentate gyrus following lesion of the entorhinal cortex. Eur J Neurosci. 2001;13:1839–1848. doi: 10.1046/j.0953-816x.2001.01559.x. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Kaltschmidt C. NF-kappaB in the nervous system. Cold Spring Harb Perspect Biol. 2009;1:a001271. doi: 10.1101/cshperspect.a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Kosugi T, Tanaka M, Nakajima T, Yamada T. Reversed operation of glutamate transporter GLT-1 is crucial to the development of preconditioning-induced ischemic tolerance of neurons in neuron/astrocyte co-cultures. Glia. 2005;49:349–359. doi: 10.1002/glia.20114. [DOI] [PubMed] [Google Scholar]
- Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem. 2008;283:13116–13123. doi: 10.1074/jbc.M707697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Lehre KP, Walaas SI, Storm-Mathisen J, Danbolt NC. Down-regulation of glial glutamate transporters after glutamatergic denervation in the rat brain. Eur J Neurosci. 1995;7:2036–2041. doi: 10.1111/j.1460-9568.1995.tb00626.x. [DOI] [PubMed] [Google Scholar]
- Li LB, Toan SV, Zelenaia O, Watson DJ, Wolfe JH, Rothstein JD, Robinson MB. Regulation of astrocytic glutamate transporter expression by Akt: evidence for a selective transcriptional effect on the GLT-1/EAAT2 subtype. J Neurochem. 2006;97:759–771. doi: 10.1111/j.1471-4159.2006.03743.x. [DOI] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Li S, Mallory M, Alford M, Tanaka S, Masliah E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol. 1997;56:901–911. doi: 10.1097/00005072-199708000-00008. [DOI] [PubMed] [Google Scholar]
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosenberg NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, Nelson N, Leite JP, Chimelli L, Born DE, Sakamoto AC, Assirati JA, Fried I, Peacock WJ, Ojemann GA, Adelson PD. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. 1999;52:453–472. doi: 10.1212/wnl.52.3.453. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107:247–254. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May MJ, Ghosh S. Rel/NF-kappa B and I kappa B proteins: an overview. Semin Cancer Biol. 1997;8:63–73. doi: 10.1006/scbi.1997.0057. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Baltimore D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005;28:37–43. doi: 10.1016/j.tins.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Dhond RP, Benz A, Xu W, Rothstein JD, Danbolt NC, Isenberg KE, Zorumski CF. Neuronal expression of the glutamate transporter GLT-1 in hippocampal microcultures. J Neurosci. 1998;18:4490–4499. doi: 10.1523/JNEUROSCI.18-12-04490.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann M, Scheidereit C. Activation of NF-kappa B in vivo is regulated by multiple phosphorylations. EMBO J. 1994;13:4597–4607. doi: 10.1002/j.1460-2075.1994.tb06781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka M, Chen XH, Pierce JE, Leoni MJ, McIntosh TK, Wolf JA, Smith DH. Prolonged activation of NF-kappaB following traumatic brain injury in rats. J Neurotrauma. 1999;16:1023–1034. doi: 10.1089/neu.1999.16.1023. [DOI] [PubMed] [Google Scholar]
- Parry GC, Mackman N. A set of inducible genes expressed by activated human monocytic and endothelial cells contain kappa B-like sites that specifically bind c-Rel-p65 heterodimers. J Biol Chem. 1994;269:20823–20825. [PubMed] [Google Scholar]
- Perego C, Vanoni C, Bossi M, Massari S, Basudev H, Longhi R, Pietrini G. The GLT-1 and GLAST glutamate transporters are expressed on morphologically distinct astrocytes and regulated by neuronal activity in primary hippocampal cocultures. J Neurochem. 2000;75:1076–1084. doi: 10.1046/j.1471-4159.2000.0751076.x. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Poitry-Yamate CL, Vutskits L, Rauen T. Neuronal-induced and glutamate-dependent activation of glial glutamate transporter function. J Neurochem. 2002;82:987–997. doi: 10.1046/j.1471-4159.2002.01075.x. [DOI] [PubMed] [Google Scholar]
- Rao VL, Başkaya MK, Doğan A, Rothstein JD, Dempsey RJ. Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J Neurochem. 1998;70:2020–2027. doi: 10.1046/j.1471-4159.1998.70052020.x. [DOI] [PubMed] [Google Scholar]
- Rawls SM, Tallarida R, Robinson W, Amin M. The beta-lactam antibiotic, ceftriaxone, attenuates morphine-evoked hyperthermia in rats. Br J Pharmacol. 2007;151:1095–1102. doi: 10.1038/sj.bjp.0707309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM, Bergles DE, Rothstein JD. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci. 2007;27:6607–6619. doi: 10.1523/JNEUROSCI.0790-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MB. The family of sodium-dependent glutamate transporters: a focus on the GLT-1/EAAT2 subtype. Neurochem Int. 1998;33:479–491. doi: 10.1016/s0197-0186(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Kern A, Gegelashvili M, Schousboe A, Zhang J, Sung L, Gegelashvili G. Beta-amyloid and brain-derived neurotrophic factor, BDNF, up-regulate the expression of glutamate transporter GLT-1/EAAT2 via different signaling pathways utilizing transcription factor NF-kappaB. Neurochem Int. 2003;43:363–370. doi: 10.1016/s0197-0186(03)00023-8. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Saura J. Microglial cells in astroglial cultures: a cautionary note. J Neuroinflammation. 2007;4:26. doi: 10.1186/1742-2094-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, Robinson MB. Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol. 1998;53:355–369. doi: 10.1124/mol.53.3.355. [DOI] [PubMed] [Google Scholar]
- Schwaninger M, Sallmann S, Petersen N, Schneider A, Prinz S, Libermann TA, Spranger M. Bradykinin induces interleukin-6 expression in astrocytes through activation of nuclear factor-kappaB. J Neurochem. 1999;73:1461–1466. doi: 10.1046/j.1471-4159.1999.0731461.x. [DOI] [PubMed] [Google Scholar]
- Shayakul C, Kanai Y, Lee WS, Brown D, Rothstein JD, Hediger MA. Localization of the high-affinity glutamate transporter EAAC1 in rat kidney. Am J Physiol. 1997;273:F1023–F1029. doi: 10.1152/ajprenal.1997.273.6.F1023. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon AL, González MI, Krizman-Genda EN, Susarla BT, Robinson MB. Ubiquitination-mediated internalization and degradation of the astroglial glutamate transporter, GLT-1. Neurochem Int. 2008;53:296–308. doi: 10.1016/j.neuint.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitcheran R, Gupta P, Fisher PB, Baldwin AS. Positive and negative regulation of EAAT2 by NF-kappaB: a role for N-myc in TNFalpha-controlled repression. EMBO J. 2005;24:510–520. doi: 10.1038/sj.emboj.7600555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susarla BT, Seal RP, Zelenaia O, Watson DJ, Wolfe JH, Amara SG, Robinson MB. Differential regulation of GLAST immunoreactivity and activity by protein kinase C: evidence for modification of amino and carboxy termini. J Neurochem. 2004;91:1151–1163. doi: 10.1111/j.1471-4159.2004.02791.x. [DOI] [PubMed] [Google Scholar]
- Sutherland ML, Delaney TA, Noebels JL. Glutamate transporter mRNA expression in proliferative zones of the developing and adult murine CNS. J Neurosci. 1996;16:2191–2207. doi: 10.1523/JNEUROSCI.16-07-02191.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997;17:932–940. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YH, Tsai RY, Wang YH, Cherng CH, Tao PL, Liu TM, Wong CS. Amitriptyline induces nuclear transcription factor-kappaB-dependent glutamate transporter upregulation in chronic morphine-infused rats. Neuroscience. 2008;153:823–831. doi: 10.1016/j.neuroscience.2008.02.055. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tong X, Yin L, Washington R, Rosenberg DW, Giardina C. The p50–p50 NF-kappaB complex as a stimulus-specific repressor of gene activation. Mol Cell Biochem. 2004;265:171–183. doi: 10.1023/b:mcbi.0000044394.66951.4d. [DOI] [PubMed] [Google Scholar]
- Watson DJ, Wolfe JH. Lentiviral vectors for gene transfer to the central nervous system: applications in lysosomal storage disease animal models. Methods Mol Med. 2003;76:383–403. doi: 10.1385/1-59259-304-6:383. [DOI] [PubMed] [Google Scholar]
- Weller ML, Stone IM, Goss A, Rau T, Rova C, Poulsen DJ. Selective overexpression of excitatory amino acid transporter 2 (EAAT2) in astrocytes enhances neuroprotection from moderate but not severe hypoxia-ischemia. Neuroscience. 2008;155:1204–1211. doi: 10.1016/j.neuroscience.2008.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Kawahara K, Kosugi T, Tanaka M. Nitric oxide produced during sublethal ischemia is crucial for the preconditioning-induced down-regulation of glutamate transporter GLT-1 in neuron/astrocyte co-cultures. Neurochem Res. 2006;31:49–56. doi: 10.1007/s11064-005-9077-4. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gozen O, Watkins A, Lorenzini I, Lepore A, Gao Y, Vidensky S, Brennan J, Poulsen D, Won Park J, Li Jeon N, Robinson MB, Rothstein JD. Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron. 2009;61:880–894. doi: 10.1016/j.neuron.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of the p50 subunit of nuclear factor-κB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenaia O, Schlag BD, Gochenauer GE, Ganel R, Song W, Beesley JS, Grinspan JB, Rothstein JD, Robinson MB. Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-κB. Mol Pharmacol. 2000;57:667–678. doi: 10.1124/mol.57.4.667. [DOI] [PubMed] [Google Scholar]