

Abstract
Product yields for the reactions of (R)-glyceraldehyde 3-phosphate (GAP) in D2O at pD 7.9 catalyzed by wildtype triosephosphate isomerase from Trypanosoma brucei brucei (Tbb TIM) and a monomeric variant (monoTIM) of this wildtype enzyme were determined by 1H NMR spectroscopy, and were compared with the yields determined in earlier work for the reactions catalyzed by TIM from rabbit and chicken muscle [O’Donoghue, A. C, Amyes, T. L. and Richard J.P. (2005), Biochemistry 44, 2610–2621]. Three products were observed from the reactions catalyzed by TIM: dihydroxyacetone phosphate (DHAP) from isomerization with intramolecular transfer of hydrogen, d-DHAP from isomerization with incorporation of deuterium from D2O into C-1 of DHAP, and d-GAP from incorporation of deuterium from D2O into C-2 of GAP. The yield of DHAP formed by intramolecular transfer of hydrogen decreases from 49% for the muscle enzymes to 40% for wildtype Tbb TIM to 34% for monoTIM. There is no significant difference in the ratio of the yields of d-DHAP and d-GAP for wildtype TIM from muscle sources and Trypanosoma brucei brucei, but partitioning of the enediolate intermediate of the monoTIM reaction to form d-DHAP is less favorable ((kC1)D/(kC2)D = 1.1) than for the wildtype enzyme ((kC1)D/(kC2)D = 1.7). Product yields for the wildtype Tbb TIM and monoTIM-catalyzed reactions of glycolaldehyde labeled with carbon-13 at the carbonyl carbon ([1-13C]-GA) at pD 7.0 in the presence of phosphite dianion and in its absence were determined by 1H NMR spectroscopy [Go, M. K., Amyes, T. L., and Richard, J. P. (2009) Biochemistry 48, 5769–5778]. There is no detectable difference in the yields of the products of wildtype muscle and Tbb TIM-catalyzed reactions of [1-13C]-GA in D2O. The kinetic parameters for phosphite dianion activation of the reactions of [1-13C]-GA catalyzed by wildtype Tbb TIM are similar to those reported for the enzyme from rabbit muscle TIM [Amyes, T. L., and Richard, J. P. (2007), Biochemistry 46, 5841–5854], but there is no detectable dianion activation of the reaction catalyzed by monoTIM. The engineered disruption of subunit contacts at monoTIM causes movement of the essential side chains of Lys-13 and His-95 away from the catalytic active positions. We suggest that this places an increased demand that the intrinsic binding energy of phosphite dianion be utilized to drive the change in the conformation of monoTIM back to the active structure for wildtype TIM, with the result that there is insufficient binding energy remaining to give a detectable stabilization of the transition state for the monoTIM-catalyzed reaction of [1-13C]-GA.
Triosephosphate isomerase (TIM) catalyzes the rapid interconversion of the triosephosphates (R)-glyceraldehyde 3-phosphate (GAP) and dihydroxyacetone phosphate (DHAP) by a stereospecific proton transfer mechanism through an enzyme-bound cis-enediolate intermediate that uses the carboxylate side chain of Glu-1672 in acid/base catalysis (Scheme 1) (1–3). More than 80% of the enzymatic rate acceleration for deprotonation of GAP is due to the utilization of the intrinsic binding energy of the remote, nonreacting phosphodianion group of the substrate (4–6). A large fraction of this binding energy is used to activate the enzyme for catalysis of deprotonation of bound substrate (5). We have proposed that this activation is achieved by using the binding interactions between TIM and the substrate phosphodianion (4), or exogenous phosphite dianion (5) to stabilize a high-energy, catalytically active loop-closed form of TIM (7, 8).
Scheme 1.

TIM catalyzes an early reaction on the remarkably successful glycolytic pathway for the metabolism of glucose. This pathway was evident in the Archean period nearly 4 billion years ago (9–11), and the amino acid sequence of the proto-TIM has undergone substantial changes during this time. There is now only ca. 32% sequence homology between TIM from modern archaebacteria and TIMs from other prokaryotes and eukaryotes (12). Despite the limited sequence homology, the overall TIM barrel fold (13), the essential active site residues (3), and the structure of the loop 6 that closes over the active site (14) are conserved; and similar kinetic parameters are observed for catalysis by TIMs from throughout the phylogenetic tree.
The mechanism of action of TIM has attracted the attention of many prominent enzymologists (14–18). This is because proton transfer at carbon is a fundamental reaction in organic chemistry and cellular metabolic pathways, which is catalyzed by an incredibly broad range of enzymes (19–22). Lessons on the mechanism for proton transfer learned through studies on TIM, an enzyme with the classic TIM barrel protein fold (23–25) that appeared early in evolution, might therefore be generalized to enzymes descended from TIM (23, 26).
Trypanosomes are parasitic unicellular protozoan homoflagellates that infect humans and cause a variety of diseases, including sleeping sickness (Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense) and Chagas disease (Trypanosoma cruzi) (27). Trypanosoma brucei brucei is morphologically and biochemically indistinguishable from the other subspecies of T. brucei (27) and is favored for laboratory study because it causes infection in many mammalian species but not in humans. Trypanosomes possess microsomal bodies called glycosomes, which contain the enzymes that catalyze the first seven steps in glycolysis (28–30). TIM from trypanosomes shows about 50% sequence homology with the enzymes from humans, chicken muscle and yeast (31), but the protein from trypanosomes is unusually basic, with an isoelectric point of 9.8 that is 3–4 units higher than that for the widely studied TIMs from yeast (29), chicken muscle (32) and rabbit muscle (29). This unusually high pI suggests that it might be possible to develop inhibitors selective for trypanosomal TIM as therapeutic reagents. Consequently, there have been extensive mechanistic (33) and crystallographic (31, 34–37) studies of trypanosomal TIM in an effort to gain insight that may help in guiding the design of Tbb TIM-specific tight-binding enzyme inhibitors (35, 38–40).
There have been several high-resolution X-ray crystal structures reported by Wierenga and coworkers for Tbb TIM and TIM from the protozoan parasite Leishmania mexicana (31, 36, 37, 41–43). These structures were used to guide the engineering of an interesting monomeric variant of TIM (monoTIM) (44) and in the design of mutagenesis studies to probe the mechanism of monoTIM (45, 46). In addition, the high quality of these structures has stimulated insightful mechanistic proposals, which have been tested in mutagenesis experiments by Wierenga and coworkers (47–49), and in ongoing investigation in our laboratory. It is tempting to generalize the results and conclusions from studies on trypanosomal TIM to TIMs from other organisms. However, there are relatively few results from classical mechanistic studies to support these generalizations, because Tbb TIM has not yet been subject to analyses using sensitive mechanistic probes, such as those pioneered by Knowles and coworkers (1, 16, 17).
We have reported, in studies on TIM from yeast, chicken muscle and rabbit muscle, three experimental protocols that provide a wealth of detailed mechanistic information: (i) Experiments to determine the yields of the three products of the TIM-catalyzed reactions of GAP (50, 51) or DHAP (52) in D2O. (ii) Experiments to determine the kinetic parameters for activation of TIM-catalyzed deprotonation of glycolaldehyde (GA) by phosphite dianion (5). (iii) Experiments to determine the yields of the three products of the unactivated and phosphite dianion-activated TIM-catalyzed reactions of [1-13C]-GA in D2O (8). We now extend these probes to this study on the mechanism of action of Tbb TIM. The results reported here are consistent with a high conservation of the catalytic properties of TIMs from across the phylogenetic tree.
The design of a monomeric variant of TIM (monoTIM) that shows significant enzyme activity represents an early and largely successful effort at protein engineering (43, 44, 46, 53). We have also examined monoTIM using our new mechanistic probes. We report that monoTIM does not catalyze any reactions of [1-13C]-GA at the enzyme active site, either in the absence or in the presence of phosphite dianion. These results show that there is a correlation between the falloff in the catalytic activity of monoTIM compared with wildtype TIM, and the loss in phosphite dianion activation of the monoTIM-catalyzed reaction of [1-13C]-GA. We suggest a structure-based explanation for the effect of the engineered changes in protein structure on phosphite dianion activation of monoTIM.
EXPERIMENTAL
Materials
Rabbit muscle glycerol 3-phosphate dehydrogenase (GPDH) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were purchased from MP Biomedicals or Sigma. Bovine serum albumin (BSA) was from Roche. CM Sepharose Fast Flow was from GE Healthcare. D,L-Glyceraldehyde 3-phosphate diethyl acetal (barium salt), DHAP (dilithium salt), NADH (disodium salt), dithiothreitol (DTT), Dowex 50WX4-200R, TEA•HCl and imidazole were from Sigma. Hydrogen arsenate heptahydrate (disodium salt) and sodium phosphite (dibasic, pentahydrate) were from Riedel-de-Haen (Fluka). NAD (free acid, oxidized form) was from MP Biomedicals. [1-13C]-GA (99% enriched with 13C at C-1, 0.09 M in water) was purchased from Omicron Biochemicals. Deuterium oxide (99% D) and deuterium chloride (35% w/w, 99.9% D) were from Cambridge Isotope Laboratories. Imidazole was recrystallized from benzene. The barium salt of D-glyceraldehyde 3-phosphate diethyl acetal was prepared according to a literature procedure (54). All other chemicals were reagent grade or better and were used without further purification.
The plasmid pTIM containing the wildtype gene for TIM from Trypanosoma brucei brucei (Tbb) (55) and the subcloned pTIM plasmid containing the gene for monoTIM (44) were generous gifts from Professor Rik Wierenga. Both wildtype and monoTIM were over expressed in E. coli BL21 pLys S grown in LB medium at 18°C and purified by ammonium sulfate precipitation followed by gradient elution on ion exchanger CM-Sepharose (44, 55). The enzymes obtained showed a single band by gel electrophoresis. The concentration of wildtype TIM was determined from the absorbance at 280 nm using the extinction coefficient of 3.50 × 104 M−1 cm−1 calculated using the ProtParam tool available on the Expasy server (32, 56). The concentration of TIM obtained using this tool is within 2% of the concentration obtained using a published value of A280 = 1.32 for a solution that contains 1 mg/mL of protein (55).
Preparation of Solutions
Solutions buffered by TEA at pH 7.5 were prepared by neutralization of the hydrochloride salt with 1 M NaOH, and solutions buffered by imidazole and phosphite were prepared as described in previous work (5, 8, 57). Solution pH was determined at 25 °C using an Orion model 720A pH meter equipped with a radiometer pHC4006-9 combination electrode that was standardized at pH 7.0, 4.0 or 10.0 at 25 °C. Values of pD were obtained by adding 0.40 to the observed reading of the pH meter (58).
Stock solutions of D,L-glyceraldehyde 3-phosphate (D,L-GAP) and GAP at pH 7.5 were prepared by hydrolysis of the diethyl acetal using Dowex 50WX4-200R (H+ form) in water at 90–100 °C as described in previous work (50). The concentration of GAP in these solutions was determined by coupled enzymatic assay (50). Solutions of [1-13C]-GA were prepared and the concentration of this compound was determined using published procedures (8, 57).
Enzyme Assays
All enzyme assays were carried out at 25 °C. The change in [NADH] was calculated from the change in the absorbance at 340 nm using an extinction coefficient of 6200 M−1 cm−1. GPDH and GAPDH were exhaustively dialyzed at 7 °C against 20 mM TEA buffer (pH 7.5). GPDH was assayed by monitoring the oxidation of NADH by DHAP at 340 nm. The assay mixture contained 100 mM TEA (pH 7.5, I = 0.1), 0.01% BSA, 0.2 mM NADH and 1 mM DHAP. GAPDH was assayed by monitoring the enzyme-catalyzed reduction of NAD by GAP in an assay mixture containing 30 mM TEA (pH 7.5, I = 0.1, NaCl), 1 mM NAD, 2 mM GAP, 5 mM disodium hydrogen arsenate, 3 mM DTT and 0.01% BSA.
The Tbb TIM-catalyzed isomerization of GAP was monitored by coupling the isomerization of GAP to the oxidation of NADH catalyzed by GPDH (50). Dilute solutions of TIM were prepared to contain 0.01% BSA in order to minimize the adsorption of enzyme to the container walls. The assay mixtures (1.0 mL) contained 100 mM TEA (pH 7.5, I = 0.1), 0.2 mM NADH, 3 mM D,L-GAP (1.5 mM GAP ≈ 6 Km), 0.01% BSA, 1 unit of GPDH and 0.05 – 0.1 nM wildtype TIM. A low background velocity, vo, due to the nonenzymatic isomerization of GAP and the isomerization catalyzed by TIM that was present in the commercial preparation of GPDH, was determined over a period of 2–4 min. An aliquot of TIM was then added, and the initial velocity, vobs, was determined by monitoring the reaction for an additional 5–10 min. The initial velocity of the TIM-catalyzed reaction was then calculated as vi = vobs – vo, where vo was ≤ 3% of the observed initial velocity, vobs. The TIM-catalyzed isomerization of DHAP to form GAP was monitored by coupling the isomerization of DHAP (0.15 – 5 mM) to the reduction of NAD catalyzed by GAPDH (59) and following the increase in absorbance at 340 nm. The assay mixtures (1.0 mL) contained 30 mM TEA (pH 7.5, I = 0.1, NaCl), 1 mM NAD, 5 mM disodium hydrogen arsenate, 3 mM DTT, 0.01% BSA, 1 unit of GAPDH and 0.8 nM wildtype TIM.
1H NMR Analyses
1H NMR spectra at 500 MHz were recorded in D2O at 25 °C using a Varian Unity Inova 500 spectrometer that was shimmed to give a line width ≤ 0.7 Hz for each peak of the doublet due to the C-1 proton of GAP hydrate, or ≤ 0.5 Hz for the most downfield peak of the double triplet due to the C-1 proton of [1-13C]-GA hydrate. Spectra (16–64 transients) were obtained using a sweep width of 6000 Hz, a pulse angle of 90°, an acquisition time of 6 s and a relaxation delay of 60 s (4T1) for experiments on the TIM-catalyzed isomerization of GAP in D2O or 120 s (> 8T1) for experiments on the TIM-catalyzed reactions of [1-13C]-GA in D2O (8, 50). Baselines were subjected to a first-order drift correction before determination of peak areas. Chemical shifts are reported relative to HOD at 4.67 ppm.
Isomerization of GAP in D2O
Wildtype TIM from Tbb was exhaustively dialyzed at 7 °C against 40 mM imidazole (70% free base) in D2O at pD 7.9 and I = 0.1 (NaCl) or 30 mM imidazole (20% free base) in D2O at pD 7.0 and I = 0.1 (NaCl) or I = 0.024, and the resulting enzyme was used in experiments to determine the product yields for reactions of GAP and [1-13C]-GA in D2O, respectively. The turnover of GAP catalyzed by Tbb TIM in D2O at 25 °C and I = 0.1 (NaCl) was followed by 1H NMR spectroscopy (8). The reaction in a volume of 750 μL was initiated by addition of enzyme to the reaction mixture containing GAP, imidazole buffer (pD 7.9) and NaCl in D2O to give final concentrations of 5–10 mM GAP, 14 mM imidazole (70% free base, I = 0.1 (NaCl)) and 0.3 or 0.6 nM TIM. The reaction at 25 °C was monitored by 1H NMR spectroscopy by recording spectra (16 transients) periodically until 70 – 80% of GAP was converted to products. The reactions of GAP (10 mM or 19 mM) catalyzed by monoTIM in D2O at 25 °C were monitored by 1H NMR in a reaction mixture that contained GAP, 14 mM imidazole (70% free base, I = 0.1 (NaCl)) and 600 or 660 nM monoTIM. Each reaction was monitored by 1H NMR spectroscopy. Spectra were recorded at hourly intervals for a period of 8 h, during which time ~ 80% of GAP was converted to products. In all experiments the fraction of the remaining substrate GAP (fGAP) and the fraction of GAP converted to DHAP (fDHAP), d-DHAP (fd-DHAP), d-GAP (fd-GAP) and methylglyoxal (MG, fMG) at time t were determined from the integrated areas of the appropriate 1H NMR signals. The peak areas were normalized using the invariant signal for the C-(4,5) protons of imidazole as an internal standard (50).
Reactions of [1-13C]-GA in D2O
The TIM-catalyzed reactions of [1-13C]-GA in D2O in the presence and absence of HPO32− at 25 °C and I = 0.1 (NaCl) were followed by 1H NMR spectroscopy (60). The reaction in the absence of phosphite was initiated by addition of enzyme to the reaction mixture, which contains [1-13C]-GA, imidazole and NaCl in D2O to give final concentrations of 20 mM [1-13C]-GA, 20 mM imidazole (20% free base, pD 7.0, I = 0.1 (NaCl)) and 0.27 mM TIM in a volume of 850 μL. The reactions in the presence of HPO32− were initiated by addition of enzyme to the reaction mixture, which contains [1-13C]-GA, HPO32−, imidazole and NaCl in D2O to give final concentrations of 20 mM [1-13C]-GA, 18–20 mM imidazole (20% free base, pD 7.0), 5 – 40 mM HPO3H- /HPO32− (50% dianion, pD 7.0) and 4–140 μM TIM in a volume of 850 μL (I = 0.1). In every case 750 μL of the reaction mixture was transferred to an NMR tube, and the NMR spectrum was recorded immediately and then at regular intervals. The reaction in the absence of HPO32− was monitored over a period of 6 days (ca. 60 % reaction of [1-13C]-GA), and the reactions in the presence of HPO32− were monitored over a period of 2–3 hours (ca. 75 % reaction of [1-13C]-GA). After collection of the last spectrum the protein was removed by ultrafiltration and the pD was determined. There was no significant change in pD (= 0.03 unit) during these reactions. The remaining reaction mixture was incubated at 25 °C and used to monitor the activity of TIM. The activity of wildtype Tbb TIM was found to remain unchanged during the time for these experiments.
The monoTIM-catalyzed reaction of [1-13C]-GA in D2O in the absence of HPO32− was monitored in a 750 μL reaction mixture that contained 20 mM [1-13C]-GA, 20 mM imidazole and 0.21 mM monoTIM at pD 7 (I = 0.1, NaCl). The monoTIM-catalyzed reaction of [1-13C]-GA in D2O in the presence of phosphite was monitored in a reaction mixture that contained 20 mM [1-13C]-GA, 40 mM HPO3H- /HPO32− (50% dianion), 20 mM imidazole and 0.092 mM monoTIM at pD 7 (I = 0.12). These reactions were monitored for the disappearance of [1-13C]-GA and for the formation of reaction products. The activity of monoTIM was monitored during the course of this experiment using a solution set aside for this purpose. Unlike wildtype Tbb TIM, a substantial falloff in activity was observed. The fraction of the remaining substrate [1-13C]-GA (fS, eq 1) and the fraction of [1-13C]-GA converted to the identifiable products [2-13C]-GA, [2-13C, 2-2H]-GA, [1-13C, 2-2H]-GA and [1-13C, 2,2-di-2H]-GA were determined from the integrated areas of the relevant 1H NMR signals as described in previous work (5, 8, 61). The observed peak areas were normalized using the signal due to the C-(4,5) protons of imidazole or the upfield peak of the doublet due to the P–H proton of phosphite as an internal standard.
| (1) |
| (2) |
Observed first-order rate constants, kobs (s−1), for the disappearance of [1-13C]-GA were determined from the slopes of linear semi-logarithmic plots of reaction progress against time that covered the first 70–80 % of the reaction using eq 1, where fS is the fraction of [1-13C]-GA that remains at time t. Observed second-order rate constants, (kcat/Km)obs, for the TIM-catalyzed reaction of [1-13C]-GA were determined from the values of kobs using eq 2, where fhyd = 0.94 is the fraction of [1-13C]-GA present as the hydrate form and [E] is the enzyme concentration (5, 8).
RESULTS
The gene for wildtype TIM from Trypanosoma brucei brucei (Tbb TIM) was expressed in E. coli and the enzyme was purified to homogeneity by following a published procedure (55). The kinetic parameters for Tbb TIM-catalyzed isomerization of GAP and DHAP at pH 7.5 and 25 °C (I = 0.1) determined in this work are reported in Table 1. The values of kcat = (2100 ± 300) s−1 determined for several preparations of Tbb TIM is smaller than the value of kcat = (3600 ± 400) s−1 reported by Wierenga and coworkers, but there is excellent agreement between the values of Km = 0.25 mM for GAP determined in our two laboratories (49). Table 1 also reports the kinetic parameters for monoTIM-catalyzed isomerization of GAP, which are in good agreement with previously published kinetic parameters (44).
Table 1.
Kinetic Parameters for Catalysis by TIM, from Different Organisms, of the Isomerization Reaction of GAP and the Phosphite-Activated and Unactivated Reactions of [1-13C]-GA.a
| GAP | DHAP | [1-13C]-GA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Organism | Km (M) | kcat (s−1) | kcat/Km (M−1 s−1) | Km (M) | kcat (s−1) | kcat/Km (M−1 s−1) | (kcat/km)E(M−1 s−1) | (kcat/km)E•HPi (M−1 s−1) | (M−2 s−1) | Kd (M) |
| T. brucei (wildtype) | 2.5 × 10−4 | 2 100 | 8.0 × 106 | 7.0 × 10−4 | 300 | 4.3 × 105 | 0.07d | 60 | 3 400 | 0.018 |
| T. brucei (monoTIM) | 5.2 × 10−3 | 5.7 | 910 | no detectable reaction | ||||||
| Rabbit muscle | 4.5 × 10−4b | 4 300b | 9.6 × 106 | 6.2 × 10−4c | 870 c | 1.4 × 106 | 0.26 e,f | 190 e 210h |
4 800 e 5 500h |
0.038 e |
Under standard assay conditions: 30 mM TEA, pH 7.5 and 25 °C (I = 0.1, NaCl).
Ref. (5).
Ref. (84).
Second-order rate constant for the TIM-catalyzed reaction of [1-13C]-GA at the enzyme active site calculated using eq 7.
Observed kinetic parameters for the reaction of unlabeled GA catalyzed by rabbit muscle TIM (5).
Observed second-order rate constant for the unactivated TIM-catalyzed reaction of unlabeled GA determined by monitoring the disappearance of GA using eq 1-2.
Kinetic parameters for the reaction of [1-13C]-GA catalyzed by rabbit muscle TIM calculated from the observed kinetic parameters for the reaction of unlabelled GA. The TIM-catalyzed reaction of [1-13C]-GA is estimated to by 12% faster than GA because there is an additional pathway for isomerization to form [2-13C]-GA in a yield of 12% (Table 4).
The coupled enzyme assay for the Tbb TIM-catalyzed reaction of DHAP was carried out in the presence of 5 mM sodium arsenate, in order to ensure the formation of an unstable acyl arsenate ester as the product of the GAPDH-catalyzed reaction. The apparent Km = 1.45 mM determined for the Tbb TIM-catalyzed reaction of DHAP under these conditions was corrected using Ki = 4.6 mM for competitive inhibition of Tbb TIM by arsenate (33) to give Km = 0.70 mM. The value of kcat = (300 ± 30) s−1 determined for the Tbb TIM-catalyzed reaction of DHAP is also smaller than an earlier published value of (730 ± 200) s−1 (49). However, the ratio of the values of kcat/Km for the Tbb TIM-catalyzed reactions of GAP and DHAP is (8.0 × 106 M−1 s−1)/(4.3 × 105 M−1 s−1) = 19 is in good agreement with the value of the equilibrium constant for the conversion of GAP to DHAP, Keq = 22, determined at 38 °C (62), as required by the Haldane relationship. Table 1 also lists kinetic parameters for the isomerization reactions catalyzed by TIMs from yeast, rabbit muscle and chicken muscle.
Tbb TIM-Catalyzed Isomerization of GAP in D2O
1H NMR spectroscopy is a powerful analytical method for monitoring proton transfer reactions in D2O at simple (63–65) and complex (66–68) carbon acids. The disappearance of substrate GAP and the appearance of the products of the nonenzymatic and TIM-catalyzed reactions of GAP in D2O was monitored by 1H NMR spectroscopy, as described in previous work (50). Figure 1A shows the decrease with time in the fraction of remaining substrate during the reaction of 10 mM GAP catalyzed by 0.6 nM Tbb TIM in D2O buffered by 14 mM imidazole at pD 8 and 25 °C (I =0.1, NaCl). The yields of the four products of this reaction, (fP)obs (P = DHAP, d-DHAP, d-GAP or methylglyoxal (MG) (69), Scheme 2), were calculated from the normalized 1H NMR peak area of a single proton for the particular product and the sum of the peak areas of single protons for all of the reaction products, as shown in eq 3 (50). The decrease in the normalized peak area of the signal for the substrate GAP agrees with the sum of the normalized peak areas of the signals for the four products to within 10% during the TIM-catalyzed reaction of 70% of GAP.
Figure 1.

Rate and product data for the reaction of GAP (10 mM) catalyzed by Tbb TIM (0.60 nM) in D2O buffered by 14 mM imidazole at pD 7.9 and 25 °C (I = 0.1, NaCl), determined by 1H NMR spectroscopy. (A) The decrease with time in the fraction of remaining GAP. (B) The change with time in the fractional yields of only the products of enzymatic reaction of GAP, normalized using the sum of the observed fraction of d-GAP, DHAP and d-DHAP according to eq 4–6. The initial product yields reported in Table 2 were obtained by making a short linear extrapolation of product yields to zero reaction time. Key: (▲) yield of DHAP; (■) yield of d-DHAP; (●) yield of d-GAP.
Scheme 2.

The fractional yields (fP)E of the products of the TIM-catalyzed reactions of GAP were calculated by correcting the observed product yields for the MG that forms by a nonenzymatic reaction (69) using eq 4 – 6 (50). Figure 1B shows the change in the product yields with time. A small increase in fd-DHAP and a decrease in fd-GAP is observed due to the TIM-catalyzed isomerization of d-GAP to form the thermodynamically favored product d-DHAP (50). The initial product yields were determined by extrapolation of the values of (fP)E (Figure 1B) to t = 0. Table 2 reports the average of the initial product yields determined in separate experiments for the reaction of 10 mM GAP catalyzed by 0.6 nM Tbb TIM (Figure 1B) and for the reaction of 5 mM GAP catalyzed by 0.3 nM TIM (data not shown).
Table 2.
The Yields of the Products from the Reaction of GAP in D2O in the Presence of Triosephosphate Isomerase from Different Organisms.a
| [TIM] | Fractional Product Yield | ||||
|---|---|---|---|---|---|
| DHAP | d-DHAP | d-GAP | MG | ||
| T brucei | fTb | 0.42c | 0.35c | 0.22c | 0.01c |
| 0.36d | 0.37d | 0.21d | 0.06d | ||
| fEe,f | 0.40± 0.02 | 0.37 ± 0.02 | 0.22 ± 0.002 | ||
|
| |||||
| T. brucei (monoTIM) | fTb | 0.31g | 0.34g | 0.32g | 0.03g |
| 0.33h | 0.33h | 0.26h | 0.09h | ||
|
| |||||
| fEe, f | 0.34 ± 0.02 | 0.35 ± 0.01 | 0.31 ± 0.02 | ||
|
| |||||
| Rabbit Muscle (50) | fEe | 0.49 | 0.31 | 0.20 | |
|
| |||||
| Chicken Muscle (50) | fEe | 0.50 | 0.31 | 0.19 | |
For the reaction of GAP at pD 8.0 (15 mM imidazole), 25 °C and I = 0.1 (NaCl).
Yield of the product of the enzymatic or nonenzymatic reactions of GAP (Scheme 2) determined by extrapolation to zero time of linear plots of product yield against time (data not shown).
Reaction of 10 mM GAP catalyzed by 0.60 nM TIM (Figure 1).
Reaction of 5 mM GAP catalyzed by 0.3 nM TIM (data not shown).
The quoted errors are the average of product yields determined in two reactions catalyzed by 0.3 nM or 0.6 nM Tbb TIM.
Reaction of 10 mM GAP catalyzed by 660 nM monoTIM in D2O (I = 0.1, NaCl).
Reaction of 19 mM GAP catalyzed by 600 nM monoTIM in D2O (I = 0.06).
| (3) |
| (4) |
| (5) |
| (6) |
The reactions of GAP (10 or 19 mM) catalyzed by 0.6 μM monoTIM in D2O at pD 8.0 (I = 0.1, NaCl) were monitored by 1H NMR for a period of 8 hours, during which time the concentration of GAP decreased by ca. 80%. The product yields were determined at hourly intervals as described above, and the initial product yields were determined by extrapolation of plots of (fP)E against time to t = 0, as shown in Figure 1B for the reaction catalyzed by wildtype Tbb TIM. The average of the product yields (DHAP, d-DHAP and d-GAP) determined in two separate experiments are reported in Table 2.
TIM-Catalyzed Reactions of [1-13C]-GA in D2O
The Tbb TIM-catalyzed reactions of [1-13C]-GA were monitored by 1H NMR spectroscopy and the product yields were determined from the normalized areas of the relevant product peaks, as described previously (8). Figure 2A shows the decrease in the fraction of the remaining substrate (fS) with time during the reaction of 20 mM [1-13C]-GA catalyzed by Tbb TIM (82 μM) in the presence of 15 mM phosphite dianion at pD 7.0, 25 °C, I = 0.1 (NaCl). The product yields were determined over the first ca. 20-30% reaction, and the disappearance of [1-13C]-GA was monitored for ca. 70–80% reaction.
Figure 2.

Rate and product data for the reaction of [1-13C]-GA catalyzed by Tbb TIM in the presence of 15 mM phosphite dianion in D2O buffered by 20 mM imidazole at pD 7.0 and 25 °C (I = 0.1, NaCl), determined by 1H NMR spectroscopy. (A) The decrease with time in the fraction of remaining [1-13C]-GA. (B) The change with time in the fractional yields of the products, fP, of the phosphite-activated TIM-catalyzed reaction of [1-13C]-GA. The initial product yields reported in Table 3 were obtained by making a short linear extrapolation of product yields to zero reaction time. Key: (▼) yield of [2-13C]-GA; (◆) yield of [2-13C, 2-2H]-GA; (■) yield of [1-13C, 2-2H]-GA.
The Tbb TIM-catalyzed reactions of [1-13C]-GA gave the same three isotopomers of GA as reported in earlier work for the chicken muscle TIM-catalyzed reaction of [1-13C]-GA: [2-13C]-GA, [2-13C, 2-2H]-GA, [1-13C, 2-2H]-GA (Chart 1) (8). The yield of a fourth product [1-13C, 2,2-di-2H]-GA, that forms by a protein-catalyzed reaction outside of the enzyme active site (8, 57, 70), is too low to be detected for the phosphite dianion-activated reactions. The fractional yields of products, fP, were calculated from the normalized 1H NMR peak area of a single proton for the particular product and the sum of the peak areas of single protons for all of the reaction products (8). Figure 2B shows the fractional yields, fP, of the products obtained during the first 30% of the reaction of 20 mM [1-13C]-GA catalyzed by Tbb TIM (82 μM) in the presence of 15 mM phosphite dianion at pD 7.0, 25 °C, I = 0.1 (NaCl). The initial fractional product yields, fE, for the phosphite-activated reactions reported in Table 4 were determined by making short extrapolations of the normalized fractional yields, fP, to zero reaction time as shown in Figure 2B. A similar protocol was followed in determining the initial product yields of the unactivated TIM-catalyzed reactions of [1-13C]-GA (Table 4).
Chart 1.

Table 4.
The Yields of the Products from the Reaction of [1-13C]-GA in D2O Catalyzed by Tbb TIM and from the Phosphite Dianion-Activated TIM-Catalyzed Reaction.a
| [HPO32−] mM | Fractional Product yield, fEb |
|||
|---|---|---|---|---|
| [2-13C]-GA | [2-13C, 2-2H]-GA | [1-13C, 2-2H]-GA | [1-13C, 2,2-di-2H]-GA | |
| 0 | 0.04 ± 0.001 c | 0.28 ± 0.01 c | 0.14 ± 0.03 c | 0.30 ± 0.05 c |
| 0 | 0.10 d | 0.61 d | 0.29 d | |
| 5 | 0.13 ± 0.01 | 0.63 ± 0.03 | 0.24 ± 0.04 | n.d |
| 10 | 0.13 ± 0.003 | 0.64 ± 0.01 | 0.23 ± 0.01 | n.d |
| 16 | 0.14 ± 0.004 | 0.63 ± 0.01 | 0.24 ± 0.01 | n.d |
| 20 | 0.13 ± 0.01 | 0.63 ± 0.04 | 0.24 ± 0.03 | n.d |
| 20 | 0.13 ± 0.004 | 0.65 ± 0.02 | 0.21 ± 0.03 | |
| Average product yield e
|
0.13 | 0.63 | 0.24 | |
| Chicken Muscle TIM (8) | 0.12 | 0.64 | 0.23 | |
For reactions of 20 mM [1-13C]-GA in 20 mM imidazole (20% free base), pD 7.0, 25 °C, and I = 0.1
The yield of the products of the reaction of [1-13C]-GA determined as described in previous work (8). The quoted error in the initial product yields is the standard deviation in the y-intercept of plots of fractional product yield against time (Figure 2).
Fractional product yields from the unactivated reaction of [1-13C]-GA catalyzed by 0.27 mM Tbb TIM.
Average yield calculated for products [2-13C]-GA, [2-13C, 2-2H]-GA and [1-13C, 2-2H]-GA that form from reactions at the enzyme active site, determined for several experiments at different concentrations of Tbb TIM (130 – 470 μM, data not shown).
Average of the experiments at four different concentrations of phosphite dianion.
The observed first-order rate constants, kobs (s−1), for the disappearance of [1-13C]-GA and the apparent second-order rate constants, (kcat/Km)obs (M−1 s−1), for the TIM-catalyzed reactions of the reactive carbonyl form of [1-13C]-GA were determined using eq 1 and 2. The kinetic data for these TIM-catalyzed reactions in the presence and absence of phosphite dianion are summarized in Table 3. There is fair mass balance observed for the phosphite dianion-activated TIM-catalyzed reactions of [1-13C]-GA, so that (kcat/Km)obs is equal to the second-order rate constant for the Tbb-TIM catalyzed reaction at the functioning enzyme active site.
Table 3.
Kinetic Data for the Reaction of [1-13C]-GA Catalyzed by Wildtype Tbb TIM in D2O in the Absence and Presence of Phosphite Dianion.a
| [HPO32−] (M) | [TIM] (M) | kobs (s−1)b | (kcat/Km)obs (M−1s−1)c |
|---|---|---|---|
| 0 | 2.72 × 10−4 | 2.15 × 10−6 | 0.13 d 0.07e |
| 2.6 × 10−3 | 1.36 × 10−4 | 7.08 × 10−5 | 8.6 |
| 5.3 × 10−3 | 1.15 × 10−4 | 1.01 × 10−4 | 14.3 |
| 7.9 × 10−3 | 1.02 × 10−4 | 1.13 × 10−4 | 18.2 |
| 9.9 × 10−3 | 9.50 × 10−5 | 1.28 × 10−4 | 22.0 |
| 12.6 × 10−3 | 9.50 × 10−5 | 1.56 × 10−4 | 26.9 |
| 15.3 × 10−3 | 8.20 × 10−5 | 1.48 × 10−4 | 29.6 |
| 17.3 × 10−3 | 6.82 × 10−5 | 1.26 × 10−4 | 30.3 |
For reactions of 20 mM [1-13C]-GA in 20 mM imidazole (20% free base), pD 7.0, 25 °C, and I = 0.1 (NaCl).
Observed first-order rate constant for the reaction of [1-13C]-GA calculated using eq 1.
Observed second-order rate constant for the TIM-catalyzed reaction of [1-13C]-GA calculated using eq 2, unless noted otherwise.
Second-order rate constant determined by monitoring the disappearance of total [1-13C]-GA.
Second-order rate constant for the reactions of [1-13C]-GA at the TIM active site, calculated from the overall reaction rate constant of 0.13 M−1 s−1 (determined from two independent experiments) and the 52% product yield (Table 4).
Only ca. 70% of [1-13C]-GA is converted to [2-13C]-GA, [2-13C, 2-2H]-GA, [1-13C, 2-2H]-GA or [1-13C, 2,2-di-2H]-GA for reactions in the absence of phosphite dianion. The other products formed were not determined. Combining the 20% yield of [1-13C, 2,2-di-2H]-GA with (kcat/Km)obs = 0.13 M−1 s−1 gives (kcat/Km) = (0.13 M−1 s−1)(0.20) = 0.03 M−1 s−1 for the unactivated Tbb TIM-catalyzed reaction of [1-13C]-GA to form [1-13C, 2,2-di-2H]-GA. The second-order rate constant for the unactivated TIM-catalyzed reaction at the enzyme active site was calculated as (kcat/Km) = (0.13 M-1 s−1)(0.52) = 0.07 M−1 s−1, where 0.52 is the fractional yield of [2-13C]-GA, [2-13C, 2-2H]-GA, and [1-13C, 2-2H]-GA from the active-site reactions.
The reaction of [1-13C]-GA catalyzed by monoTIM at 25 °C was monitored in both the absence and presence of 40 mM phosphite (50% dianion) in solutions that contain 20 mM imidazole (20% free base) at pD 7.0 and I = 0.1 (NaCl). The reaction in the absence of phosphite (0.21 mM monoTIM) was monitored for 25 h, during which time there was 12% reaction of [1-13C]-GA. A 65% falloff of the initial monoTIM activity was observed during the first 12 h of the reaction. The reaction in the presence of phosphite dianion (0.09 mM monoTIM) was monitored for 14 h during which time there was 10% reaction of [1-13C]-GA. A 90% loss in the activity of monoTIM was observed during the first 9 h of the reaction. In both cases, the reaction remained first-order in the concentration of [1-13C]-GA during this substantial loss of enzymatic activity. Apparent second-order rate constants (kcat/Km)obs = 0.1 and 0.4 M−1 s−1 were determined, respectively, for the monoTIM-catalyzed reactions of [1-13C]-GA in the absence and presence of phosphite by monitoring the disappearance of [1-13C]-GA using eq 1 and 2.
The 1H NMR analysis of the products of the monoTIM-catalyzed reaction of [1-13C]-GA showed no detectable [2-13C]-GA or [2-13C, 2-2H]-GA (< 1%) from reactions at a functioning enzyme active site. The only observed products were [1-13C, 2-2H]-GA and [1-13C, 2,2-di-2H]-GA, from the direct deuterium exchange reaction at carbon-2 of [1-13C]-GA. The reaction of [1-13C]-GA in the absence and in the presence of phosphite gave ca. 40% and 55% yields of [1-13C, 2-2H]-GA and [1-13C, 2,2-di-2H]-GA, respectively, with [1-13C, 2,2-di-2H]-GA being the major product that forms in a yield of 75% of total products.
DISCUSSION
Reactions of GAP in D2O
DHAP binds at a cavity of TIM that is exposed to solvent. The carbon acid fragment of DHAP projects towards the cavity bottom, and the phosphodianion lies at the protein surface, where it is covered by the flexible phosphate-gripper loop 6 (3, 7). This loop closure shields the bound carbon acid from interaction with the bulk solvent. Proton transfer from bound triosephosphate to the carboxylate side chain of Glu-167 in a solvent of D2O gives an enediolate intermediate (57) and a protonated acid side chain of Glu-167. The substrate-derived hydrogen partitions between intramolecular transfer to form DHAP, and effectively irreversible exchange with solvent D2O to form the D-labeled carboxylic acid (Scheme 3). This D-labeled side chain next partitions between protonation of the enediolate intermediate at C-1 and C-2 (Scheme 3) to form d-DHAP and d-GAP, respectively.
Scheme 3.

Table 2 compares the product yields of the Tbb and chicken TIM-catalyzed isomerization reactions of GAP in D2O determined by 1H NMR analyses. Two rate constant ratios can be determined from these product yields:
The ratio kex/(kC1)H for partitioning of hydrogen-labeled TIM between protonation of the enediolate at carbon-1 and exchange with -D from solvent was determined as the ratio of the yields of H-labeled (DHAP) and D-labeled (d-DHAP and d-GAP) products. The 40 ± 2% yield of DHAP for Tbb TIM is detectably smaller than the 50% yield of DHAP from the chicken and rabbit muscle TIM-catalyzed reactions (Table 2); and, the derived rate constant ratio for Tbb TIM, kex/(kC1)H = 1.44 is larger than for the muscle enzymes, kex/(kC1)H = 1.04.
The ratio (kC1)D/(kC2)D for partitioning of D-labeled TIM between D-transfer to carbon-1 and to carbon-2 is determined as the ratio of the yields of d-DHAP and d-GAP. The rate constant ratio (kC1)D/(kC2)D = 1.7 ± 0.2 is the same, within experimental error, as the values of (kC1)D/(kC2)D = 1.5 for the reactions catalyzed by TIM from muscle sources (50).
Previous studies on TIM have shown that: (i) The exchange reaction of the protonated carboxylic acid side chain of Glu-167 involves deuterons that are sequestered from interactions Scheme 3 with bulk solvent at the enzyme active site (51). (ii) The O-1 and O-2 enediolate intermediates of deprotonation of GAP and DHAP, respectively, do not achieve chemical equilibrium with one another during the isomerization reaction (52). (iii) The substrate-derived -H undergoes exchange between the carboxylic acid side chain of Glu-167 and a relatively large pool of -D that are sequestered from solvent at the enzyme active site (71). In other words, these exchange reactions are fast and involve an undetermined number of hydrons at an active site that is inaccessible to solvent on the time scale for the exchange reaction.
The similarity in the product yields for the reactions catalyzed by TIM from several sources shows that the dynamics and mechanism for these exchange reactions are unaffected by the relatively large differences in the sequences of these different TIMs. The small differences in the very large rate constants for the fast exchange reactions between the electronegative carboxylic acid oxygen (72) and basic atoms at the TIM-active site correspond to a small 0.2 kcal/mol difference in relative barriers to (kC1)H and kex, that we are unable to rationalize.
The monomers of wildtype TIM are held together in the dimer by interactions between loop 3 (residues 65 – 79) and residues at a deep crevice of the second subunit near loops 1 and 4, which contain the active site residues Lys-13 and His-95, respectively. The monomeric form of TIM was engineered as shown in Figure 4 by shortening the long fifteen amino acid-residue loop 3 by seven residues and making judicious substitutions at the remaining residues to reduce hydrophobicity and extend the β-strand 3 by one turn (44). These sequence changes reduce the interactions between the TIM subunits, and produce a monomeric form of TIM with lower, but still significant, enzymatic activity (44, 47).
Figure 4.

A comparison of the amino acid sequences for wildtype Tbb TIM and the engineered monoTIM. The first amino acid residue in loop 3 that follows β-strand 3 is Gln-65. Residues 69 – 79 (red) from loop 3 were replaced by four amino acids (N A D A) that extend the β-strand 3 by one turn. Additional amino acid substitutions for the wildtype enzyme are shown in blue.
The monoTIM-catalyzed reaction of GAP in D2O gives a lower 34 ± 2% yield of DHAP from the reaction with intramolecular transfer of -H than the 40 ± 2% yield observed for wildtype Tbb TIM. A decrease in (kC1)D/(kC2)D for partitioning of the D-labeled enzyme is also observed, from (kC1)D/(kC2)D = 1.7 for the wildtype enzyme to 1.2 for monoTIM. The relatively larger yield of D-labeled products observed here for the reaction catalyzed by monoTIM might, nominally, reflect the increased exposure of the active site to solvent due to the elimination of hydrophobic interactions at the subunit interface. We do not have an explanation for the difference in the rate constant ratios for partitioning of deuterium-labeled enzyme between protonation of the enediolate reaction intermediate at C-1 and C-2 at the active sites for wildtype and monoTIM.
Wildtype Tbb TIM-Catalyzed Reactions of [1-13C]-GA
Three products form from the phosphite-activated TIM-catalyzed reactions of [1-13C]-GA in D2O (Scheme 4): (a) [2-13C]-GA from intramolecular transfer of the substrate-derived hydrogen to the 13C-labeled carbon of the enediolate reaction intermediate; (b) [1-13C, 2-2H]-GA from transfer of deuterium from solvent to the 12C-labeled carbon of the enediolate; and (c) [2-13C, 2-2H]-GA from transfer of deuterium from solvent to the 13C-labeled carbon of the enediolate. We observe the same yields of [2-13C]-GA, [1-13C, 2-2H]-GA and [2-13C, 2-2H]-GA from the reactions of [1-13C]-GA catalyzed by TIM from trypanosomes and from chicken muscle (Table 4). Note that these products may form with phosphite bound in two different conformations [not shown in Scheme 4] (8). The dianion may bind next to 2-12C, to mimic the binding of GAP, or next to 1-13C to mimic the binding of DHAP.
Scheme 4.

A ca. 52% total yield of [2-13C]-GA, [2-13C, 2-2H]-GA and [1-13C, 2-2H]-GA was observed from the slow Tbb TIM-catalyzed reactions of [1-13C]-GA in the absence of exogenous phosphite dianion in a ratio of 0.10:0.61:0.29, respectively (Table 4). These products form by the reactions of [1-13C]-GA at the active site of TIM. A similar overall product yield in a ratio of 0.10:0.60:0.30 for [2-13C]-GA, [2-13C, 2-2H]-GA and [1-13C, 2-2H]-GA, respectively, was determined for the unactivated chicken muscle TIM-catalyzed reaction of [1-13C]-GA (8).
Similar ca. 20% yields of the dideuterium-labeled [1-13C, 2,2-di-2H]-GA were determined for the unactivated Tbb and chicken muscle TIM-catalyzed reactions (8). [1-13C, 2,2-di-2H]-GA is the only product detected from the reaction of [1-13C]-GA catalyzed by the severely crippled K12G mutant of TIM from yeast (57) and, bovine serum albumin (BSA) was also found to catalyze the conversion of [1-13C]-GA to [1-13C, 2,2-di-2H]-GA in D2O (70). These results show that this activity is intrinsic to some globular proteins. We have proposed that [1-13C, 2,2-di-2H]-GA forms by the reaction of [1-13C]-GA with the alkylamino side chain of surface lysine residues to form a Schiff base that undergoes base-catalyzed deuterium exchange of the C-2 protons through an enamine intermediate (Scheme 5) (8, 70).
Scheme 5.

Phosphite Activation of Tbb TIM-Catalyzed Reaction of [1-13C]-GA in D2O
The observed second-order rate constant (kcat/Km)obs = 0.13 M−1 s−1 for the Tbb TIM-catalyzed reaction of [1-13C]-GA in the absence of exogenous phosphite is similar to the values of (kcat/Km)obs = 0.26 M−1 s−1 and 0.19 M−1 s−1 reported for rabbit muscle TIM (5) and chicken muscle TIM (8), respectively. These observed rate constants were corrected using eq 7 to obtain the second-order rate constant (kcat/Km)E for the reactions to form [2-13C]-GA, [1-13C, 2-2H]-GA and [2-13C, 2-2H]-GA that occur at the enzyme active site. The sum of the fractional yields of these products, (fE)T = 0.52, determined for wildtype Tbb TIM gives (kcat/Km)E = 0.07 M−1 s−1, which is similar to (kcat/Km)E = 0.1 M−1 s−1 determined for TIM from chicken muscle (8).
| (7) |
Figure 3 shows the dependence on the concentration of exogenous HPO32− for turnover of the carbonyl form of [1-13C]-GA by Tbb TIM in D2O at pD 7.0 and 25 °C (I = 0.1, NaCl). The phosphite-activated TIM-catalyzed reactions of [1-13C]-GA are very fast relative to the nonspecific protein-catalyzed reaction, and [2-13C]-GA, [1-13C, 2-2H]-GA and [2-13C, 2-2H]-GA account for 100% of the observed reaction products. Figure 3 includes earlier data for the rabbit muscle TIM-catalyzed deuterium exchange reaction of GA under the same reaction conditions.
Figure 3.

Dependence of the observed second-order rate constant (kcat/Km)obs (M−1 s−1) for the turnover of the free carbonyl form of [1-13C]-GA by Tbb TIM and of unlabeled GA by rabbit muscle TIM in D2O on the concentration of added phosphite dianion at pD 7 and 25 °C (I = 0.10). Key: (●) Reaction of 20 mM [1-13C]-GA catalyzed by Tbb TIM; (▲) Reaction of 20 mM unlabeled GA catalyzed by rabbit muscle TIM (5); (▼) Reaction of 10 mM unlabeled GA catalyzed by rabbit muscle TIM (5).
| (8) |
The fit of the data for the Tbb TIM-catalyzed reaction to eq 8, derived for Scheme 6, gave Kd = 19 ± 3 mM for binding of HPO32− to the free enzyme and (kcat/Km)E•HPi = 64 ± 6 M−1 s−1 as the second-order rate constant for turnover of [1-13C]-GA by enzyme that is saturated by HPO32− (Table 1). By comparison, values of Kd = 38 mM and (kcat/Km)E•HPi = 185 M−1 s−1 were reported in an earlier study (5) of rabbit muscle TIM-catalyzed deuterium exchange reaction of unlabeled GA (Table 1). We estimate that the TIM-catalyzed reaction of [1-13C]-GA in D2O is ca. 10% faster than the reaction of GA, because the 10% yield of [2-13C]-GA from the reaction of the carbon-13 labeled substrate can not be detected in the reaction of the unlabeled substrate. This small 10% correction in the previously reported kinetic parameters for the rabbit-muscle TIM-catalyzed reaction of GA is given in Table 1.
Scheme 6.

In comparison to rabbit muscle TIM, there is a smaller Kd (19 compared to 38 mM) and limiting second order rate constant ((kcat/Km)E•HPi = 64 compared to 210 M−1 s−1) for phosphite dianion activation of the wildtype Tbb TIM-catalyzed reactions of [1-13C]-GA, but more similar third order rate constants for activation by low concentrations of phosphite dianion ((kcat/Km)E•HPi/Kd = 3400 compared to 4900 M−2 s−1). There is a similar trend for values of Km and kcat (smaller for Tbb compared to rabbit muscle TIM), and kcat/Km (similar for Tbb and rabbit muscle TIM) for enzyme-catalyzed isomerization of GAP (Table 1). These trends show that the total catalytic efficiency for TIM catalysis at low substrate or activator concentration is similar, but that the rabbit muscle enzyme shows a larger activity for catalysis of the reaction of saturating concentrations of substrate or activator. The results are consistent with the notion that a slightly greater fraction of the total substrate or ligand binding energy is expressed at the ground-state for the Tbb TIM-catalyzed reaction, where it is expressed in the parameter kcat/Km and Km, while a larger fraction of this binding energy is expressed at the transition state for the rabbit muscle TIM-catalyzed reaction, where it is expressed specifically in kcat but not in Km.
MonoTIM-Catalyzed Reactions of [1-13C]-GA
It is possible to monitor the very slow wildtype TIM-catalyzed reactions of [1-13C]-GA at 25 °C, because the enzyme remains fully active for at least one week. By contrast, monoTIM loses substantial activity over a period of several hours. For example there was 90% loss in enzyme activity during a 9-hour room temperature reaction of monoTIM (0.09 mM) in the presence of 20 mM phosphite dianion at pD 7.0. The value of (kcat/Km)obs = 0.4 M−1 s−1 for this monoTIM-catalyzed reaction in the presence of 20 mM phosphite dianion is 80-fold smaller than the calculated value of (kcat/Km)obs = 33 M−1 s−1 for the wildtype Tbb TIM-catalyzed reaction of [1-13C]-GA under the same reaction conditions.3
The only product detected of monoTIM catalyzed reactions is [1-13C, 2,2-di-2H]-GA. The observation that these reactions remain first-order in the concentration of [1-13C]-GA as monoTIM loses more than 90% of its activity for catalysis of isomerization at a functioning active site confirms that [1-13C, 2,2-di-2H]-GA is formed in a protein-catalyzed reaction (8, 70), with a velocity that is independent of enzymatic activity. Therefore, the value of kcat/Km for the monoTIM-catalyzed reaction of [1-13C]-GA at the enzyme active site must be smaller than (kcat/Km)obs = 0.4 M−1 s−1. We conclude that the structural differences between wildtype Tbb TIM and monoTIM result in a > 80-fold decrease in the second-order rate constant for the reaction of [1-13C]-GA determined for enzyme activation in the presence of 20 mM phosphite dianion.
A comparison of the X-ray crystal structures for wildtype and monoTIM shows that elimination of the monomer contacts results in large movements of loop 1 and loop 4, which lie at the monomer interface (43). Loop 1 has much larger B-factors for monoTIM compared with the wildtype enzyme, and there is no detectable electron density for Lys-13, Cys-14 and Asn-15 for monoTIM, consistent with a high degree of loop mobility. The shift in the position of loop 4 results in a 12 Å displacement in the imidazole side chain of His-95 (43).
Both Lys-13 (57, 73–75) and His-95 (76, 77) play critical roles in the wildtype TIM-catalyzed isomerization reaction. It is notable and surprising that such large changes in the position of these essential side chains do not eliminate the enzymatic activity for engineered monoTIM. The observation that K13A and H95A mutations of monoTIM (46) cause further falloffs in catalytic activity show that these side chains play an important role in the reaction catalyzed by the engineered protein. This suggests that the position of these side chains moves towards that for active wildtype Tbb TIM on proceeding to the transition state for the monoTIM catalyzed reaction. Indeed, X-ray crystal structures of complexes between point mutation variants of monoTIM [kinetic parameters nearly identical to parent monoTIM studied here] and 2-phosphoglycolohydroxamate (PGH) or 2-phosphoglycolate (PGA) provide direct evidence that ligand binding causes the side chains of Lys-13 and His-95 to adopt conformations similar to those observed in wildtype Tbb TIM and very different from the position at unliganded monoTIM (45).
We have proposed that phosphite activation of the TIM-catalyzed reaction of [1-13C]-GA is due to the utilization of the intrinsic binding energy (78) of the exogenous dianion to stabilize a rare (Kc ≪ 1, Scheme 7), but catalytically active, closed form of TIM (5, 7, 8). This change in conformation from inactive Eo to active Ec is a complex process that involves the dramatic movement of loop 6, smaller changes in the positions of loop 7 and 8, and movement of catalytic side chains at the enzyme active site (42, 79–81). The elimination of contacts between the subunits of TIM in monoTIM causes additional large movements observed for loops 1 and 4 away from the catalytically active conformation. We propose that the effect of these engineered changes in the conformation of free monoTIM is to cause an increase in the thermodynamic barrier for the change in conformation from inactive Eo to active Ec (decrease in Kc, Scheme 7). This places an additional requirement that the phosphodianion binding energy be used to drive the enzyme conformational change and reduces the binding energy available to provide a net stabilization of the transition state for TIM-catalyzed isomerization reaction (78).
Scheme 7.

We propose that the binding of phosphite dianion to monoTIM drives a change in enzyme conformation similar to that observed for the binding of phosphate dianion (82), PGH (83) and PGA (80) to wildtype TIM. The failure to detect activation of the monoTIM-catalyzed reaction of [1-13C]-GA by phosphite dianion would then reflect the overall larger barrier to this conformational change and the resulting reduction in the concentration of the catalytically active binary complex Ec•HPO32−. We suggest that this concentration is too low to give a detectable monoTIM-catalyzed reaction of [1-13C]-GA by our analytical methods.
Summary and Conclusions
Figure 5 shows the X-ray crystal structure in the region of the active site of Tbb TIM complexed to the inhibitor phosphoglycolohydroxamate (PGH) (PDB entry 1TRD) (37) superimposed on the X-ray crystal structure of the chicken TIM-PGH complex (PDB entry 1TPH) (81). This comparison shows that, while the sequences of Tbb and chicken TIM are only 50% homologous, the active site architecture for the two enzymes is nearly identical. The results of several sensitive probes for the mechanism of action of Tbb TIM reported in this work show a similar high degree of similarity to results from studies on TIM from yeast, rabbit muscle and chicken muscle. Taken as a whole, these data demonstrate a high degree of structural and mechanistic homology between TIMs from different organisms, so that the results of studies on triosephosphate isomerase from one organism may be readily generalized to all TIMs.
Figure 5.
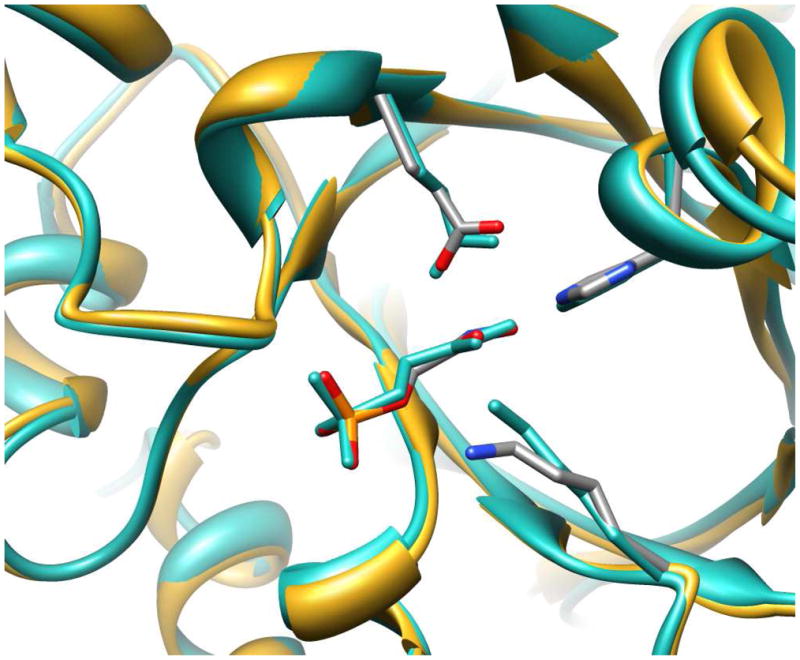
Superimposed crystal structures of the loop-closed Tbb TIM-PGH complex, shown in gold ribbons and in atoms colored by element (PDB entry 1TRD) and chicken muscle TIM-PGH complex, shown in sea green atoms and ribbons (PDB entry 1TPH).
By comparison, the engineering of monoTIM causes large changes in the structure of the unliganded enzyme. However, there is sufficient conformational mobility in monoTIM to allow ligand binding to induce a change to the conformation of the active wildtype structure, which shows optimal interactions with bound ligands. We propose that a reduction in the catalytic activity of monoTIM compared to the wildtype enzyme is observed because the intrinsic binding energy of the substrate ligand used to drive this additional change in protein conformation is no longer available to be used in the stabilization of the transition state for the isomerization reaction (78). The smaller intrinsic binding energy of phosphite dianion is presumably used exclusively to drive the protein conformational change, so that there is insufficient total binding energy to give a detectable phosphite activation of the monoTIM-catalyzed reaction of [1-13C]-GA.
Glossary
- TIM
triosephosphate isomerase
- DHAP
dihydroxyacetone phosphate
- DTT
dithiothreitol; GAP, (R) -glyceraldehyde 3-phosphate
- GA
glycolaldehyde
- GPDH
glycerol 3-phosphate dehydrogenase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- BSA
bovine serum albumin
- NMR
nuclear magnetic resonance
- D,L-GAP
D,L-glyceraldehyde 3-phosphate
- d-DHAP
[1(R)-2H]-dihydroxyacetone phosphate
- d-GAP
[2(R)-2H]-glyceraldehyde 3-phosphate
- MG
methylglyoxal
- NADH
nicotinamide adenine dinucleotide, reduced form
- NAD
nicotinamide adenine dinucleotide, oxidized form
- PGH
2-phosphoglycolohydroxamate
- PGA
2-phosphoglycolate
- TEA
triethanolamine
- Tbb TIM
triosephosphate isomerase from Trypanosoma brucei brucei
Footnotes
This work was supported by Grant GM39754 from the National Institutes of Health
Unless noted otherwise, residues are numbered according to the sequence for the enzyme from Trypanosoma brucei.
The value of (kcat/Km)obs for the Tbb TIM-catalyzed reaction in the presence of 20 mM HPO32− was calculated using eq 8 and the kinetic parameters for Tbb TIM reported in this paper.
References
- 1.Knowles JR, Albery WJ. Perfection in enzyme catalysis: the energetics of triosephosphate isomerase. Acc Chem Res. 1977;10:105–111. [Google Scholar]
- 2.Rieder SV, Rose IA. Mechanism of the triose phosphate isomerase reaction. J Biol Chem. 1959;234:1007–1010. [PubMed] [Google Scholar]
- 3.Wierenga RK. Triosephosphate isomerase: a highly evolved biocatalyst. Cell Mol Life Sci. 2010;67:3961–3982. doi: 10.1007/s00018-010-0473-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amyes TL, O’Donoghue AC, Richard JP. Contribution of phosphate intrinsic binding energy to the enzymatic rate acceleration for triosephosphate isomerase. J Am Chem Soc. 2001;123:11325–11326. doi: 10.1021/ja016754a. [DOI] [PubMed] [Google Scholar]
- 5.Amyes TL, Richard JP. Enzymatic catalysis of proton transfer at carbon: Activation of triosephosphate isomerase by phosphite dianion. Biochemistry. 2007;46:5841–5854. doi: 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrow JR, Amyes TL, Richard JP. Phosphate Binding Energy and Catalysis by Small and Large Molecules. Acc Chem Res. 2008;41:539–548. doi: 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malabanan MM, Amyes TL, Richard JP. A role for flexible loops in enzyme catalysis. Curr Opin Struct Biol. 2010;20:702–710. doi: 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Go MK, Amyes TL, Richard JP. Hydron Transfer Catalyzed by Triosephosphate Isomerase, Products of the Direct and Phosphite-Activated Isomerization of [1–13C]-Glycolaldehyde in D2O. Biochemistry. 2009;48:5769–5778. doi: 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gebbia JA, Backenson PB, Coleman JL, Anda P, Benach JL. Glycolytic enzyme operon of Borrelia burgdorferi: characterization and evolutionary implications. Gene. 1997;188:221–228. doi: 10.1016/s0378-1119(96)00811-6. [DOI] [PubMed] [Google Scholar]
- 10.Webster KA. Evolution of the coordinate regulation of glycolytic enzyme genes by hypoxia. J Exp Biol. 2003;206:2911–2922. doi: 10.1242/jeb.00516. [DOI] [PubMed] [Google Scholar]
- 11.Wierenga RK, Borchert TV, Noble MEM. Crystallographic binding studies with triosephosphate isomerases: conformational changes induced by substrate and substrate-analogs. FEBS Lett. 1992;307:34–39. doi: 10.1016/0014-5793(92)80897-p. [DOI] [PubMed] [Google Scholar]
- 12.Feng DF, Cho G, Doolittle RF. Determining divergence times with a protein clock: update and reevaluation. Proc Natl Acad Sci. 1997;94:13028–13033. doi: 10.1073/pnas.94.24.13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wierenga RK. The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett. 2001;492:193–198. doi: 10.1016/s0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- 14.Kursula I, Salin M, Sun J, Norledge BV, Haapalainen AM, Sampson NS, Wierenga RK. Understanding protein lids: structural analysis of active hinge mutants in triosephosphate isomerase. Prot Eng Des & Sel. 2004;17:375–382. doi: 10.1093/protein/gzh048. [DOI] [PubMed] [Google Scholar]
- 15.Rose IA. Mechanism of C-H bond cleavage in aldolase and isomerase reactions. Brookhaven Symp Biol. 1962;15:293–309. [PubMed] [Google Scholar]
- 16.Knowles JR. To build an enzyme. Philos Trans R Soc London, Ser B. 1991;332:115–121. doi: 10.1098/rstb.1991.0039. [DOI] [PubMed] [Google Scholar]
- 17.Knowles JR. Enzyme catalysis: not different, just better. Nature. 1991;350:121–124. doi: 10.1038/350121a0. [DOI] [PubMed] [Google Scholar]
- 18.Banner DW, Bloomer AC, Petsko GA, Phillips DC, Pogson CI. Crystallographic studies of chicken triose phosphate isomerase. Cold Spring Harbor Symp Quant Biol. 1971;36:151–155. doi: 10.1101/sqb.1972.036.01.021. [DOI] [PubMed] [Google Scholar]
- 19.Richard JP, Amyes TL. Proton transfer at carbon. Curr Opin Chem Biol. 2001;5:626–633. doi: 10.1016/s1367-5931(01)00258-7. [DOI] [PubMed] [Google Scholar]
- 20.Gerlt JA, Kozarich JW, Kenyon GL, Gassman PG. Electrophilic Catalysis Can Explain the Unexpected Acidity of Carbon Acids in Enzyme-Catalyzed Reactions. J Am Chem Soc. 1991;113:9667–9669. [Google Scholar]
- 21.Gerlt JA, Gassman PG. Understanding the rates of certain enzyme-catalyzed reactions: Proton abstraction from carbon acids, acyl transfer reactions, and displacement reactions of phosphodiesters. Biochemistry. 1993;32:11943–11952. doi: 10.1021/bi00096a001. [DOI] [PubMed] [Google Scholar]
- 22.Richard JP, Amyes TL. On the importance of being zwitterionic: enzymic catalysis of decarboxylation and deprotonation of cationic carbon. Bioorg Chem. 2004;32:354–366. doi: 10.1016/j.bioorg.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Gerlt JA, Raushel FM. Evolution of function in (β/α)8-barrel enzymes. Curr Opin Chem Biol. 2003;7:252–264. doi: 10.1016/s1367-5931(03)00019-x. [DOI] [PubMed] [Google Scholar]
- 24.Wise E, Yew WS, Babbitt PC, Gerlt JA, Rayment I. Homologous (β/α)8-Barrel Enzymes That Catalyze Unrelated Reactions: Orotidine 5′-Monophosphate Decarboxylase and 3-Keto-L-Gulonate 6-Phosphate Decarboxylase. Biochemistry. 2002;41:3861–3869. doi: 10.1021/bi012174e. [DOI] [PubMed] [Google Scholar]
- 25.Nagano N, Orengo CA, Thornton JM. One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J Mol Biol. 2002;321:741–765. doi: 10.1016/s0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- 26.Sterner R, Hocker B. Catalytic versatility, stability, and evolution of the (β/α)8-barrel enzyme fold. Chem Rev. 2005;105:4038–4055. doi: 10.1021/cr030191z. [DOI] [PubMed] [Google Scholar]
- 27.Hoet S, Opperdoes F, Brun R, Quetin-Leclercq J. Natural products active against african trypanosomes: A step towards new drugs. Nat Prod Rep. 2004;21:353–364. doi: 10.1039/b311021b. [DOI] [PubMed] [Google Scholar]
- 28.Opperdoes FR, Baudhuin P, Coppens I, De Roe C, Edwards SW, Weijers PJ, Misset O. Purification, morphometric analysis, and characterization of the glycosomes (microbodies) of the protozoan hemoflagellate Trypanosoma brucei. J Cell Biol. 1984;98:1178–1184. doi: 10.1083/jcb.98.4.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misset O, Bos OJ, Opperdoes FR. Glycolytic enzymes of Trypanosoma brucei, Simultaneous purification, intraglycosomal concentrations and physical properties. Eur J Biochem. 1986;157:441–453. doi: 10.1111/j.1432-1033.1986.tb09687.x. [DOI] [PubMed] [Google Scholar]
- 30.Misset O, Opperdoes FR. Simultaneous purification of hexokinase, class I fructose-bisphosphate aldolase, triosephosphate isomerase and phosphoglycerate kinase from Trypanosoma brucei. Eur J Biochem. 1984;144:475–483. doi: 10.1111/j.1432-1033.1984.tb08490.x. [DOI] [PubMed] [Google Scholar]
- 31.Wierenga RK, Noble MEM, Vriend G, Nauche S, Hol WGJ. Refined 1.83 Å structure of trypanosomal triosephosphate isomerase crystallized in the presence of 2.4 M-ammonium sulfate, A comparison with the structure of the trypanosomal triosephosphate isomerase-glycerol-3-phosphate complex. J Mol Biol. 1991;220:995–1015. doi: 10.1016/0022-2836(91)90368-g. [DOI] [PubMed] [Google Scholar]
- 32.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. Proteomics Protocol Handbook. 2005:571–607. [Google Scholar]
- 33.Lambeir AM, Opperdoes FR, Wierenga RK. Kinetic properties of triose-phosphate isomerase from Trypanosoma brucei brucei, A comparison with the rabbit muscle and yeast enzymes. Eur J Biochem. 1987;168:69–74. doi: 10.1111/j.1432-1033.1987.tb13388.x. [DOI] [PubMed] [Google Scholar]
- 34.Wierenga RK, Kalk KH, Hol WGJ. Structure determination of the glycosomal triosephosphate isomerase from Trypanosoma brucei brucei at 2.4 √Ö resolution. J Mol Biol. 1987;198:109–121. doi: 10.1016/0022-2836(87)90461-x. [DOI] [PubMed] [Google Scholar]
- 35.Noble MEM, Wierenga RK, Lambeir AM, Opperdoes FR, Thunnissen AMWH, Kalk KH, Groendijk H, Hol WGJ. The adaptability of the active site of trypanosomal triosephosphate isomerase as observed in the crystal structures of three different complexes. Proteins Struct Funct, Genet. 1991;10:50–69. doi: 10.1002/prot.340100106. [DOI] [PubMed] [Google Scholar]
- 36.Wierenga RK, Noble MEM, Davenport RC. Comparison of the refined crystal structures of liganded and unliganded chicken, yeast and trypanosomal triosephosphate isomerase. J Mol Biol. 1992;224:1115–1126. doi: 10.1016/0022-2836(92)90473-w. [DOI] [PubMed] [Google Scholar]
- 37.Alahuhta M, Wierenga RK. Atomic Resolution crystallography of a complex of triosephosphate isomerase with a reaction intermediate analog: new insight in the proton transfer reaction mechanism. Prot: Struct Funct Bioinform. 2010;78:1878–1888. doi: 10.1002/prot.22701. [DOI] [PubMed] [Google Scholar]
- 38.Kessler H, Matter H, Geyer A, Diehl HJ, Koeck M, Kurz G, Opperdoes FR, Callens M, Wierenga RK. Selective inhibition of trypanosomal triose phosphate isomerase with a thiopeptide. Angew Chem, Int Ed Engl. 1992;104:328–330. [Google Scholar]
- 39.Kuntz DA, Osowski R, Schudok M, Wierenga RK, Mueller K, Kessler H, Opperdoes FR. Inhibition of triosephosphate isomerase from Trypanosoma brucei with cyclic hexapeptides. Eur J Biochem. 1992;207:441–447. doi: 10.1111/j.1432-1033.1992.tb17069.x. [DOI] [PubMed] [Google Scholar]
- 40.Callens M, Van Roy J, Zeelen JP, Borchert TV, Nalis D, Wierenga RK, Opperdoes FR. Selective interaction of glycosomal enzymes from Trypanosoma brucei with hydrophobic cyclic hexapeptides. Biochem Biophys Res Commun. 1993;195:667–672. doi: 10.1006/bbrc.1993.2097. [DOI] [PubMed] [Google Scholar]
- 41.Kishan KVR, Zeelen JP, NOble MEM, Borchert TV, Wierenga RK. Comparison of the structures and the crystal contacts of trypanosomal triosephosphate isomerase in four different crystal forms. Protein Science. 1994;3:779–787. doi: 10.1002/pro.5560030507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kursula I, Wierenga RK. Crystal structure of triosephosphate isomerase complexed with 2-phosphoglycolate at 0.83-Å resolution. J Biol Chem. 2003;278:9544–9551. doi: 10.1074/jbc.M211389200. [DOI] [PubMed] [Google Scholar]
- 43.Borchert TV, Abagyan R, Kishan KVR, Zeelen JP, Wierenga RK. The crystal structure of an engineered monomeric triosephosphate isomerase, monoTIM: the correct modeling of an eight-residue loop. Structure (London) 1993;1:205–213. doi: 10.1016/0969-2126(93)90021-8. [DOI] [PubMed] [Google Scholar]
- 44.Borchert TV, Abagyan R, Jaenicke R, Wierenga RK. Design, creation, and characterization of a stable, monomeric triosephosphate isomerase. Proc Natl Acad Sci. 1994;91:1515–1518. doi: 10.1073/pnas.91.4.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borchert TV, Kishan KVR, Zeelen JP, Schliebs W, Thanki N, Abagyan R, Jaenicke R, Wierenga RK. Three new crystal structures of point mutation variant of mono TIM: conformational flexibility of loop-1, loop-4 and loop-8. Structure. 1995;3:669–679. doi: 10.1016/s0969-2126(01)00202-7. [DOI] [PubMed] [Google Scholar]
- 46.Schliebs W, Thani N, Eritja R, Wierenga R. Active site properties of monomeric triosephosphate isomerase (monoTIM) as deduced from mutational and structural studies. Protein Science. 1996;5:229–239. doi: 10.1002/pro.5560050206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schliebs W, Thanki N, Jaenicke R, Wierenga RK. A double mutation at the tip of the dimer interface loop of triosephosphate isomerase generates active monomers with reduced stability. Biochemistry. 1997;36:9655–9662. doi: 10.1021/bi963086a. [DOI] [PubMed] [Google Scholar]
- 48.Alahuhta M, Casteleijn MG, Neubauer P, Wierenga RK. Structural studies show that the A178L mutation in the C-terminal hinge of the catalytic loop-6 of triosephosphate isomerase (TIM) induces a closed-like conformation in dimeric and monomeric TIM. Acta Crystallogr D Biol Crystallogr. 2008;64:178–188. doi: 10.1107/S0907444907059021. [DOI] [PubMed] [Google Scholar]
- 49.Casteleijn MG, Alahuhta M, Groebel K, El-Sayed I, Augustyns K, Lambeir AM, Neubauer P, Wierenga RK. Functional role of the conserved active site proline of triosephosphate isomerase. Biochemistry. 2006;45:15483–15494. doi: 10.1021/bi061683j. [DOI] [PubMed] [Google Scholar]
- 50.O’Donoghue AC, Amyes TL, Richard JP. Hydron Transfer Catalyzed by Triosephosphate Isomerase, Products of Isomerization of (R)-Glyceraldehyde 3- Phosphate in D2O. Biochemistry. 2005;44:2610–2621. doi: 10.1021/bi047954c. [DOI] [PubMed] [Google Scholar]
- 51.O’Donoghue AC, Amyes TL, Richard JP. Slow proton transfer from the hydrogen-labelled carboxylic acid side chain (Glu-165) of triosephosphate isomerase to imidazole buffer in D2O. Org Biomol Chem. 2008;6:391–396. doi: 10.1039/b714304d. [DOI] [PubMed] [Google Scholar]
- 52.O’Donoghue AC, Amyes TL, Richard JP. Hydron Transfer Catalyzed by Triosephosphate Isomerase, Products of Isomerization of Dihydroxyacetone Phosphate in D2O. Biochemistry. 2005;44:2622–2631. doi: 10.1021/bi047953k. [DOI] [PubMed] [Google Scholar]
- 53.Salin M, Kapetaniou EG, Vaismaa M, Lajunen M, Casteleijn MG, Neubauer P, Salmon L, Wierenga RK. Crystallographic binding studies with an engineered monomeric variant of triosephosphate isomerase. Acta Crystallogr D Biol Crystallogr. 2010;66:934–944. doi: 10.1107/S0907444910025710. [DOI] [PubMed] [Google Scholar]
- 54.Bergemeyer HU, Haid E, Nelboeck-Hochstetter M Office, U. P. Proocess for preparing open ring tetrose and triosephosphate acetals and phosphate ketals. US: 1972. [Google Scholar]
- 55.Borchert TV, Pratt K, Zeelen JP, Callens M, Noble MEM, Opperdoes FR, Michels PAM, Wierenga RK. Overexpression of trypanosomal triosephosphate isomerase in Escherichia coli and characterization of a dimer-interface mutant. Eur J Biochem. 1993;211:703–710. doi: 10.1111/j.1432-1033.1993.tb17599.x. [DOI] [PubMed] [Google Scholar]
- 56.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nuc Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Go MK, Koudelka A, Amyes TL, Richard JP. Role of Lys-12 in Catalysis by Triosephosphate Isomerase: A Two-Part Substrate Approach. Biochemistry. 2010;49:5377–5389. doi: 10.1021/bi100538b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glasoe PK, Long FA. Use of glass electrodes to measure acidities in deuterium oxide. J Phys Chem. 1960;64:188–190. [Google Scholar]
- 59.Plaut B, Knowles JR. pH-Dependence of the triose phosphate isomerase reaction. Biochem J. 1972;129:311–320. doi: 10.1042/bj1290311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Go MDK. Chemistry. University at Buffalo; Buffalo: 2009. Studies on enzymatic and non-enzymatic proton transfer in aqueous solution; p. 181. [Google Scholar]
- 61.George WO, Collins GCS. Nuclear magnetic resonance spectra of glycolaldehyde. J Chem Soc B. 1971:1352–1355. [Google Scholar]
- 62.Veech RL, Raijman L, Dalziel K, Krebs HA. Disequilibrium in the triose phosphate isomerase system in rat liver. Biochem J. 1969;115:837–842. doi: 10.1042/bj1150837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amyes TL, Richard JP. Generation and stability of a simple thiol ester enolate in aqueous solution. J Am Chem Soc. 1992;114:10297–10302. [Google Scholar]
- 64.Amyes TL, Richard JP. Determination of the pKa of ethyl acetate: Brønsted correlation for deprotonation of a simple oxygen ester in aqueous solution. J Am Chem Soc. 1996;118:3129–3141. [Google Scholar]
- 65.Richard JP, Williams G, O’Donoghue AC, Amyes TL. Formation and Stability of Enolates of Acetamide and Acetate Anion: An Eigen Plot for Proton Transfer at α-Carbonyl Carbon. J Am Chem Soc. 2002;124:2957–2968. doi: 10.1021/ja0125321. [DOI] [PubMed] [Google Scholar]
- 66.Rios A, Richard JP, Amyes TL. Formation and Stability of Peptide Enolates in Aqueous Solution. J Am Chem Soc. 2002;124:8251–8259. doi: 10.1021/ja026267a. [DOI] [PubMed] [Google Scholar]
- 67.Toth K, Richard JP. Covalent Catalysis by Pyridoxal: Evaluation of the Effect of the Cofactor on the Carbon Acidity of Glycine. J Am Chem Soc. 2007;129:3013–3021. doi: 10.1021/ja0679228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Crugeiras J, Rios A, Riveiros E, Amyes TL, Richard JP. Glycine Enolates: The Effect of Formation of Iminium Ions to Simple Ketones on 〈-Amino Carbon Acidity and a Comparison with Pyridoxal Iminium Ions. J Am Chem Soc. 2008;130:2041–2050. doi: 10.1021/ja078006c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Richard JP. Acid-base catalysis of the elimination and isomerization reactions of triose phosphates. J Am Chem Soc. 1984;106:4926–4936. [Google Scholar]
- 70.Go MK, Malabanan MM, Amyes TL, Richard JP. Bovine Serum Albumin-Catalyzed Deprotonation of [1–13C]Glycolaldehyde: Protein Reactivity toward Deprotonation of the α-Hydroxy α-Carbonyl Carbon. Biochemistry. 2010;49:7704–7708. doi: 10.1021/bi101118g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rose IA, Fung WJ, Warms JVB. Proton diffusion in the active site of triosephosphate isomerase. Biochemistry. 1990;29:4312–4317. doi: 10.1021/bi00470a008. [DOI] [PubMed] [Google Scholar]
- 72.Eigen M. Proton Transfer, Acid-Base Catalysis, and Enzymatic Hydrolysis. Angew Chem Int Ed Engl. 1964;3:1–72. [Google Scholar]
- 73.Go MK, Amyes TL, Richard JP. Rescue of K12G mutant TIM by NH4+ and alkylammonium cations: The reaction of an enzyme in pieces. J Am Chem Soc. 2010;132:13525–13532. doi: 10.1021/ja106104h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Joseph-McCarthy D, Lolis E, Komives EA, Petsko GA. Crystal structure of the K12M/G15A triosephosphate isomerase double mutant and electrostatic analysis of the active site. Biochemistry. 1994;33:2815–2823. doi: 10.1021/bi00176a010. [DOI] [PubMed] [Google Scholar]
- 75.Lodi PJ, Chang LC, Knowles JR, Komives EA. Triosephosphate isomerase requires a positively charged active site: The role of lysine-12. Biochemistry. 1994;33:2809–2814. doi: 10.1021/bi00176a009. [DOI] [PubMed] [Google Scholar]
- 76.Komives EA, Chang LC, Lolis E, Tilton RF, Petsko GA, Knowles JR. Electrophilic catalysis in triosephosphate isomerase: the role of histidine-95. Biochemistry. 1991;30:3011–3019. doi: 10.1021/bi00226a005. [DOI] [PubMed] [Google Scholar]
- 77.Nickbarg EB, Davenport RC, Petsko GA, Knowles JR. Triosephosphate isomerase: removal of a putatively electrophilic histidine residue results in a subtle change in catalytic mechanism. Biochemistry. 1988;27:5948–5960. doi: 10.1021/bi00416a019. [DOI] [PubMed] [Google Scholar]
- 78.Jencks WP. Binding energy, specificity and enzymic catalysis: The Circe effect. Adv Enzymology Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 79.Jogl G, Rozovsky S, McDermott AE, Tong L. Optimal alignment for enzymatic proton transfer: structure of the Michaelis complex of triosephosphate isomerase at 1.2-Å resolution. Proc Natl Acad Sci. 2003;100:50–55. doi: 10.1073/pnas.0233793100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lolis E, Petsko GA. Crystallographic analysis of the complex between triosephosphate isomerase and 2-phosphoglycolate at 2.5-Å resolution: implications for catalysis. Biochemistry. 1990;29:6619–6625. doi: 10.1021/bi00480a010. [DOI] [PubMed] [Google Scholar]
- 81.Zhang Z, Sugio S, Komives EA, Liu KD, Knowles JR, Petsko GA, Ringe D. Crystal Structure of Recombinant Chicken Triosephosphate Isomerase-Phosphoglycolohydroxamate Complex at 1.8-Å Resolution. Biochemistry. 1994;33:2830–2837. doi: 10.1021/bi00176a012. [DOI] [PubMed] [Google Scholar]
- 82.Verlinde CLMJ, Noble MEM, Kalk KH, Groendijk H, Wierenga RK, Hol WGJ. Anion binding at the active site of trypanosomal triosephosphate isomerase, Monohydrogen phosphate does not mimic sulfate. Eur J Biochem. 1991;198:53–57. doi: 10.1111/j.1432-1033.1991.tb15985.x. [DOI] [PubMed] [Google Scholar]
- 83.Davenport RC, Bash PA, Seaton BA, Karplus M, Petsko GA, Ringe D. Structure of the triosephosphate isomerase-phosphoglycolohydroxamate complex: an analog of the intermediate on the reaction pathway. Biochemistry. 1991;30:5821–5826. doi: 10.1021/bi00238a002. [DOI] [PubMed] [Google Scholar]
- 84.Krietsch WKG, Pentchev PG, Klingenbuerg H, Hofstaetter T, Buecher T. Isolation and crystallization of yeast and rabbit liver triose phosphate isomerase and a comparative characterization with the rabbit muscle enzyme. Eur J Biochem. 1970;14:289–300. doi: 10.1111/j.1432-1033.1970.tb00289.x. [DOI] [PubMed] [Google Scholar]