

Synopsis
The EGFR/ErbB/HER family of kinases contains four homologous receptor tyrosine kinases that are important regulatory elements in key signaling pathways. To elucidate the atomistic mechanisms of dimerization-dependent activation in the ErbB family, we have performed molecular dynamics simulations of the intracellular kinase domains of three members of the ErbB family (those with known kinase activity), namely EGFR, ErbB2 (HER2) and ErbB4 (HER4), in different molecular contexts: monomer vs. dimer, wildtype vs. mutant. Using bioinformatics and fluctuation analyses of the molecular dynamics trajectories, we relate sequence similarities to correspondence of specific bond-interaction networks and collective dynamical modes. We find that in the active conformation of the ErbB kinases, key subdomain motions are coordinated through conserved hydrophilic interactions: activating bond-networks consisting of hydrogen bonds and salt bridges. The inactive conformations also demonstrate conserved bonding patterns (albeit less extensive) that sequester key residues and disrupt the activating bond network. Both conformational states have distinct hydrophobic advantages through context-specific hydrophobic interactions. We show that the functional (activating) asymmetric kinase dimer interface forces a corresponding change in the hydrophobic and hydrophilic interactions that characterize the inactivating bond network, resulting in motion of the αC-helix through allostery. Several of the clinically identified activating kinase mutations of EGFR act in a similar fashion to disrupt the inactivating bond network. Our molecular dynamics study reveals a fundamental difference in the sequence of events in EGFR activation compared with that described for the Src kinase Hck.
Keywords: Molecular Dynamics, Homology Modeling, Principal Component Analysis, Kinase Activation, Epidermal Growth Factor Receptor
Introduction
Receptor tyrosine kinases (RTKs) are transmembrane glycoproteins important in intercellular communication and oncogenesis [1]; they comprise a large ligand-binding extracellular domain, a single transmembrane α-helix, an intracellular tyrosine kinase domain and a C-terminal tail that harbors regulatory tyrosine autophosphorylation sites (reviewed in [2], [3]). The ErbB family consists of four homologous RTKs: the epidermal growth factor receptor (EGFR/ErbB1/HER1), ErbB2 (HER2/Neu), ErbB3 (HER3), and ErbB4 (HER4). Binding of growth factors to the extracellular ligand-binding domains of ErbB receptors promotes their homo and/or heterodimerization, which in turn leads to activation of the cytoplasmic kinase domain. This triggers a multi-layered signaling network of crucial pathways regulating cell proliferation, differentiation, migration, etc [4]. Aberrant signaling by EGFR and ErbB2 is correlated with a variety of diseases, from psoriasis to cancer [5]. In particular, clinically identified mutations in EGFR have been shown to increase the basal activity of the EGFR kinase domain, and non-small-cell lung cancer (NSCLC) patients carrying these mutations respond remarkably to the EGFR RTK inhibitor gefitinib [6-8]. ErbB2 is the target of the therapeutic Herceptin antibody, and its amplification and overexpression in breast cancer correlates with a poor prognosis [9, 10]. The loss of ErbB4 signaling in mice has been shown to result in defective heart, nervous system, and mammary gland function [11-13].
In RTK signaling, the intracellular kinase domain catalyzes transfer of the γ-phosphate of ATP to tyrosines on both the RTK itself and in other target substrates (reviewed in [2]). Regulation of the RTK kinase domain is thought to involve contributions from several conserved subregions: the catalytic loop (C-loop), the activation loop (A-loop), the glycine-rich nucleotide binding loop (P-loop), and the αC-helix, which together define the active site in the cleft between the β strand-rich N-lobe and the helical C-lobe. The catalytic loop residues directly participate in phosphoryl transfer. The A-loop, P-loop and αC-helix (marked in Figure 1A) modulate the activity of the kinase domain by regulating accessibility of the active site to binding and coordinating both ATP and the substrate tyrosine. The ∼20 amino acid A-loop in ErbB kinases contains one phosphorylatable tyrosine (Y845 in EGFR, Y877 in ErbB2, Y850 in ErbB4). In many kinases, e.g. the insulin receptor kinase (IRK), phosphorylation of this A-loop tyrosine triggers conformational changes that allow substrate access to the active site. Thus, A-loop conformational changes constitute a key event in RTK regulation (Figure 1B). The αC-helix and P-loop must also be re-positioned to coordinate the ATP and the substrate tyrosine for effective phosphoryl transfer. The four regulatory subdomains are highly conserved across the ErbB family, with 88% sequence identity between EGFR and ErbB2 and 77% sequence identity between EGFR and ErbB4 (Figure 1A). ErbB3 is unique in the ErbB family as it lacks specific conserved residues considered to be required for full kinase activity.
Figure 1.
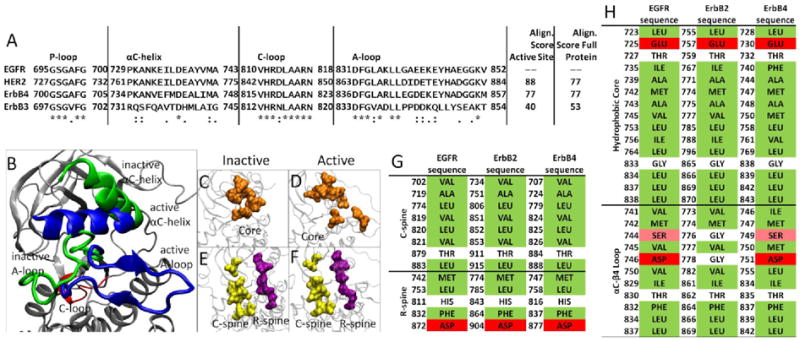
(A) Alignment of the four catalytic subdomains comprising the active site in the ErbB kinases, with accompanying alignment scores for the subdomains alone and for the entire kinase. (B-F) Comparison of the inactive and active conformations of the ErbB kinases highlighting the conserved features. (B) The C-loop is highlighted in red with the active conformation shown in blue with the inactive in green. (C,D) The hydrophobic core is shown in orange, (E,F) while the C-spine is shown in yellow and the R-spine is in purple. (G) The residues comprising the C-spine and R-spine for EGFR, ErbB2 and ErbB4 kinase. The residues highlighted in green are hydrophobic, the residues in red are hydrophilic and those in white are neutral. (H) The residues comprising the Hydrophobic core and the αC-βM region for EGFR, ErbB2 and ErbB4 kinase. Serine is shown in pink as it is slightly hydrophilic, though it is still considered neutral.
Recent structural studies have revealed highly conserved hydrophobic “spines” within kinases that are considered important for defining their catalytic state [14, 15], shown in Figure 1E,F. The regulatory spine (R-spine) consists of four hydrophobic side chains (M742, L753, H811, F832 in EGFR) anchored by an aspartic acid in the αF-helix (D872 in EGFR). The R-spine spans several key regulatory subdomains, and coordinates the motion of the N- and C-lobes of the kinase [14]. The catalytic spine (C-spine) involves eight hydrophobic side-chains (V702, A719, L774, V819, L820, V821, T879, L883 in EGFR) that help support and coordinate the adenine ring of ATP in the active state [15]. Similarly, in the inactive state there is a small hydrophobic ‘core’ formed between the αC-helix and the A-loop, which maintains the kinase in the inactive conformation (Figure 1C-F). Disruption of this hydrophobic core by single point mutations has been shown to activate EGFR [6, 8, 16-18].
EGFR stands out among RTKs in appearing not to require A-loop phosphorylation for its activity [19]. Mutation of the EGFR A-loop tyrosine Y845 to phenylalanine does not appear to change the level of receptor auto-phosphorylation in cells or in vitro [20], in contrast to other kinases such as IRK (although Y845 is phosphorylated by Src in EGFR signaling [21]). Crystal structures have confirmed that the EGFR and ErbB4 kinase domains can adopt active-like conformations even without Y845 (Y850 in ErbB4) phosphorylation [16, 22], and have revealed an allosteric mechanism for kinase domain activation [20]. Activation of the EGFR TKD involves the formation of an asymmetric ‘head-to-tail’ dimer in which one kinase domain (the ‘receiver’) becomes activated through allosteric changes arising from contacts between its N-lobe and the C-lobe of its neighbor (the ‘activator’). The C-lobe of the activator kinase appears to play a cyclin-like function in activating its dimerization partner (the receiver). The importance of the asymmetric dimer interface was confirmed by mutational studies in EGFR and ErbB4 [20, 22]. More recent studies have shown that the intracellular juxtamembrane region of the receptor also contributes to formation of the asymmetric dimer interface, in a manner that is necessary for maximal activation [23-25].
Considering the high degree of sequence similarity and structural homology across the ErbB family members (Figure 1A,G,H), we sought to identify the degree to which molecular mechanisms of activation are conserved across the ErbB family, and to identify differences in overall function that arise from variability in primary structure. Recently, we and others have hypothesized the existence of distinct networks of intramolecular non-covalent bonds that characterize the active and inactive conformations of kinases (for Lyn [26, 27], Abl [28], EGFR [28-30] and ErbB2 [31]), with transitions between the states necessitating a shift in these bond networks. Here, we present bioinformatics and fluctuation analyses of molecular dynamics trajectories of ErbB kinase domains and relate sequence similarities to correspondence of specific bond-interaction networks and resemblances in collective dynamical modes. We investigate how the various stimuli/perturbations such as dimerization, phosphorylation of the A-loop tyrosine, and mutations seen in cancer patients impact both the active and inactive conformations of the ErbB family kinase domains. The solvated systems of the truncated ErbB family kinases we present even have a physiological relevance to cell studies. The protein tyrosine kinases, Src and Abl, have a highly similar active structure to those in receptor tyrosine kinases [2, 32]. Furthermore, ErbB4 is cleaved from the membrane into the s80 protein, a fully active soluble form of the ErbB4 kinase domain [13].
Methods
Molecular Dynamics (MD) Simulation
Models for ErbB1 (EGFR) kinase were derived from the 1M14 (active) and 2GS7 (inactive) structures [16, 20]. Models for ErbB4 were derived from the structures of Qiu et al., PDB ID: 3BCE and 3BBW [22]. Structures for ErbB2 were constructed using homology modeling following the procedure described in [31]. Models for kinase dimers were constructed based on the asymmetric dimer interface described in [20]. Each system was simulated as a fully atomistic, explicitly solvated-system in NAMD [33], using the CHARMM 27 forcefield [34]. The missing hydrogens in the protein were added using the hbuild plugin in the VMD algorithm [35]. To simulate a physiological pH of 7.0 the histidines were constructed with +1 protonation state on the δ-nitrogen. The entire system consisted of the protein, water molecules (TIP3P model [36]) and ions at 75 mM ionic strength to simulate physiological conditions; the water molecules and ions were placed at locations of electrostatic extrema (maxima for negative Cl- ions and minima for positive Na+ ions) as determined by a Debye-Huckel potential using Solvate 1.0 [37]. The placement of the counter-ions was also restricted to 8 Å away from any protein residue. The Rattle algorithm [38] was employed to constrain the hydrogens to allow for a 2 fs timestep of integration. Periodic boundaries were implemented in all three dimensions and long-ranged electrostatics interactions were accounted for using the Particle Mesh Ewald Algorithm [39]. A conjugate gradient algorithm was employed for all energy minimizations. The solvated system was energy minimized and the volume of each system subsequently equilibrated with constant pressure and temperature (CPT) simulations. Both the temperature and pressure were constrained using a Langevin algorithm [40]. The pressure Langevin piston was set with a reference pressure of 1 atm, a mass of 2000 amu and a collision frequency of 5 (1/ps), while the temperature Langevin piston was set with a reference temperature of 300 K and a mass of 10000 kcal·ps2. Following volume equilibration, the energy of the system was equilibrated using constant volume and temperature (NVT) simulations. Each system was simulated for at least 10 ns (see Table S1 for simulation times). For the dimer systems restraints of 1 kcal/Å2 were placed on each kinase to their initial state in the inactive asymmetric dimers to maintain the equilibrated structures while removing steric clashes. The kinase systems were equilibrated and then the restraints were reduced to 0.5 kcal/Å2 and 0.1 kcal/Å2. Following energy and volume equilibration (see Figure S1), the production simulations were performed.
Analyses of MD Simulations
Root-mean-squared deviation (RMSD) calculations were performed using the RMSD tool plugin in VMD by first removing global translation and rotation, and then computing the RMSD of the selected sub-regions (A-loop, C-loop, P-loop and αC-helix) relative to a reference structure (the respective active or inactive crystal structure). Principal component analysis (PCA) was performed using the software Carma [41], by constructing the covariance matrix of atomic fluctuations σ̱ in Cartesian space and then diagonalizing σ̱ to obtain the principal components (eigenvalues and eigenvectors). An analysis of hydrogen bond (H-bond) patterns in the production simulations was performed using CHARMM in conjunction with VMD. The trajectories were first analyzed in CHARMM with a hydrogen bond cutoff of 3.4 Å and a cutoff angle of 150 degrees. CHARMM was used to generate a list of hydrogen bonds that were present in at least 60% of the trajectory; we note that the threshold of 60% was varied from 50-80% in our sensitivity analysis and the results were nor altered significantly. These hydrogen bonds were then visualized in VMD and any bonds not providing a consistent, sustained bond were removed to reveal the persistent H-bonds. During the H-bond analysis, acidic and basic residues formed strong hydrogen bonds. Such hydrogen bonds were considered salt bridges if they were between the side chain of an acidic and basic residue, with a bond length less than 1.6 Å and present in the majority (>60% of the trajectory) of the simulation. Statistical hydrogen bonding criteria was chosen to highlight the contributions of specific residues within the protein regardless of its surrounding energetic environment [42]. Solvent accessible surface area (SASA) values were calculated in VMD using the measure SASA module using a probe radius of 1.4 Å larger than the van der Waals radius. The SASA was calculated for each step in the trajectory from which the mean and standard deviation were computed. As an alternative measure of hydrophobicity in heterogeneous environments, following Garde et al. [43, 44], normalized water density fluctuations were computed by recording the ratio of VolN*(〈N2〉-〈N〉2)/〈 N 〉2, where VolN is the volume of interest, 〈N2 〉-〈N〉2 is the variance in the fluctuation of water number in VolN, and 〈 N 〉 is mean associated with the number of water molecules. We choose the region of interest to be within 5 Å of a specified hydrophobic sub-region in the EGFR monomer kinase. The results are then divided by the water density fluctuations of bulk water for normalization. Although results are presented for a cutoff of 5 Å, other cutoffs ranging from 3 Å-15 Å were investigated and similar trends were recorded.
Results
Following molecular dynamics simulation of each active or inactive monomeric kinase system for at least 10 ns, the time evolution of the RMSD was used to monitor equilibration and to track any reorganization of the A-loop and αC-helix conformations; no conformational switching towards active or inactive states was observed (Figure S2, supplementary material). Whereas the majority of the protein backbone, including the C-loop and the P-loop, aligns closely between the inactive and active states, the A-loop and αC-helix conformations differ considerably. In transitioning from the inactive to the active conformation, the αC-helix rotates toward the C-lobe, with the rotating end shifting by ∼9 Å toward the base of the cleft between the N- and C-lobes (Figure 1B). This helix is also extended by 2 turns (involving residues 728-732 in EGFR) in the active conformation compared with the inactive conformation. In the inactive kinase, the A-loop maintains a ‘closed’ conformation (mainly through inter-region hydrogen bonds) and partially blocks the catalytic site. By contrast, the A-loop appears ‘unfurled’ in the active kinase, and lies against the C-lobe (blue in Figure 1B).
PCA reveals a tightly coordinated motion in all active ErbB members
Principal component analysis (PCA) revealed that motions occurring within the active sites of the inactive and active EGFR monomers differ significantly, particularly in the A-loop (see Figure 2). The inactive EGFR monomer exhibits large-amplitude motion in both the αC-helix and the A-loop (4.95 and 7.34 Å, respectively), with smaller fluctuations in the P-loop and C-loop (4.17 and 3.65 Å, respectively). We attribute the larger amplitudes to a more flexible protein segment; this is consistent with the observation that part of the activation loop is unresolved in almost all the crystal structures of ErbB kinases to date. Our previous work with the loop modeling program MODELLER [45] discusses the sensitivity of the modeled region (which is absent in the crystallographic structures). In particular, several candidate structures do not show significant differences in dynamics [31]. Moreover, alternate structures of ErbB4 also using MODELLER are remarkably close to initial simulation structures (data not shown). In contrast, the active EGFR monomer demonstrates a uniform level of motion across all four subdomains of the active site with only low-amplitude fluctuations (2-3Å), and shows no significant local deformations. This implies that the motions are more tightly coordinated across the active site in the active conformation than in the inactive conformation. The kinase monomers of the other ErbB family members (modeled ErbB2 and the ErbB4 structure) demonstrate similar motions, as shown in Figure 2. The inactive ErbB4 kinase exhibits a dominant motion in the A-loop (6.46 Å), while the P-loop, C-loop, and the αC-helix undergo smaller lateral motions (4.45 Å, 2.64 Å, and 2.30 Å, respectively). In the active conformation, the ErbB4 kinase presents a fluctuation profile similar to EGFR: the subdomains all have similar small-amplitude motions (2-3 Å) with no large local deformations. Thus, for the three homologous members of the ErbB family, which have a high-degree of sequence similarity, not only are the principal motions conserved across the systems, but the characteristic differences between the inactive and active kinase conformations are maintained in character. This finding suggests large (and possibly similar) differences in the internal network of bonds between the two activity states of each kinase. Indeed, the identified bonding network (Table 1, Table S2) shows clear conservation across the members of the ErbB family, and also reflects the differences in principal motions between the inactive and active conformations. In general, the inactive conformations have significantly fewer persistent bonds in this network when compared to the active conformations, consistent with the larger amplitude motion.
Figure 2.
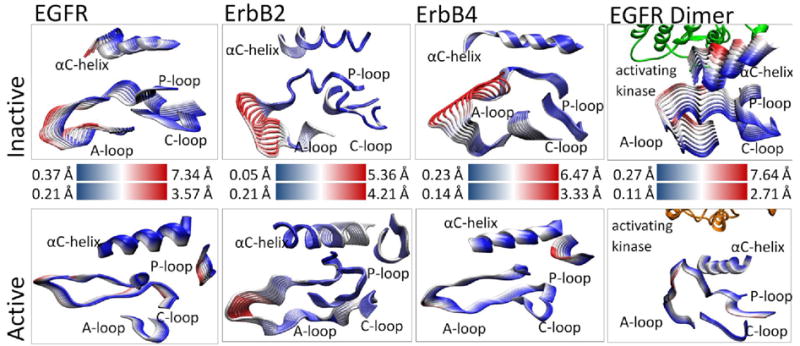
Visualization of the first principal component for the four key subdomains in the inactive and active conformation of the ErbB kinases. The motions are overlaid sequentially where the large-amplitude motion in each frame is highlighted in red and the low-amplitude motion is highlighted in blue. For the two dimer systems, the activating dimer is shown in orange and green. The inactive conformations exhibit large, localized motion while the active conformations demonstrate smaller, coordinated motions.
Table 1.
Persistent H-bonds and salt bridges in the three homologous ErbB kinase monomer systems. The salt bridges are in bold and homologous bonds are aligned. The table presents the subdomains with key conserved bonds across the ErbB family, see Table S1 for an exhaustive list.
| EGFR active | HER2 active | ErbB4 active | EGFR inactive | HER2 inactive | ErbB4 inactive |
|---|---|---|---|---|---|
| aC-helix A-loop bonds | |||||
|
| |||||
| – – | – – | – – | – – | – – | E739,R841 |
| E734,K851 | E766,K883 | E739,K856 | – – | – – | – – |
| D737,K836 | D769,R868 | – – | – – | – – | D742,R841 |
| E738,F832 | – – | E743,F837 | – – | – – | – – |
| – – | – – | – – | E738,K836 | – – | E743,R841 |
|
| |||||
| aC-helix C-loop bonds | |||||
|
| |||||
| – – | – – | – – | – – | – – | E743,R817 |
|
| |||||
| aC-helix bonds | |||||
|
| |||||
| – – | A763,S760 | – – | – – | – – | – – |
| – – | E766,R756 | – – | – – | – – | – – |
| E738,K721 | E770,K753 | E743,K726 | – – | – – | – – |
| – – | – – | – – | M742,L753 | M774,L785 | M747,L758 |
| A743,L679 | – – | A748,Q684 | – – | – – | – – |
| – – | – – | – – | – – | – – | A748,R757 |
|
| |||||
| C-loop C-loop bonds | |||||
|
| |||||
| – – | H843,D845 | – – | – – | – – | – – |
| – – | – – | – – | R812,D813 | R844,D845 | – – |
| D813,R817 | D845,R849 | D818,R822 | – – | – – | – – |
| D813,N818 | – – | – – | – – | – – | – – |
| A815,N818 | A847,N850 | A820,N823 | A815,N818 | A847,N850 | – – |
| – – | A848,V851 | – – | – – | – – | – – |
|
| |||||
| A-loop C-loop bonds | |||||
|
| |||||
| – – | – – | – – | – – | G865,V842 | – – |
| – – | – – | – – | – – | – – | G838,R817 |
| L834,R812 | L866,R844 | L839,R817 | – – | – – | – – |
| – – | – – | – – | L834,D813 | – – | – – |
| K836,V810 | R868,V842 | R841,V815 | – – | – – | – – |
| E848,R812 | – – | – – | – – | – – | – – |
| – – | – – | – – | K851,R812 | – – | – – |
|
| |||||
| A-loop bonds | |||||
|
| |||||
| – – | – – | D836,K726 | – – | D863,K753 | D836,K726 |
| – – | – – | D836,T835 | – – | – – | – – |
| L838,R808 | L870,R840 | L843,R813 | – – | – – | – – |
| – – | D871,R840 | – – | – – | – – | – – |
| A840,G672 | – – | – – | – – | – – | – – |
| – – | – – | – – | – – | D873,R897 | – – |
| – – | – – | K848,T873 | – – | – – | – – |
| K843,D932 | – – | K848,D937 | – – | – – | – – |
| – – | E876,R898 | – – | – – | – – | – – |
| – – | – – | E849,K871 | – – | – – | – – |
| Y845,Y867 | – – | Y850,F872 | – – | – – | – – |
| – – | – – | – – | H846,R865 | – – | – – |
| – – | – – | A852,R870 | – – | – – | – – |
| – – | D880,R897 | D853,R870 | E848,R865 | D880,R897 | D853,R870 |
| – – | – – | – – | – – | – – | G855,E730 |
| – – | – – | – – | – – | K883,E757 | – – |
| – – | – – | – – | – – | – – | K856,E844 |
Activating bond-network
We find several bonds are conserved across all members of the ErbB family in the active state (EGFR numbering is used in this discussion: see Table 1): two salt bridges: E734-K851 and E738-K721, three H-bonds: L834-R812, K836-V810, and L838-R808, as well as the bond D813-R817 which is a salt bridge in EGFR and ErbB4, but an H-bond in ErbB2. The E738-K721 salt bridge is highly conserved across all active kinases and helps coordinate the α and β phosphates of ATP bound in the active site. The E734-K851 salt bridge connects the A-loop and the αC-helix, coordinating the movements of these two sub-domains and dampening larger fluctuations. Similarly, three conserved H-bonds link the A-loop and the C-loop, coupling the motions of these two loops. These can be regarded as “fastening” H-bonds that maintain the N-terminal side of the A-loop open in its active state – the alternative (in the inactive state) being steric hindrance to the binding of ATP and peptide substrates. A fourth H-bond is seen only in ErbB2: E876-R898 fastens the C-terminal side of the A-loop open [31]. Although there is no analogous bond in EGFR and ErbB4, the Y845-Y867 bond in EGFR and the homologous Y850-F872 bond in ErbB4 serve a similar role. The conserved D813-R817 bond positions the sidechain of the catalytic aspartate D813 in the active site and thus likely facilitates the preorganization of the catalytic site in the active kinase system.
Inactivating bond-network
In contrast with the case for the active configurations, few intramolecular bonds in the inactive kinase conformation are conserved across the ErbB family (Table 1 and Table S2). In fact, only one such bond is conserved across EGFR, ErbB2 and ErbB4: a hydrogen bond between M742-L753 that pins the C-terminal end of the αC-helix in its location away from the active conformation. The inactive kinases do share an autoinhibitory pattern in which key residues required for kinase activation are sequestered. Similar to the situation described for Lck [26, 27], E738 of EGFR is salt bridged to K836 in a manner that ‘sequesters’ this glutamate and prevents it from forming the highly conserved E738-K721 salt bridge [31] required for full activation. For ErbB2 and ErbB4, the homologous residue K836 is replaced with an arginine. In ErbB4, this arginine contacts additional residues (as well as the E738 equivalent), apparently dampening the fluctuations of the αC-helix. In ErbB2 the arginine that replaces the K836 equivalent has flipped away from the αC-helix, so cannot sequester the E738 residue equivalent. In ErbB2 and ErbB4, the other half of the highly conserved salt bridge is sequestered; namely, the homologous K721 interacts with the D831 side chain, and this interaction in turn prevents K721 from forming the coordinating salt bridge.
Activation in the asymmetric dimer occurs through disruption of the inactivating bond-network
We also analyzed fluctuations in the EGFR kinase within the context of the asymmetric dimer described by Kuriyan and colleagues [20]. The fluctuations recorded for active EGFR in this context are very similar to those seen in the active EGFR monomer (Figure 2), with the conserved bonds described above being mostly preserved (Table S3). Slight variations in bond patterns in the active configuration include a shift from E734-K851 and L838-R808 in the monomer to E734-K836 and A840-R808 in the dimer, but dimerization has little overall effect on the activating bond network. In contrast, in the inactive dimer the first principal component reveals substantial motion of the αC-helix that is much greater than seen in the inactive monomer system (7.49Å shift in dimer compared to 4.14 Å in the monomer, see Figure 2). Simulations of the symmetric dimer interface of the inactive kinase in the unphosphorylated and phosphorylated states does not result in any conformational switching (see Figure S3), each system is stable in the inactive state. Furthermore, in previous simulations of ErbB2 [31] we used loop modeling to reduce the effect of steric clashes with no significant differences in dynamical motions. Therefore we attribute the motion of the αC-helix uniquely to the introduction of the asymmetric dimer interface. Even in the short timescale of 30 ns of the EGFR dimer trajectory, we observe a rearrangement of the αC-helix position towards the active conformation. Consistent with the allosteric activation mechanism proposed by Zhang et al. [20], several interactions in the inactivating bond network surrounding the A-loop and the αC-helix are indeed disrupted in the dimer trajectory, including Y740-S744, L834-D813, H846-R865, and K851-R812 interactions (Table S3). Some bonds (e.g. E738-K836) are still present, although the population statistics indicate that their survival percentage (fraction present in the trajectory) has decreased from >90% in the inactive monomer trajectory to ∼60% in the inactive dimer trajectory over 30ns.
When compared with their monomeric counterparts, the ErbB2 and ErbB4 inactive dimers demonstrate a similar loss of bonds surrounding the αC-helix and the A-loop (see Figure S4, Table S3 supplementary information and [31]). For ErbB4, a list of bonds disrupted upon dimerization includes: E739-R841, D742-R841, E743-R817, G838-R817, G855-E730, and K856-E844. Similar to the E738-K836 salt bridge in EGFR, the E743-R841 salt bridge in ErbB4 shows a marked decrease in survival time from >90% in the monomer trajectory to ∼70% in the dimer trajectory. Moreover, two of the bonds broken (E739-R841 and D742-R841) involve the R841 residue, resulting in a significant weakening of the bonds in the inactivating bond-network discussed above that sequester key side-chains in the inactive state.
EGFR mutations activate the kinase by disrupting the inactivating bond network
In light of the allosteric activation mechanism described in the context of the EGFR dimer system, we examined the effect of EGFR-activating mutations clinically identified in non-small cell lung cancer (NSCLC). The two mutants we examined are those most commonly found in NSCLC: a small in-frame deletion in the αC-helix (del 723-729 ins S) and a point mutation in the A-loop (L834R), see Figure 3. The deletion mutant physically shifts the αC-helix, removing residues 723-729 and leading to the shortening of a disordered segment that adjoins the αC-helix. This shifts the αC-helix toward the active state. The shortening of the αC-helix also alters the overall movement of the αC-helix, with a distinctly different motion from all other systems (Figure 3). The bond patterns we recorded in the dimer of the deletion mutant are similar to those of the wildtype (WT) inactive EGFR dimer (see Table S3, supplementary information).
Figure 3.
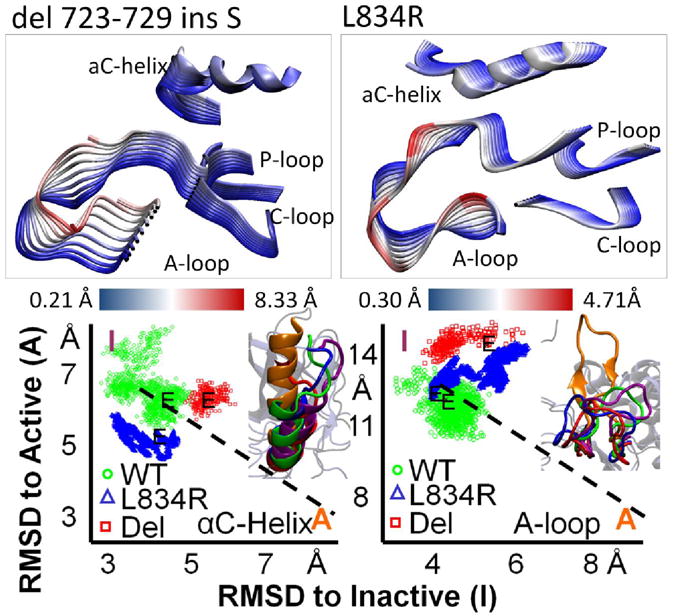
(Top) Comparison of the first principal component for the EGFR inactive mutant dimer systems: del 723-729 ins S and L834R (Bottom) Mapping of the RMSD of the inactive EGFR dimers as a scatter plot with RMSD from inactive on one axis and RMSD from active on the other. The end structures are visualized with an active reference (orange) and an inactive reference (purple). The conformational switching between active and inactive for EGFR of the A-loop and αC-helix are separable.
The majority of the differences between the two systems are seen in the αC-helix: the deletion mutant loses two bonds seen in the WT inactive dimer between the αC-helix and the A-loop: E738-F832 & E738-G833. The L834R mutation alters the conformation of the A-loop slightly; however, our trajectory shows the mutant kinase is still in a distinctly inactive conformation. The L834R mutant dimer system shares a similar bond pattern (Table S3) with the WT inactive EGFR dimer system with some exceptions: namely, G833 has H-bonded with H811, similar to the N-terminal fastening bonds that couple the A-loop and C-loop in the active state. Moreover, R834 has H-bonded to R865, representing a fastening bond to the C-lobe.
Hydrophobic interactions help differentiate active and inactive conformations
To investigate the effect of hydrophobic interactions on the ErbB kinase conformations, we analyzed the hydrophobicity as well as the solvent accessible surface area (SASA) of relevant hydrophobic sub-regions, namely, the C-spine, R-spine, hydrophobic core, and the αC-β4 region (Figure 1). The four regions have a high percentage of hydrophobic side chains; however, some minor differences between members of the ErbB family exist (Figure 1), particularly in the αC-β4 region. The hydrophobicity of the sub-region is a non-additive quantity, dependent not only upon the primary structure but also on the surrounding environment. Garde et al. [43, 44] have recently proposed an approach for quantifying the hydrophobicity of heterogeneous surfaces using normalized water density fluctuations (see Methods), according to which increased normalized water density fluctuations are used as a signature of a more hydrophobic surface.
The active conformations in the C-spine are noted by the elevated normalized water density fluctuations with the exception of EGFR monomer (where the difference is minor), see Figure 4A. The active conformations also consistently expose a mean surface area of around 500 Å2 across the members of the ErbB family (represented as a horizontal line), which is consistently less than the exposed surface area in the inactive conformations (mean value of over 700Å2), see Figure 4B. Together, these trends reflect a more hydrophobic C-spine region in the active compared to the inactive conformations. The hydrophobicity of the R-spine is not significantly different between the active and inactive conformations (Figure 4C). However, the R-spine analysis does reveal that the active conformations have a lower mean SASA value than the inactive conformations (Figure 4D); for some systems, the difference is not so clearly delineated in part because it consists of just four residues, so SASA is subject to larger fluctuations. With similar levels of water density fluctuations between active and inactive conformations, the decreased mean SASA values for the R-spines in the active conformations imply a more stable hydrophobic context, consistent with the finding that the well-formed spines are characteristic of the active kinase conformations. Interestingly, all ErbB2 systems present a higher fluctuation in water density, which is consistent with the notion that hydrophobicity is particularly important in the context of ErbB2 owing to its interaction with Hsp90 known to be mediated by hydrophobic contacts; see also the analysis surrounding the αC-β4 region, below. We also record a decrease of the R-spine SASA for inactive EGFR in the dimeric form compared to the monomeric form, with similar hydrophobicity (Figure 4C,D). The addition of the dimer interface in inactive EGFR reduces the mean SASA for the R-spine from 140Å2 to 80Å2, implying that dimerization can provide additional stabilization due to hydrophobic interactions in EGFR.
Figure 4.
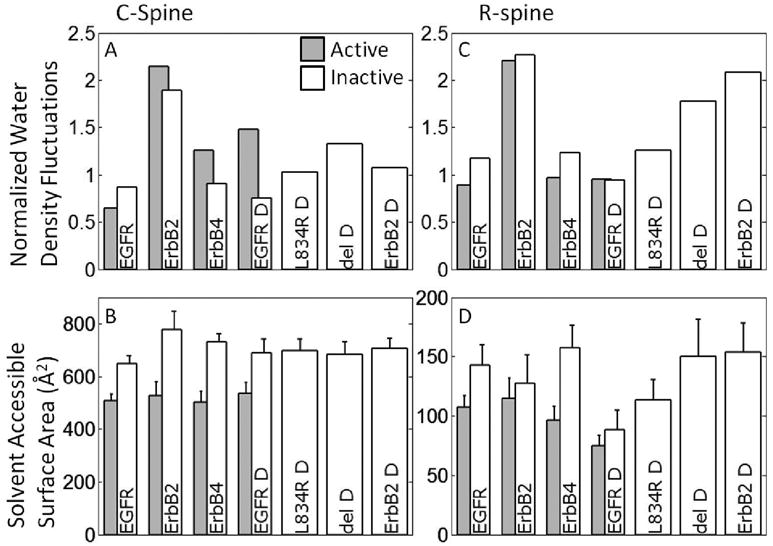
Normalized water density fluctuations and the Mean surface accessible (exposed) surface area (SASA) values for functionally important sub-regions, specifically: C-spine (panel A & B), and R-spine. For the normalized water density fluctuations (panels A, C, E) a higher value is correlated with a higher hydrophobicity, which would be expected to bury more SASA. See also Figure 5.
With respect to the water density fluctuations in the hydrophobic core, the inactive conformation is more hydrophobic than the active conformation (with the exception of ErbB2), see Figure 5A. This difference is consistent with the hydrophobic stabilization of the inactive conformation, especially for EGFR and ErbB4 but not for ErbB2. Notably, mutations of hydrophobic residues in the hydrophobic core are reported for EGFR and ErbB4 in clinical studies, (see Discussion section), whereas, for ErbB2, such mutations are found surrounding the αC-β4 region. The analysis in Figure 5A also implies that the difference in hydrophobicity between the inactive and active conformations is reduced in the dimer EGFR compared to monomer. This is consistent with the allosteric activation mechanism, namely that dimerization significantly reduces the hydrophobic advantage and provides a stimulus for activation. As for the SASA results, the hydrophobic core does not show a clear separation between the active and inactive conformations (Figure 5B). However, to describe the highly irregular nature of the surface of the hydrophobic core, the water density analysis may be more suitable that the SASA.
Figure 5.
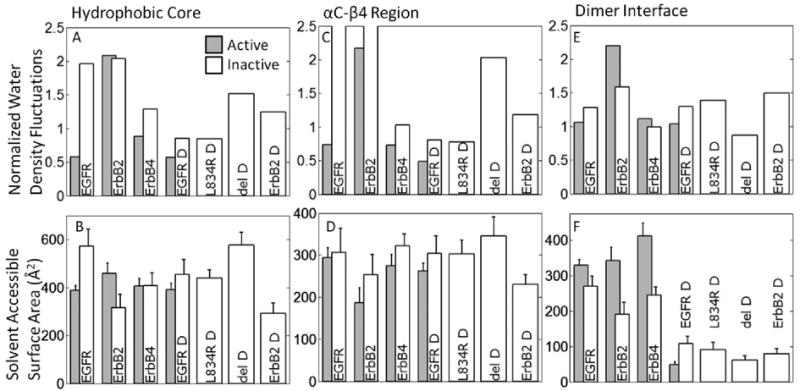
Normalized water density fluctuations and the Mean surface accessible (exposed) surface area (SASA) values for functionally important sub-regions, specifically: hydrophobic core (panel A & B), αC-β4 region (panel C & D) and dimer interface (panel E & F). For the normalized water density fluctuations (panels A, C, E) a higher value is correlated with a higher hydrophobicity, which would be expected to bury more SASA.
The αC-β4 region is an unstructured span between the αC-helix and the β4 sheet in RTKs. From a sequence perspective, only in ErbB2 is the αC-β4 region predominantly hydrophobic (Figure 1, see primary sequence; G776 in ErbB2 corresponds to S744 and S749 in EGFR and ErbB4, respectively; G778 in ErbB2 corresponds to D746 and D751 in EGFR and ErbB4, respectively). The water density analysis clearly reflects this trend by singling out the ErbB2 monomer systems are particularly hydrophobic (Figure 5C); the mean SASA for the αC-β4 region in ErbB2 systems is also consistently lower than in other members of the ErbB family (Figure 5D). As discussed in [31], this unique feature of ErbB2 is thought to be responsible for its preferential association with the molecular chaperone Hsp90.
The asymmetric dimer interface consists largely of hydrophobic side-chains in the N-lobe of the receiver kinase (L680, I682, L736, L758, and V762) and the C-lobe of the activator kinase (I917, Y920, M921, V924, and M928) [20]. With respect to water density analysis, the dimer interface presents a similar hydrophobicity across all members, with the ErbB2 monomers a little more hydrophobic, particularly in the active conformation (Figure 5E). The monomeric systems record a mean SASA value of approximately 350 Å2 for the dimer interface residues in the active conformations versus 200 Å2 in the inactive conformations (Figure 5F). This decrease for the inactive monomers may imply a preference for the inactive state in the monomer context. Notably, the dimeric systems record much lower SASA values of about 75 Å2, implying that hydrophobic stabilization provides a dominant driving force for dimerization; interestingly, for EGFR dimer, the SASA for the inactive state is greater than that for the active case and hence the preference for the inactive conformation is not implied in the context of the dimer. Thus, dimerization also provides a stimulus for activation.
Discussion
The analysis described here identifies conserved intramolecular non-covalent bond networks across the ErbB family kinases that emerge from sequence homology and lead to conserved dynamic characteristics as well as function. The hydrophilic and hydrophobic networks are cooperative interactions which help differentiate the active and inactive conformations while modulating key loop fluctuations and bonds. Investigation of the networks allows identification of network fragilities which can be exploited through mutation to alter the basal kinase activity.
The bond-networks highlight key conserved residues and bonds that are characteristic to each conformational state. Some of the bonds of been highlighted previously, the highly conserved salt bridge E738-K721 [16] and the fastening bonds L834-R812, K836-V810 and L838-R808 [31]. We add to this another salt bridge coordinating the αC-helix and the A-loop, E734-K851 salt bridge and the H-bond D813-R817 which facilitates the placement of the D813 aspartate side-chain in a catalytically competent orientation. Overall, the six conserved interactions tightly coordinate the N-lobe to the αC-helix, the αC-helix to the A-loop, and the A-loop to the C-loop. The nature of this bond network suggests that interactions among the nearest key subdomains in the ErbB family helps in maintaining the proximity of important catalytic residues, and positioning them so they can contribute directly to kinase activity. The conserved bond network in the inactive conformations of the ErbB family kinases is less extensive (in EGFR, E738-K836 and D831-K721), but appears to serve a crucial role in sequestering key catalytic residues and thus preventing activity.
Hydrophobic interactions appear to provide context-specific contributions to stability to the active and inactive conformations of ErbB kinases. Since a single amino acid change can alter the hydrophobicity of a region, we considered the water fluctuation analysis in conjunction with SASA to present the contributions from hydrophobicity for irregular surfaces characteristic of sub-regions in the kinase. Both SASA and fluctuation analyses of local water densities imply that the inactive monomer conformations of ErbB kinases are preferentially stabilized through hydrophobic interactions associated with the dimer interface. Moreover, consistent across the ErbB family, kinase domain dimerization further reduces the SASA of the dimer interface residues, implying that hydrophobic interactions provide a dominant impetus for dimerization.
The hydrophobic interactions with respect to the C-spine and R-spine regions are found to benefit the active conformations, overall; in particular, the SASA analysis suggests the active conformations benefit from a larger buried surface area). Interestingly, we found ErbB2 to possess a greater hydrophobicity in the spine regions. Thus, the αC-β4 region as well as the C-spine and R-spine are found to display uniquely hydrophobic character in ErbB2 consistent with its association with Hsp90. Also, in the case of the ErbB2 dimer interface, the active conformation is more hydrophobic than the inactive conformation, but also exposes more surface area, implying a destabilizing hydrophobic force in the active conformation; considering that ErbB2 does not have a known ligand, this could serve as an auto-inhibitory mechanism against activation. It is also intriguing to note that in the hydrophobic core, there is a large differential between the inactive and active conformations in the water fluctuations, consistent with preferential benefit for the inactive conformation in EGFR and ErbB4, but not for ErbB2. Thus, the analyses for the spine regions and the hydrophobic core collectively lead to the remarkable prediction that while the αC-β4 region and the R-spine confer hydrophobic benefit to the inactive conformation of ErbB2, the hydrophobic core has the same effect for EGFR and ErbB4. Indeed, this correlates well with clinical studies, where activating point mutations in the hydrophobic core have been found in EGFR and ErbB4 but those in the αC-β4 region are found in ErbB2, suggesting that the hydrophobic analysis enables the context-specific identification of fragile sub-regions.
The active kinase conformations have a greater number of conserved intramolecular persistent bonds (hydrophilic specific interactions) than the inactive systems, whereas the inactive kinase conformations appear to have a distinct hydrophobic advantage in the dimer interface region. This observation is fully consistent with the view that, for ErbB kinases, the dominant stimuli that activate them operate by modulating the dimer interface (activation mechanism in the wildtype) and the hydrophobic core regions (mode of activation in clinically identified mutation) to destabilize the inactive conformation, which we discuss below.
For the wildtype systems, this finding provides strong support for the allosteric mechanism associated with formation of the asymmetric dimer interface reported by Zhang et al. [20] and Qiu et al. [22], and the recent studies of juxtamembrane domain associations [23-25]. In this respect, the emerging view on allosteric EGFR regulation posits that, rather than forcing the protein to a new conformation, the allosteric interface guides the kinase domain to the active state through pre-existing conformational ensembles [46]. Our results for the EGFR dimer simulations (Figures 2, 3) are consistent with this view; namely, the allosteric dimer interface causes the αC-helix to alter the conformational space it samples and thus biases it towards a more active conformation. This leads to a metastable intermediate characterized by the αC-helix adopting an active-like conformation, albeit still remaining partially molten. We note that while we have captured the initial conformation change associated with the introduction of the allosteric asymmetric dimer interface which suggests a shift towards the active conformation, we are unable to capture the entire conformational shift because of limited timescales accessed in our simulations. We have also not attempted to compute the free energy landscape, which will be pursued in future studies.
We hypothesize a sequence of events that characterize the EGFR kinase activation pathway (see section S4, supplementary material): (1) Formation of the dimer interface triggers a shift of the αC-helix conformation toward the active state. (2) Formation of the additional helical turns in the αC-helix and the initiation of the A-loop movement, either happen concurrently or sequentially, but the A-loop does not complete its conformational switching until the αC-helix is fully formed. This sequence differs from the mapped pathway of the Src kinase Hck [47-49]; in Hck, following A-loop tyrosine phosphorylation the A-loop can adopt an open (active) conformation while the αC-helix is still in the inactive conformation. In the EGFR the opposite is true, namely, upon dimerization the αC-helix moves towards the active conformation with the A-loop in the closed (inactive) conformation (section S4).
Intriguingly, kinase activation in the two clinically identified EGFR activating mutants studied here (L834R and del 723-729 ins S) also appears to be governed by the same principles. The L834R mutation, apart from directly disrupting the hydrophobic core in-lieu of the hydrophilic Arg, leads to additional coupling of the A-loop to the C-loop (through the G833-H811 H-bond) and the A-loop to the C-lobe (through the R834-R865 interaction) not seen in other systems. Hence, the activating stimulus for this system possibly stems from a disruption of some of the hydrophobic interactions within the protein kinase responsible for stabilizing the inactive conformation. The deletion mutation, however, directly alters the αC-helix to cause a shift toward its active conformation, although it also disrupts the normal dynamics and potential extension of the αC-helix. Hydrophobic analysis also reveals that the L834R and deletion mutant function through different mechanisms. The deletion mutant has increased normalized water density fluctuations compared to L834R and the wildtype systems, particularly around the hydrophobic core but also increased SASA (see Figure 5A,B), indicating that the hydrophobic region is not shielded from water exposure. Moreover, based on Figure 5E,F, the dimer interface for the deletion mutant does not show a pronounced hydrophobicity indicated by a lower normalized water density fluctuations. Consistent with these salient observations for the deletion mutant, in the functional studies of Choi et al. [8], the deletion mutant showed an increase in basal phosphorylation, but a decrease in the overall phosphorylation under EGF stimulation. By interfering with the key interactions surrounding the helix, it can potentially stabilize the active conformation of the monomer kinase. However, upon dimerization the active conformation is destabilized because the deformation disrupts both the dimer interface due to its proximity and because the αC-helix cannot fully form. This justifies the dual response of the deletion mutant on kinase phosphorylation activity in the presence/absence of EGF stimulation.
The hydrophilic and hydrophobic bond networks we have identified also enable us to propose a set of mechanisms that increase the basal level of kinase activity for the other EGFR mutations in NSCLC [6-8]: E685S, G695S, S744I and L837Q, as well as a set of mutations in ErbB4 found in melanoma [50], namely, E836K, E872K and G936R. Similar to L834R, in the L837Q mutation, replacing the hydrophobic leucine side-chain with the hydrophilic glutamine side-chain is likely to cause it to rotate away from the small hydrophobic core between the αC-helix and the A-loop, thereby disrupting the hydrophobic core and inducing a local steric effect. This reconfiguration may also disrupt the interaction of K836 with E738. The S744I mutation in NSCLC most likely disrupts the M742-L753 bond, allowing transition of the αC-helix into its active position. E685G and G695S are in the N-lobe, close to the asymmetric dimer interface, so mutations here are likely to alter kinase activity either by increasing the dimerization affinity or by reconfiguring the EGFR RTK monomer by partially mimicking the formation of the asymmetric dimer interface. Two of the ErbB4 mutations seen in melanoma, E836K and E872K, are situated around the small hydrophobic core and the change from a negatively charged side chain to a positively charged chain is poised to disrupt the core's stability, thereby providing an activating stimulus. The G936R mutant in ErbB4 is located close to the asymmetric dimer interface in the C-lobe and potentially alters the dimerization affinity. Analogous mutations in ErbB2 have not been reported. Instead, the activating clinical mutations in ErbB2 are in αC-β4 region [51, 52], shown here to be uniquely hydrophobic in the ErbB family. Disruption of the hydrophobicity of the αC-β4 region would alter the binding to Hsp90 [53] and increase heterodimerization [54] and thus, activity of ErbB2. Characterization of another set of mutations in ErbB4 found in NSCLC, show two inactivating mutations G802dup and D861Y [55]. D861 is the start of the highly conserved DFG motif in RTKs and mutation of it would reduce activity, while the G802dup is spatially located near the P-loop and likely affects ATP binding affinity. Similarly in ErbB4, two of the point mutations are located near the active site: E836 is located next to the C-loop, whereas E872 is situated in the A-loop and hence owing to their proximity to the active site, the mutations are also poised to alter substrate binding and phosphorylation; such mechanisms are discussed in a recent computational study in the context of EGFR [56].
As mentioned in the Introduction, A-loop phosphorylation is thought not to be required for ErbB family kinase activation [19]. Consistent with this notion, although we record some context-specific structural changes between systems in which the A-loop is phosphorylated versus unphosphorylated (see section S1-S3, supplementary material), our results indicate that this phosphorylation does not provide a dominant activating effect. We reached a similar conclusion in recent studies of HER2 using a free energy perturbation approach [31].
In conclusion, our results help to establish the molecular context governing the stability and activation stimuli of ErbB kinases. Given that receptor tyrosine kinases are important signaling elements in cells, and mutations within these kinases cause subtle changes in molecular behavior that result in substantial alterations of downstream signaling [29], this work helps establish the crucial link between molecular mechanisms of kinase activation and the ensuing signaling response.
Supplementary Material
Acknowledgments
The authors thank members of the Lemmon and Radhakrishnan laboratories for helpful discussions. We acknowledge funding from NSF grants CBET- 0730955, CBET-0853389, CBET-0853539. Shannon E. Telesco received support through the NSF Graduate Research Fellowship, a GAANN Award, and a NIH T32 training grant. Computational resources were provided in part by NPACI under grant MCB060006.
Footnotes
Supplementary information for this manuscript is available online.
References
- 1.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 2.Hubbard SR, Till JH. Protein Tyrosine Kinase Structure and Function. Annual Review of Biochemistry. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 3.Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 4.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 5.Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Critical Reviews in Oncology/Hematology. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 6.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 7.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 8.Choi SH, Mendrola JM, Lemmon MA. EGF-independent activation of cell-surface EGF receptors harboring mutations found in gefitinib-sensitive lung cancer. Oncogene. 2007;26:1567–1576. doi: 10.1038/sj.onc.1209957. [DOI] [PubMed] [Google Scholar]
- 9.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2//neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 10.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE. Studies of the HER-2//neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 11.Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, Klein R, Lemke G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378:390–394. doi: 10.1038/378390a0. [DOI] [PubMed] [Google Scholar]
- 12.Tidcombe H, Jackson-Fisher A, Mathers K, Stern DF, Gassmann M, Golding JP. Neural and mammary gland defects in ErbB4 knockout mice genetically rescued from embryonic lethality. Proceedings of the National Academy of Sciences. 2003;100:8281–8286. doi: 10.1073/pnas.1436402100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linggi B, Carpenter G. ErbB-4 s80 Intracellular Domain Abrogates ETO2-dependent Transcriptional Repression. J Biol Chem. 2006;281:25373–25380. doi: 10.1074/jbc.M603998200. [DOI] [PubMed] [Google Scholar]
- 14.Kornev AP, Haste NM, Taylor SS, Ten Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proceedings of the National Academy of Sciences. 2006;103:17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proceedings of the National Academy of Sciences. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 17.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 18.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I. EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gotoh N, Tojo A, Hino M, Yazaki Y, Shibuya M, Gotoh N, Tojo A, Muroya K, Hashimoto Y, Hattori S, Nakamura S, Takenawa T, Yazaki Y, Shibuya M. EGFR mutant lacking the auto-phosphorylation sites induces phosphorylation of Shc protein and Shc-Grb2/Ash association and retains mitogenic activity. Biochem Biophys Res Commun. 1992;186:768–774. [Google Scholar]
- 20.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 21.Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated Phosphorylation of the Epidermal Growth Factor Receptor on Tyr845 and Tyr1101 Is Associated with Modulation of Receptor Function. J Biol Chem. 1999;274:8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 22.Qiu C, Tarrant MK, Choi SH, Sathyamurthy A, Bose R, Banjade S, Pal A, Bornmann WG, Lemmon MA, Cole PA, Leahy DJ. Mechanism of Activation and Inhibition of the HER4/ErbB4 Kinase. Structure. 2008;16:460–467. doi: 10.1016/j.str.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thiel KW, Carpenter G. Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proceedings of the National Academy of Sciences. 2007;104:19238–19243. doi: 10.1073/pnas.0703854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Red Brewer M, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA, Carpenter G. The Juxtamembrane Region of the EGF Receptor Functions as an Activation Domain. Molecular Cell. 2009;34:641–651. doi: 10.1016/j.molcel.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, Kuriyan J. Mechanism for Activation of the EGF Receptor Catalytic Domain by the Juxtamembrane Segment. Cell. 2009;137:1293–1307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozkirimli E, Post CB. Src kinase activation: A switched electrostatic network. Protein Science. 2006;15:1051–1062. doi: 10.1110/ps.051999206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozkirimli E, Yadav SS, Miller WT, Post CB. An electrostatic network and long-range regulation of Src kinases. Protein Science. 2008;17:1871–1880. doi: 10.1110/ps.037457.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dixit A, Verkhivker GM. Hierarchical Modeling of Activation Mechanisms in the ABL and EGFR Kinase Domains: Thermodynamic and Mechanistic Catalysts of Kinase Activation by Cancer Mutations. PLoS Comput Biol. 2009;5:e1000487. doi: 10.1371/journal.pcbi.1000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shih AJ, Purvis J, Radhakrishnan R. Molecular Systems Biology of ErbB1 Signaling: Bridging the Gap through Multiscale Modeling and High-Performance. Computing Molecular Biosystems. 2008;4:1151–1159. doi: 10.1039/b803806f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papakyriakou A, Vourloumis D, Tzortzatou-Stathopoulou F, Karpusas M. Conformational dynamics of the EGFR kinase domain reveals structural features involved in activation. Proteins: Structure, Function, and Bioinformatics. 2009;76:375–386. doi: 10.1002/prot.22353. [DOI] [PubMed] [Google Scholar]
- 31.Telesco SE, Radhakrishnan R. Atomistic Insights into Regulatory Mechanisms of the HER2 Tyrosine Kinase Domain: A Molecular Dynamics Study. Biophysical Journal. 2009;96:2321–2334. doi: 10.1016/j.bpj.2008.12.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 33.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. Journal of Physical Chemistry B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 35.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 36.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 1983;79:926–935. [Google Scholar]
- 37.Grubmuller H. Solvate 1.0. Theoretische Biophysik. Institut für Medizinische Optik, Universität München; Theresienstr: [Google Scholar]
- 38.Andersen HC. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. Journal of Computational Physics. 1983;52:24–34. [Google Scholar]
- 39.Essmann U. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 40.Scott EF, Yuhong Z, Richard WP, Bernard RB. Constant pressure molecular dynamics simulation: The Langevin piston method. The Journal of Chemical Physics. 1995;103:4613–4621. [Google Scholar]
- 41.Glykos NM, Kokkinidis M. Structural polymorphism of a marginally stable 4-alpha-helical bundle. Images of a trapped molten globule? Proteins. 2004;56:420–425. doi: 10.1002/prot.20167. [DOI] [PubMed] [Google Scholar]
- 42.Buck M, Karplus M. Hydrogen Bond Energetics: A Simulation and Statistical Analysis of N-Methyl Acetamide (NMA), Water, and Human Lysozyme. The Journal of Physical Chemistry B. 2001;105:11000–11015. [Google Scholar]
- 43.Godawat R, Jamadagni SN, Garde S. Characterizing hydrophobicity of interfaces by using cavity formation, solute binding, and water correlations. Proceedings of the National Academy of Sciences. 2009;106:15119–15124. doi: 10.1073/pnas.0902778106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acharya H, Vembanur S, Jamadagni SN, Garde S. Mapping hydrophobicity at the nanoscale: Applications to heterogeneous surfaces and proteins. Faraday Discuss. 2010 doi: 10.1039/b927019a. In Press. [DOI] [PubMed] [Google Scholar]
- 45.Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 46.Tsai CJ, Sol Ad, Nussinov R. Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms. Molecular BioSystems. 2009;5:207–216. doi: 10.1039/b819720b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang S, Banavali NK, Roux Bt. Proceedings of the National Academy of Sciences. Vol. 106. 2009. Mapping the conformational transition in Src activation by cumulating the information from multiple molecular dynamics trajectories; pp. 3776–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banavali NK, Roux B. Anatomy of a structural pathway for activation of the catalytic domain of Src kinase Hck. Proteins: Structure, Function, and Bioinformatics. 2007;67:1096–1112. doi: 10.1002/prot.21334. [DOI] [PubMed] [Google Scholar]
- 49.Gan W, Yang S, Roux B. Atomistic View of the Conformational Activation of Src Kinase Using the String Method with Swarms-of-Trajectories. Biophysical Journal. 2009;97:L8–L10. doi: 10.1016/j.bpj.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA, Samuels Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41:1127–1132. doi: 10.1038/ng.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, Stevens C, O'Meara S, Smith R, Parker A, Barthorpe A, Blow M, Brackenbury L, Butler A, Clarke O, Cole J, Dicks E, Dike A, Drozd A, Edwards K, Forbes S, Foster R, Gray K, Greenman C, Halliday K, Hills K, Kosmidou V, Lugg R, Menzies A, Perry J, Petty R, Raine K, Ratford L, Shepherd R, Small A, Stephens Y, Tofts C, Varian J, West S, Widaa S, Yates A, Brasseur F, Cooper CS, Flanagan AM, Knowles M, Leung SY, Louis DN, Looijenga LH, Malkowicz B, Pierotti MA, Teh B, Chenevix-Trench G, Weber BL, Yuen ST, Harris G, Goldstraw P, Nicholson AG, Futreal PA, Wooster R, Stratton MR. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–526. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 52.Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, Carpenter G, Gazdar AF, Muthuswamy SK, Arteaga CL. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10:25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 53.Xu W, Yuan X, Xiang Z, Mimnaugh E, Marcu M, Neckers L. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat Struct Mol Biol. 2005;12:120–126. doi: 10.1038/nsmb885. [DOI] [PubMed] [Google Scholar]
- 54.Citri A, Gan J, Mosesson Y, Vereb G, Szollosi J, Yarden Y. Hsp90 restrains ErbB-2/HER2 signalling by limiting heterodimer formation. EMBO Rep. 2004;5:1165–1170. doi: 10.1038/sj.embor.7400300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tvorogov D, Sundvall M, Kurppa K, Hollmen M, Repo S, Johnson MS, Elenius K. Somatic Mutations of ErbB4. Journal of Biological Chemistry. 2009;284:5582–5591. doi: 10.1074/jbc.M805438200. [DOI] [PubMed] [Google Scholar]
- 56.Liu Y, Radhakrishnan R. Computational Delineation of the Tyrosyl-Substrate Binding, Pre-Catalytic, and Catalytic Landscapes in the Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Domain. 2010 doi: 10.1039/c3mb70620f. To be submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.