

Abstract
Mitochondria have long been recognized for their central role in energy transduction and apoptosis. More recently, extensive work in multiple laboratories around the world has significantly extended the role of cardiac mitochondria from relatively static arbitrators of cell death and survival pathways to highly dynamic organelles that form interactive functional networks across cardiomyocytes. These coupled networks were shown to strongly affect cardiomyoyte responses to oxidative stress by modulating cell signaling pathways that strongly impact physiological properties. Of particular importance is the role of mitochondria in modulating key electrophysiological and calcium cycling properties in cardiomyocytes, either directly through activation of a myriad of mitochondrial ion channels or indirectly by affecting cell signaling cascades, ATP levels, and the over-all redox state of the cardiomyocyte. This important recognition has ushered a renewed interest in understanding, at a more fundamental level, the exact role that cardiac metabolism, in general and mitochondria, in particular, play in both health and disease. In this article, we provide an overview of recent advances in our growing understanding of the fundamental role that cardiac mitochondria play in the genesis of lethal arrhythmias.
Keywords: Ischemia-reperfusion injury, mitochondria, reactive oxygen species. Arrhythmias
INTRODUCTION
Mitochondria are well recognized for their importance in energy production and apoptosis (Gustafsson & Gottlieb, 2008). They generate ATP through oxidative phosphorylation, driven by electron transport across the electron transport chain. In addition, mitochondria generate reactive oxygen species (ROS) which have diverse cell signaling functions (Droge, 2002; Becker, 2004). A key metric of mitochondrial function is the mitochondrial membrane potential (Δψm) which forms the proton-motive force used to produce ATP (O’Rourke, 2007). In normal hearts, Δψm is tightly regulated such that the production of ATP is maintained within a physiological range that matches energy production to demand (O’Rourke, 2007). This limits ROS generation and oxidative stress. In ischemia-reperfusion injury, Δψm is disrupted, altering over-all energy and redox balance within cardiac myocytes (Honda et al., 2005).
Seminal work has extended the role of mitochondria from static arbitrators of cell death and survival pathways to highly dynamic organelles that form interactive networks across cardiomyocytes. These coupled networks strongly affect cardiomyocyte responses to oxidative stress by modulating cell signaling pathways. This important recognition has ushered a renewed interest in understanding, at a more fundamental level, the exact role that cardiac mitochondria play in both health and disease (Michelakis, 2008). In this review, we focus on the role of mitochondrial dysfunction in promoting cardiac electrophysiological abnormalities at the cellular level and malignant arrhythmias at the tissue-network level.
Mitochondrial Criticality and Metabolic Oscillations
Zorov et al (Zorov et al., 2000; Zorov et al., 2006) advanced the notion of ROS-induced ROS-release (RIRR) to explain how local ROS injury within a discrete region of a cardiomyocyte can rapidly accumulate across a critical mass of the mitochondrial network to cause cellular oxidative stress. In these studies, RIRR was described as a fundamental mechanism by which cardiac mitochondria respond to elevated ROS levels by stimulating endogenous ROS production in a regenerative, autocatalytic process that ultimately results in cellular dysfunction and death (Zorov et al., 2006).
Distinct modes of RIRR have been postulated based on their dependence on various mitochondrial ion channels (Yang et al.). Specifically, Zorov et al (Zorov et al., 2000; Zorov et al., 2006) demonstrated a convincing relationship between the destabilization of Δψm upon mitochondrial oxidation and the induction of the mitochondrial permeability transition which causes apoptosis (Zorov et al., 2000). On the other hand, studies by Aon et al. (Aon et al., 2003) provided strong evidence in support of the inner membrane anion channel (IMAC) as a mediator of RIRR and associated electrophysiological and metabolic instabilities. In these studies, photo-induced oxidation of a discrete region within the cardiac myocyte unleashed a regenerative process of RIRR that was dependent on IMAC activation and not the mPTP. Once a threshold level of ROS was exceeded across a critical mass of the mitochondrial network (ie mitochondrial criticality), sustained Δψm oscillations were initiated (Aon et al., 2006; Aon et al., 2009). Similar Δψm oscillations are also generated in isolated myocytes subjected to oxidative stress via substrate deprivation (Romashko et al., 1998), ATP depletion (Ryu et al., 2005), diamide (Aon et al., 2007), and respiratory inhibition (Ryu et al., 2005). Recent evidence using two-photon microscopy confirmed these cellular data as reversible collapses in Δψm were observed in intact hearts exposed to global ischemia/reperfusion or diamide administration (Slodzinski et al., 2008). As will be discussed next, these mitochondrial oscillations can result in cellular electrophysiological oscillations via cyclical activation of sarcolemmal K-ATP (sarcKATP) channels providing compelling evidence of a mechanistic link between mitochondrial dynamics and cellular electrical dysfunction. In what follows, we describe the downstream ionic mediator of electrical inexcitability caused by mitochondrial dysfunction, followed by a discussion of key upstream mechanisms that regulate arrhythmias, including mitochondrial ion channels and the redox state of the cardiomyocyte.
Down-stream mediator of metabolic stress: Role of sarcolemmal KATP channels
SarcKATP channels link membrane excitability to metabolism (Nichols, 2006). They are regulated by intracellular nucleotides, membrane phospholipids, protein kinases and phosphatases (Nichols, 2006). SarcKATP channel activation can precede cellular ATP depletion because the open probability of these channels is increased when cofactors like ADP, pH and Mg2+ begin to rise. SarcKATP channels activate rapidly when mitochondria uncouple because the drop in Δψm due to increased proton leak causes the reversal of the ATP synthase, thus consuming cytoplasmic ATP and decreasing the phosphorylation potential. Tight coupling between the mitochondrial energy state and sarcKATP channel activation is facilitated by the high energy phosphoryl transfer reactions of the cytoplasm (Sasaki et al., 2001).
Due to their abundance in the plasma membrane, the opening of sarcKATP channels causes rapid action potential shortening, loss of intracellular K+, and reduction in myocyte excitability (Billman, 2008). In fact, increased K+ conductance through sarcKATP channels can effectively lock the resting membrane potential close to the equilibrium potential for K+ (Kleber, 1983). Indeed, sarcKATP channel activation accounts for most of the action potential shortening during ischemia, as evidenced by the ability of KATP channel blockers (ie, glibenclamide) to prevent the decrease in action potential duration during early ischemia (Akar et al., 2005).
The dynamic relationship between sarcKATP channel activation and the metabolic status of the cardiomyocyte was first observed by O’Rourke and colleagues (O’Rourke et al., 1994; O’Rourke et al., 1995). Following metabolic stress either by substrate deprivation or increased ADP levels, sarcKATP currents were activated in phase with NADH fluctuations. In these experiments, sustained Δψm oscillations occurred in phase with cellular electrophysiological (namely action potential) oscillations that were driven by ‘out-of-phase’ sarcKATP current activation (Aon et al., 2003).
While sarcKATP channel activation is thought to protect the viability of ischemic tissue by limiting calcium cycling and force generation during periods of reduced energy supply, increased potassium conductance through these channels predisposes to electrical dysfunction and arrhythmias (Billman, 1994; Billman et al., 1998; Billman, 2008). The pro-arrhythmic potential of sarcKATP channel activation during ischemia-reperfusion could be attributed to increased dispersion of repolarization and shortening of the effective refractory period, and therefore the cardiac wavelength, at a time when calcium mediated triggers are known to arise. Moreover, the opening of sarcKATP channels creates a current sink which can slow or block conduction wavefronts in local regions where the open probability of sarcKATP channels is high (i.e. where the energetic status of the cell is compromised), a phenomenon that we previously termed ‘metabolic sink’ (Akar et al., 2005).
This pro-arrhythmic potential of sarcKATP channel activation has been confirmed in multiple studies. Preventing sarcKATP channel activation by pharmacological blockade of the channel decreased the incidence of ventricular arrhythmias in rat (Vajda et al., 2007), rabbit (Fischbach et al., 2004), pig (Wirth et al., 1999), dog (Billman et al., 1998), and man (Cacciapuoti et al., 1991; Lomuscio et al., 1994; Aronson et al., 2003). On the other hand, sarcKATP channel blockade with glibenclamide failed to delay the onset of inexcitability during late ischemia or the initiation of arrhythmias upon reperfusion in the ex vivo perfused guinea pig heart. In order to understand the factors driving the opening of sarcKATP channels during metabolic stress, an overview of key mitochondrial ion channels and bioenergetic properties are discussed below.
Mitochondrial ion channels as root causes of mitochondrial dysfunction and arrhythmias
The mitochondrial membrane is a highly resistive structure that maintains a large voltage gradient and proton-motive force, required for electron transport and ATP production (Brown et al., 2010). Nonetheless, a rich diversity of ion channels and transporters has been discovered in the inner and outer membranes of mitochondria. Of note to arrhythmia mechanisms are various ion channels (Figure 1) that modulate Δψm and also promote apoptosis (mPTP), cellular inexcitability (IMAC), cardioprotection (mitoKATP), and mitochondrial calcium influx (MCU). The interested reader is referred to excellent reviews that exclusively cover mitochondrial ion channel targets in a more comprehensive manner (Peixoto et al.).
Figure 1.
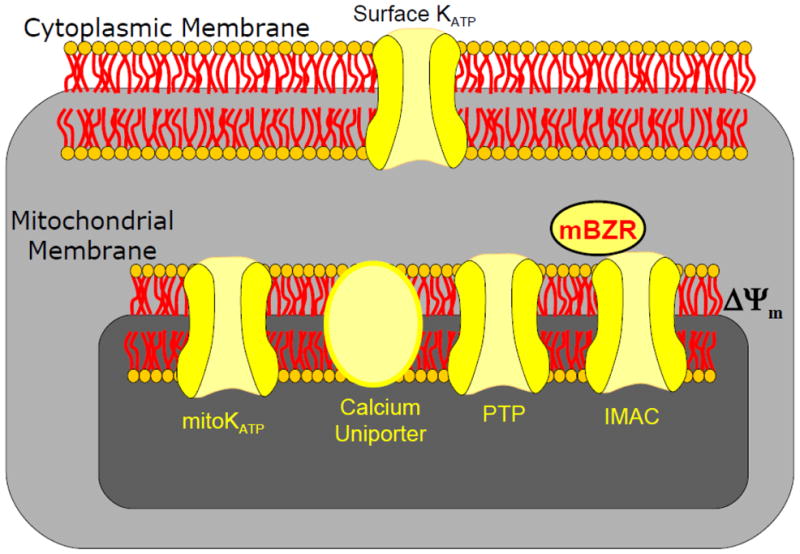
Schematic of key energy sensitive ion channels that can promote cell survival, death, or arrhythmias.
Inner Membrane Anion Channel
Anion flux across the inner mitochondrial membrane was observed in early studies in which anion movement was shown to regulate mitochondrial volume (Azzi & Azzone, 1966, 1967; Brierley, 1970). Since then, the existence of IMAC has been confirmed in multiple studies demonstrating its importance in anion efflux from energized mitochondria (Garlid & Beavis, 1986; Beavis, 1992). Although the exact structure and molecular identity of IMAC remain elusive, the tight regulation of this channel by benzodiazepine compounds (Beavis, 1989) suggests a strong association between a partially anion selective pore-forming subunit in the inner membrane and a peripheral benzodiazepine receptor in the outer membrane.
The importance of IMAC in modulating Δψm was first noted when several distinct IMAC ligands were shown to prevent pathological Δψm oscillations in isolated cardiac myocytes (Aon et al., 2003). Importantly, blocking Δψm oscillations by targeting the IMAC also inhibited action potential oscillations and prevented myocyte inexcitability (Aon et al., 2003). This provided indirect evidence that targeting the IMAC may be an effective strategy for preventing arrhythmias, at least at the cellular level.
Indeed, IMAC blockade successfully prevented post-ischemic arrhythmias in intact myocardium (Akar et al., 2005; Brown et al., 2008b; Brown et al., 2010). Optical mapping of the epicardial surface of guinea pig hearts revealed that IMAC blockade decreased ischemia-induced action potential shortening and markedly suppressed the incidence of ventricular tachycardia/fibrillation during the early onset of reperfusion (Akar et al., 2005). Cardioprotection mediated by IMAC blockade was also observed in isolated rabbit hearts and accompanied by improved left ventricular function (Brown et al., 2008b). Of notable clinical interest, reperfusion arrhythmias in both studies were also prevented when the IMAC blocker was delivered as a bolus injection at the onset of reperfusion (Akar et al., 2005; Brown et al., 2008b).
Mitochondrial Permeability Transition Pore
The role of the mitochondrial permeability transition pore (mPTP) in ischemia/reperfusion injury has received considerable attention (Halestrap et al., 2004; Murphy & Steenbergen, 2008; Halestrap, 2009; Halestrap & Pasdois, 2009). It is clear that the opening of the mPTP plays a significant role in the generation of necrotic and apoptotic cell death, both of which are involved in the etiology of myocardial infarction (McCully et al., 2004). Administration of cyclosporin-A or sanglifehrin-A, both blockers of the mPTP, attenuates myocardial infarction (Weinbrenner et al., 1998; Minners et al., 2000; Hausenloy et al., 2002; Argaud et al., 2004), left ventricular dysfunction (Griffiths & Halestrap, 1993; Clarke et al., 2002; Hausenloy et al., 2004; Oka et al., 2008a), cardiomyocyte death (Nazareth et al., 1991; Duchen et al., 1993; Kim et al., 2006), and ischemia-reperfusion injury (Di Lisa et al., 2001; Oka et al., 2008b). The translation of these findings was recently supported in a clinical study, in which administration of cyclosporin-A immediately prior to percutaneous coronary intervention decreased the extent of short-term injury in a small clinical trial (Piot et al., 2008).
While the role of the mPTP in cell death is well established, its involvement in the generation of arrhythmias remains controversial. While some studies showed moderate protection against arrhythmias, other studies confirmed a lack of protection in rat (Dow et al., 2009), guinea pig (Akar et al., 2005), and rabbit (Brown et al., 2008b) hearts. Moreover, delivery of a cyclosporin-A bolus prior to stenting did not seem to influence the incidence of ventricular fibrillation in humans (Piot et al., 2008). Lack of protection against arrhythmias by mPTP blockade is also supported by mechanistic studies in isolated myocytes that demonstrate that Δψm depolarization caused by substrate deprivation or photo-induced oxidation is not prevented by cyclosporin-A (Romashko et al., 1998; Huser & Blatter, 1999; Zorov et al., 2000; Aon et al., 2003). It is very important to note, however, that by protecting against apoptosis and reducing the size of myocardial infarction following an ischemic insult, mPTP blockade may suppress scar related arrhythmias that are associated with healed myocardial infarction. Also, by inhibiting myocyte loss and improving left ventricular function, this strategy may confer an anti-arrhythmic effect through beneficial mechano-electrical feedback or by hindering the progression of adverse electrical remodeling.
MitoKATP Channel
Evidence for a mitochondrial ATP-sensitive potassium (mitoKATP) channel was first observed in rat liver mitochondria (Inoue et al., 1991), and later confirmed in heart (Paucek et al., 1992). The opening of mitoKATP channels may underlie the cardioprotective effects of preconditioning stimuli by partial dissipation of Δψm, reduction in the driving force for calcium entry into the mitochondrial matrix, inhibition of apoptosis, and overall improvement in cellular respiration ((O’Rourke, 2000; Gross & Peart, 2003).
Numerous studies have examined the role of mitoKATP channel activation/blockade in altering infarct size (Takashi et al., 1999; O’Rourke, 2004). In general, mitoKATP channel blockade with 5-hydroxydecanoate (5-HD) abolished the ability of the cardioprotective stimulus to reduce infarct size (Takashi et al., 1999). While these studies have yielded important mechanistic insights, it is noteworthy that mitoKATP channel opening also fails to evoke a cardioprotective response when repetitive preconditioning stimuli, such as multiple cycles of ischemia/reperfusion (Schwartz et al., 2002) or chronic exercise (Brown et al., 2005) are administered prior to the main insult, confounding the translation of this strategy to clinical use.
Few studies have examined the role of mitoKATP channels in the genesis of cardiac arrhythmias. A protective role for mitoKATP channel activation against arrhythmias has been inferred by experiments demonstrating that mitoKATP channel blockers consistently abolished the anti-arrhythmic phenotype provided by preconditioning stimuli, such as ischemic preconditioning (Vegh & Parratt, 2002; Rajesh et al., 2004), adenosine (Headrick et al., 2003), delta opioid agonists (Fryer et al., 2000; Fischbach et al., 2003), estrogen (Das & Sarkar, 2006), 3-nitropropionic acid (Basgut et al., 2008), nitroglycerin (Baharvand et al., 2009), noradrenaline (Imani et al., 2008), or endothelin receptor agonists (Das et al., 2007). It is important to note, however, that mitoKATP channel blockade during other preconditioning stimuli; namely, bradykinin (Driamov et al., 2004), low-flow ischemia (Driamov et al., 2004), peroxynitrite (Kiss et al., 2008), and estradiol (Tsai et al., 2002) failed to attenuate the anti-arrhythmic protection of these stimuli.
Studies investigating the efficacy of direct mitoKATP channel activation on the suppression of post-ischemic arrhythmias have yielded discrepant results (Schwartz et al., 2002; Headrick et al., 2003). One putative explanation for the discordant findings is that various pharmacological agents used to open mitoKATP channels are confounded by non-specific action. In fact, the non-specificity of mitoKATP channel openers (such as diazoxide) and blockers (such as 5-HD) has received considerable attention in recent years (Hanley et al., 2003; Suzuki et al., 2003; O’Rourke, 2004; Brown et al., 2005; Hanley et al., 2005). Moreover, mitoKATP channel activity is largely dependent on complex signaling cascades, including phosphorylation by protein kinase C (Ohnuma et al., 2002), which may be differentially altered in various studies.
While the preconditioning literature provides interesting mechanistic insights regarding anti-arrhythmic strategies administered before index ischemia, the clinical relevance of these strategies should be put into question. To the clinician, arrhythmia suppression must often be attempted after, not before, the onset of the ischemic insult. Targeting mitoKATP channels after the onset of metabolic stress seemed promising based on cellular studies, in which the administration of mitoKATP channel openers effectively inhibited ongoing Δψm oscillations that were evoked by halting respiration (Ryu et al., 2005). This strategy also improved cellular survival and mitochondrial integrity during cellular reoxygenation (Ozcan et al., 2007). Despite these encouraging cellular findings, post-ischemic administration of mitoKATP channel openers failed to decrease the incidence of arrhythmias (Das & Sarkar, 2005).
Mitochondrial Calcium Uniporter
Although altered intracellular calcium cycling and cytosolic calcium overload are well established sources of arrhythmia triggers and beat-to-beat repolarization abnormalities (Wilson et al., 2006), the role of mitochondrial calcium fluxes in the generation of arrhythmias remains unclear. Mitochondrial calcium homeostasis is achieved by balanced calcium influx into the matrix via the mitochondrial calcium uniporter (MCU) and efflux out of the matrix through the mitochondrial sodium–calcium exchanger. MCU blockade with ruthenium compounds has shown some promise in suppressing the incidence of arrhythmias. Specifically, pre-ischemic administration of both ruthenium red and Ru360 decreased the incidence of ventricular fibrillation upon reperfusion in rats (Garcia-Rivas Gde et al., 2006). Moreover, both compounds converted ongoing ventricular fibrillation to ventricular tachycardia when administered after the onset of arrhythmias, although neither compound led to sinus rhythm (Kawahara et al., 2003).
Mechanisms by which MCU blockade protects against arrhythmias are not well understood but may involve a decrease in the open channel probability of the mPTP by maintaining relatively low matrix calcium concentrations (Garcia-Rivas Gde et al., 2006). While this is largely expected to confer an anti-apoptotic effect, it seems unlikely to play a major role in arrhythmogenesis since blockers of the mPTP have not been particularly effective in preventing arrhythmias, as discussed previously. Indeed, these findings are supported by cellular experiments in which the reversible collapse in Δψm induced during RIRR was not prevented by either ruthenium red (Romashko et al., 1998) or Ru360 (Zorov et al., 2000).
The exact role of the mitochondrial calcium uniporter in arrhythmogenesis remains unclear because of major confounding effects of the ruthenium compounds on intracellular calcium fluxes (Griffiths, 2000). For example, Ruthenium red blocks calcium entry through L-type calcium channels (Vassilev et al., 1987) and release from the sarcoplasmic reticulum (Gupta et al., 1989), suggesting that the anti-arrhythmic efficacy of this compound may be related to its prevention of intracellular calcium overload and not to its primary mitochondrial target (Griffiths & Rutter, 2009). Ru360 appears to be more specific for the MCU, but whole heart experiments are confounded by permeability issues, with some investigators showing successful drug entry into myocytes (Kawahara et al., 2003) and others arguing against it (Robert et al., 2001; Bell et al., 2006). Consistent with their ability to reduce cytosolic calcium transients, both ruthenium compounds are potent negative inotropes at concentrations that protect against arrhythmias (Gupta et al., 1988; Kimura et al., 2005), an undesirable side effect when the overall purpose of administering the compound is to improve cardiac function. Future research using novel compounds that lack these pleiotropic/permeability issues will provide better insights into the role of the MCU in post-ischemic arrhythmias.
Anti-oxidant depletion as a mechanism of mitochondrial dysfunction and arrhythmias
Oxidative stress in cardiomyocytes is caused by either increased ROS production and/or reduced scavenging capacity. In fact, myocardial Glutatione (GSH), a main anti-oxidant defense system in myocytes, is a key regulator of RIRR and mitochondrial stability. Interestingly, depletion of the intracellular antioxidant GSH pool with diamide effectively triggers Δψm oscillations that are similar in nature to those generated by photo-induced oxidation of the myocyte (Aon et al., 2007). These observations were extended to the level of the whole heart, in which diamide treatment of ex vivo perfused hearts resulted in heterogeneous ROS production, Δψm depolarization (Slodzinski et al., 2004) and ventricular fibrillation (Brown et al., 2008a). Interestingly, reduced-to-oxidized glutathione ratio (GSH/GSSG) in whole heart homogenates following diamide administration was similar to that in isolated cells undergoing RIRR and Δψm oscillations (Aon et al., 2007). These findings are corroborated by human data, where low GSH/GSSG ratios were observed in human heart samples from patients with heart failure (Damy et al., 2009) and type 2 diabetes (Anderson et al., 2009), both important risk factors for cardiac arrhythmias and sudden death. Consistent with this notion, administration of N-acetylcycsteine significantly decreased the incidence of cardiac arrhythmias in patients following cardiac surgery (Ozaydin et al., 2008). While promising, N-acetylcycsteine itself is confounded by limited bioavailability (Holdiness, 1991) and anaphylactoid-like reactions (Holdiness, 1991). This clearly highlights the need for alternative compounds that can more effectively and safely restore GSH levels.
Finally, the redox state of the cardiomyocyte can also modulate its excitability properties through mitochondria-independent mechanisms. For example, increased oxidation has been shown to directly activate sarcKATP channels (Tokube et al., 1996), alter the inactivation kinetics of L-type calcium channels, decrease sodium current density (Liu et al., 2010), increase ryanodine receptor calcium ‘leak’ (Belevych et al., 2009), and modulate the activation state of mitochondrial inner membrane ion channels. Attempts to improve the redox status of the cardiomyocyte by scavenging ROS with superoxide dismutase mimetics (Konya et al., 1992) or mitochondria-targeted anti-oxidant peptides (Cho et al., 2007) were successful in decreasing the incidence of arrhythmias. Future experiments that optimize effective delivery of ROS-scavenging agents to mitochondria have clear potential in abrogating electrical abnormalities caused by metabolic dysfunction.
Spatio-temporal dynamics of mitochondrial function across the intact heart
As mentioned above, Δψm depolarization is triggered by opening of mitochondrial ion channels under conditions of oxidative stress (Weiss et al., 2003; O’Rourke, 2007; Brown et al., 2010). Specifically, during metabolic insults, increased mitochondrial ROS production from complex III of the electron transport chain triggers the opening of IMAC and/or mPTP (Weiss et al., 2003). This results in ROS release from mitochondria and Δψm depolarization. In isolated cardiomyocytes, ROS diffusion within the cytosol triggers further ROS release from neighboring mitochondria, initiating a feedback cycle of RIRR and Δψm depolarization (Zhou et al.,; Zorov et al., 2000; Aon et al., 2003; Brady et al., 2004).
Despite major advances in our understanding of mitochondrial biochemistry at the subcellular/molecular levels, the pathophysiological consequences of mitochondrial dysfunction at the level of the intact heart remained unclear. Since mitochondrial function of individual cells is highly influenced by network properties, it is critical to investigate mitochondrial function within the milieu of the intact heart (Weiss et al., 2006). We recently found that the metabolic substrate of the heart during the early onset of ischemia is spatially and temporally heterogeneous (Lyon et al., 2010b). These spatio-temporal heterogeneities in mitochondrial function may ultimately dictate myocardial excitability and contribute to the formation of zones of conduction block by heterogeneous activation of surface KATP channels, as we had previously speculated (Akar et al., 2005).
Δψm depolarization
A semi-quantitative approach of optical Δψm imaging in the ex vivo perfused heart allowed the identification of waves of Δψm depolarization that actively propagate across the myocardium with a mean velocity of ~20μm/sec (Figure 2), several orders of magnitude slower than myocardial action potential propagation (Lyon et al., 2010a). We further elucidated complex spatio-temporal metabolic instabilities that preceded and accompanied the formation of these organized waves (Figure 2). Furthermore, we identified at the tissue level the presence of Δψm ripples prior to mitochondrial collapse during ischemia. These data suggested patterns of wave behavior spreading across the myocardium ahead of the main wave of Δψm depolarization, with propagation reflecting the direct interaction between adjacent cells within the intact ischemic tissue. Although we did not directly image ROS levels, it is conceivable that ROS diffusion at the interface between depolarized (acting as ROS sources) and polarized (ROS sinks) regions can drive the propagation of Δψm collapse, in a manner that extends the notion of RIRR from a subcellular to a multi-cellular phenomenon. The amplification and propagation of Δψm depolarization across the electrically coupled syncytium may present novel opportunities to limit injury by potentially targeting areas of early Δψm collapse that form the origin of the organized propagating wavefront of mitochondrial dysfunction.
Figure 2. Spatio-temporal fluctuations of Δψm during global ischemia (Adapted from Figure 4, Lyon et al. J Mol Cell Cardiol. 2010, PMID: 20624394).

Successive contour maps of normalized Δψm (above) and its first derivative (below) acquired at 10, 40, 70, and 180 seconds following the onset of global no-flow ischemia in a representative rat heart. These data illustrate the presence of spatially and temporally discordant kinetics of Δψm that exist ahead of the main depolarization wave of Δψm collapse, which actively propagates across the heart. Color scale: a) Δψm contour maps: baseline (black), depolarization (red), hyperpolarization (yellow); b) δΔψm/δt contour maps: baseline (black), positive slopes (turquoise), negative slopes (purple).
The importance of Δψm kinetics at the tissue level was also highlighted in a recent study in which cardiac arrhythmias induced by GSH oxidation were effectively inhibited by preventing Δψm depolarization using IMAC blockade (Brown et al., 2010). Paradoxically, we also recently found that Δψm depolarization was completely prevented in hypertrophied hearts that were challenged with short episodes of ischemia (Jin et al., 2010). Protection against Δψm depolarization in this rat model of ascending aortic banding was not, however, associated with protection against arrhythmias (Jin et al., 2010).
Finally, in embryonic mouse hearts, Chen et al (Chen et al., 2007) elegantly investigated the differential effects of inhibiting glycolysis versus oxidative phosphorylation on Δψm depolarization and arrhythmia propensity. While inhibition of oxidative phosphorylation but not glycolysis caused a major depolarization in Δψm, both strategies led to comparable slowing of heart rate, shortening of the action potential duration, blunting of the intracellular calcium transients, and promotion of arrhythmias (Chen et al., 2007). Of note is the fact that the developing myocardium is more dependent on glycolysis than is the adult heart.
Δψm Recovery
Prompt reperfusion is required for preventing irreversible cell damage and death. Unfortunately, restoration of blood flow, in itself, results in additional cardiac damage, known as reperfusion injury, which results from large bursts of ROS (Bolli et al., 1989). ROS-mediated oxidative damage is more severe when reperfusion therapy is delayed. Effective strategies to limit or prevent reperfusion injury have proven elusive. Despite an improved understanding of the pathophysiology of this process, the vast majority of clinical trials aimed at preventing reperfusion injury have been quite disappointing. We recently demonstrated that the successful recovery of Δψm upon reperfusion is indeed highly dependent on the duration of the preceding ischemic episode. Despite a comparable degree of Δψm depolarization following 7.5 and 15 minutes of global no-flow ischemia in the rat, reperfusion led to recovery of Δψm only following the short (7.5 min) but not longer episodes of ischemia (Lyon et al., 2010b). Interestingly, sustained Δψm recovery was also predictive of post-ischemic functional and electrical recovery (Lyon et al., 2010b). These findings reinforce the notion that reperfusion is a highly complex phenomenon which could either reverse or exacerbate ischemia mediated changes in Δψm. In fact, additional Δψm depolarization upon reperfusion following long episodes of ischemia is consistent with ROS induced damage during this phase (Lyon et al., 2010b). Strategies aimed at promoting rapid recovery of Δψm during the early (first 5 minutes) phase of reperfusion, potentially by ischemic or pharmacologic post-conditioning strategies, may be an effective strategy for avoiding the genesis of ventricular fibrillation (Lyon et al., 2010b).
Metabolic sinks and reperfusion arrhythmias
Spatio-temporal heterogeneities in mitochondrial function may be associated with local changes in sarcKATP current density which could potentially create areas of depressed excitability to form conduction block through a mechanism we termed “metabolic sink” (Akar et al., 2005). The presence of metabolic sinks may promote the genesis of arrhythmias by shortening the effective refractory period and slowing myocardial conduction in the area of the sink; thereby, shortening the excitation wavelength. Moreover, presence of heterogeneous metabolic sinks is expected to promote heterogeneous action potential repolarization across the tissue. Finally, having a discrete region or dispersed loci of metabolic sinks may predispose to arrhythmias either by forming unidirectional conduction block or causing heterogeneous conduction, respectively. In support of the concept of metabolic sink, IMAC activation using agonists of the mitochondrial benzodiazepine receptor led to an accelerated shortening of the action potential and an early form of conduction failure during ischemia. In contrast, IMAC blockade delayed action potential shortening and the onset of inexcitability (Akar et al., 2005). In this guinea pig model, sustained ventricular tachyarrhythmias were readily generated upon reperfusion in ~90% of hearts (Akar et al., 2005). Remarkably, IMAC blockade, which stabilizes Δψm in vitro, markedly suppressed the formation of these arrhythmias. Indeed, these data suggest that mitochondrial depolarization is the primary factor driving KATP channel activation in ischemia and arrhythmias upon reperfusion. The protective effect of IMAC blockade on electrical and contractile post-ischemic function was further demonstrated in a rabbit model of ischemia reperfusion injury (Brown et al., 2008b). This anti-arrhythmic effect was not evident in hearts treated with the mPTP blocker, cyclosporine A, reinforcing IMAC as the primary mitochondrial mediator of post-ischemic arrhythmias. This concept of metabolic sinks is strengthened by our Δψm imaging studies, which revealed complex spatio-temporal dynamics of Δψm properties that were closely related to post-ischemic electrical and contractile recovery (Lyon et al., 2010a). Finally, the dependence of electrical dysfunction on Δψm was recently argued in hearts that did not undergo ischemia-reperfusion injury, but rather, were challenged with diamide-induced glutathione oxidation. Again, IMAC blockade was effective in preventing both Δψm depolarization and arrhythmias in this model of metabolic stress (Brown et al., 2010).
Mitochondria as therapeutic targets
Cardiac mitochondria form a compact three dimensional lattice structure that is tightly packed between myofilaments and surrounding t-tubules. This spatial organization places mitochondria in close proximity to the major sites of energy consumption (myofilaments) and excitation-contraction coupling (diads). By being the major source of ROS production, mitochondria can intricately alter the activity of multiple ion channel, Ca2+ handling and contractile proteins. Moreover, the generation of metabolic intermediates within mitochondria provides the reducing equivalents required to maintain the negative redox potential of cellular antioxidant pathways. As such, mitochondria clearly represent an attractive target for altering myocyte function, including electrophysiological properties.
Uncovering mechanisms by which mitochondrial dysfunction predisposes to arrhythmias will allow us to design novel strategies. Targeting root causes (ie mitochondria) rather than downstream consequences (cell surface membrane transporters, calcium cycling proteins, etc) is expected to be advantageous as mitochondria represent a main hub of myocyte function that controls energetics, cell signaling, calcium handling and electrical function.
The development of effective therapeutic strategies targeting the mitochondrial network is currently hampered by a lack of solid molecular information regarding the identity of key mitochondrial ion channels and transporters. For example, none of the proteins involved in mitochondrial Ca2+ homeostasis have thus far been completely resolved. Pharmacological studies point us towards promising targets such as the IMAC, mPTP, mitoKATP, and MCU, but actual mitochondrial structures and macromolecular complexes that mediate changes in Δψm remain a subject of intensive debate and active investigation. Indeed, this field of mitochondrial biology is ripe for discovery as powerful proteomic and genomic tools become more readily available. Meanwhile, integrative multi-scale investigation, involving complementary in vivo, ex vivo, in vitro, and in silico approaches is essential for understanding how metabolic failure at the level of the organelle can scale to produce arrhythmias in the whole heart.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. The Journal of clinical investigation. 2005;115:3527–3535. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. Journal of the American College of Cardiology. 2009;54:1891–1898. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Akar FG, Brown DA, Zhou L, O’Rourke B. From mitochondrial dynamics to arrhythmias. Int J Biochem Cell Biol. 2009;41:1940–1948. doi: 10.1016/j.biocel.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Akar FG, O’Rourke B. Mitochondrial criticality: a new concept at the turning point of life or death. Biochimica et biophysica acta. 2006;1762:232–240. doi: 10.1016/j.bbadis.2005.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Maack C, O’Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. The Journal of biological chemistry. 2007;282:21889–21900. doi: 10.1074/jbc.M702841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Marban E, O’Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. The Journal of biological chemistry. 2003;278:44735–44744. doi: 10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- Argaud L, Gateau-Roesch O, Chalabreysse L, Gomez L, Loufouat J, Thivolet-Bejui F, Robert D, Ovize M. Preconditioning delays Ca2+-induced mitochondrial permeability transition. Cardiovascular research. 2004;61:115–122. doi: 10.1016/j.cardiores.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Aronson D, Mittleman MA, Burger AJ. Effects of sulfonylurea hypoglycemic agents and adenosine triphosphate dependent potassium channel antagonists on ventricular arrhythmias in patients with decompensated heart failure. Pacing Clin Electrophysiol. 2003;26:1254–1261. doi: 10.1046/j.1460-9592.2003.t01-1-00177.x. [DOI] [PubMed] [Google Scholar]
- Azzi A, Azzone GF. Metabolism-dependent mitochondrial shrinkage coupled to ion movement. Biochimica et biophysica acta. 1966;120:466–468. doi: 10.1016/0926-6585(66)90316-5. [DOI] [PubMed] [Google Scholar]
- Azzi A, Azzone GF. Swelling and shrinkage phenomena in liver mitochondria. VI. Metabolism-independent swelling coupled to ion movement. Biochimica et biophysica acta. 1967;131:468–478. doi: 10.1016/0005-2728(67)90006-0. [DOI] [PubMed] [Google Scholar]
- Baharvand B, Dehaj ME, Rasoulian B, Namdari M, Shikhani Y, Kiani AA. Delayed anti-arrhythmic effect of nitroglycerin in anesthetized rats: involvement of CGRP, PKC and mK ATP channels. International journal of cardiology. 2009;135:187–192. doi: 10.1016/j.ijcard.2008.03.048. [DOI] [PubMed] [Google Scholar]
- Basgut B, Aypar E, Basgut E, Akin KO, Kilic N, Cakici I. The mechanism of the late preconditioning effect of 3-nitropropionic acid. Archives of pharmacal research. 2008;31:1257–1263. doi: 10.1007/s12272-001-2104-3. [DOI] [PubMed] [Google Scholar]
- Beavis AD. On the inhibition of the mitochondrial inner membrane anion uniporter by cationic amphiphiles and other drugs. The Journal of biological chemistry. 1989;264:1508–1515. [PubMed] [Google Scholar]
- Beavis AD. Properties of the inner membrane anion channel in intact mitochondria. Journal of bioenergetics and biomembranes. 1992;24:77–90. doi: 10.1007/BF00769534. [DOI] [PubMed] [Google Scholar]
- Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovascular research. 2004;61:461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, Wilson LD, Cardounel AJ, Laurita KR, Carnes CA, Billman GE, Gyorke S. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovascular research. 2009;84:387–395. doi: 10.1093/cvr/cvp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CJ, Bright NA, Rutter GA, Griffiths EJ. ATP regulation in adult rat cardiomyocytes: time-resolved decoding of rapid mitochondrial calcium spiking imaged with targeted photoproteins. The Journal of biological chemistry. 2006;281:28058–28067. doi: 10.1074/jbc.M604540200. [DOI] [PubMed] [Google Scholar]
- Billman GE. Role of ATP sensitive potassium channel in extracellular potassium accumulation and cardiac arrhythmias during myocardial ischaemia. Cardiovascular research. 1994;28:762–769. doi: 10.1093/cvr/28.6.762. [DOI] [PubMed] [Google Scholar]
- Billman GE. The cardiac sarcolemmal ATP-sensitive potassium channel as a novel target for anti-arrhythmic therapy. Pharmacology & therapeutics. 2008;120:54–70. doi: 10.1016/j.pharmthera.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Billman GE, Englert HC, Scholkens BA. HMR 1883, a novel cardioselective inhibitor of the ATP-sensitive potassium channel. Part II: effects on susceptibility to ventricular fibrillation induced by myocardial ischemia in conscious dogs. The Journal of pharmacology and experimental therapeutics. 1998;286:1465–1473. [PubMed] [Google Scholar]
- Bolli R, Jeroudi MO, Patel BS, DuBose CM, Lai EK, Roberts R, McCay PB. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:4695–4699. doi: 10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady NR, Elmore SP, van Beek JJ, Krab K, Courtoy PJ, Hue L, Westerhoff HV. Coordinated behavior of mitochondria in both space and time: a reactive oxygen species-activated wave of mitochondrial depolarization. Biophys J. 2004;87:2022–2034. doi: 10.1529/biophysj.103.035097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley GP. Energy-linked alteration of the permeability of heart mitochondria to chloride and other anions. Biochemistry. 1970;9:697–707. doi: 10.1021/bi00806a001. [DOI] [PubMed] [Google Scholar]
- Brown D, Aon M, Akar F, O’Rourke B. A ligand to the mitochondrial benzodiazepine receptor prevents ventricular arrhythmias and LV dysfunction after ischemia or glutathione depletion. FASEB J. 2008a;22:747–747. [Google Scholar]
- Brown DA, Aon MA, Akar FG, Liu T, Sorarrain N, O’Rourke B. Effects of 4′-chlorodiazepam on cellular excitation-contraction coupling and ischaemia-reperfusion injury in rabbit heart. Cardiovascular research. 2008b;79:141–149. doi: 10.1093/cvr/cvn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, O’Rourke B. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. Journal of molecular and cellular cardiology. 2010;48:673–679. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Chicco AJ, Jew KN, Johnson MS, Lynch JM, Watson PA, Moore RL. Cardioprotection afforded by chronic exercise is mediated by the sarcolemmal, and not the mitochondrial, isoform of the KATP channel in the rat. The Journal of physiology. 2005;569:913–924. doi: 10.1113/jphysiol.2005.095729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacciapuoti F, Spiezia R, Bianchi U, Lama D, D’Avino M, Varricchio M. Effectiveness of glibenclamide on myocardial ischemic ventricular arrhythmias in non-insulin-dependent diabetes mellitus. The American journal of cardiology. 1991;67:843–847. doi: 10.1016/0002-9149(91)90617-t. [DOI] [PubMed] [Google Scholar]
- Chen F, De Diego C, Xie LH, Yang JH, Klitzner TS, Weiss JN. Effects of metabolic inhibition on conduction, Ca transients, and arrhythmia vulnerability in embryonic mouse hearts. American journal of physiology. 2007;293:H2472–2478. doi: 10.1152/ajpheart.00359.2007. [DOI] [PubMed] [Google Scholar]
- Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, Hong MK. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coronary artery disease. 2007;18:215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. The Journal of biological chemistry. 2002;277:34793–34799. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- Damy T, Kirsch M, Khouzami L, Caramelle P, Le Corvoisier P, Roudot-Thoraval F, Dubois-Rande JL, Hittinger L, Pavoine C, Pecker F. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PloS one. 2009;4:e4871. doi: 10.1371/journal.pone.0004871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Sarkar C. Is the sarcolemmal or mitochondrial K(ATP) channel activation important in the antiarrhythmic and cardioprotective effects during acute ischemia/reperfusion in the intact anesthetized rabbit model? Life sciences. 2005;77:1226–1248. doi: 10.1016/j.lfs.2004.12.042. [DOI] [PubMed] [Google Scholar]
- Das B, Sarkar C. Similarities between ischemic preconditioning and 17beta-estradiol mediated cardiomyocyte KATP channel activation leading to cardioprotective and antiarrhythmic effects during ischemia/reperfusion in the intact rabbit heart. Journal of cardiovascular pharmacology. 2006;47:277–286. doi: 10.1097/01.fjc.0000202563.54043.d6. [DOI] [PubMed] [Google Scholar]
- Das B, Sarkar C, Shankar PR. Pretreatment with sarafotoxin 6c prior to coronary occlusion protects against infarction and arrhythmias via cardiomyocyte mitochondrial K(ATP) channel activation in the intact rabbit heart during ischemia/reperfusion. Cardiovascular drugs and therapy/sponsored by the International Society of Cardiovascular Pharmacotherapy. 2007;21:243–251. doi: 10.1007/s10557-007-6031-5. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. The Journal of biological chemistry. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- Dow J, Bhandari A, Kloner RA. The mechanism by which ischemic postconditioning reduces reperfusion arrhythmias in rats remains elusive. Journal of cardiovascular pharmacology and therapeutics. 2009;14:99–103. doi: 10.1177/1074248408329606. [DOI] [PubMed] [Google Scholar]
- Driamov S, Bellahcene M, Ziegler A, Barbosa V, Traub D, Butz S, Buser PT, Zaugg CE. Antiarrhythmic effect of ischemic preconditioning during low-flow ischemia. The role of bradykinin and sarcolemmal versus mitochondrial ATP-sensitive K(+) channels. Basic research in cardiology. 2004;99:299–308. doi: 10.1007/s00395-004-0468-5. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiological reviews. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Duchen MR, McGuinness O, Brown LA, Crompton M. On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovascular research. 1993;27:1790–1794. doi: 10.1093/cvr/27.10.1790. [DOI] [PubMed] [Google Scholar]
- Fischbach PS, Barrett TD, Reed NJ, Lucchesi BR. SNC-80-induced preconditioning: selective activation of the mitochondrial adenosine triphosphate-gated potassium channel. Journal of cardiovascular pharmacology. 2003;41:744–750. doi: 10.1097/00005344-200305000-00011. [DOI] [PubMed] [Google Scholar]
- Fischbach PS, White A, Barrett TD, Lucchesi BR. Risk of ventricular proarrhythmia with selective opening of the myocardial sarcolemmal versus mitochondrial ATP-gated potassium channel. The Journal of pharmacology and experimental therapeutics. 2004;309:554–559. doi: 10.1124/jpet.103.060780. [DOI] [PubMed] [Google Scholar]
- Fryer RM, Hsu AK, Nagase H, Gross GJ. Opioid-induced cardioprotection against myocardial infarction and arrhythmias: mitochondrial versus sarcolemmal ATP-sensitive potassium channels. The Journal of pharmacology and experimental therapeutics. 2000;294:451–457. [PubMed] [Google Scholar]
- Garcia-Rivas Gde J, Carvajal K, Correa F, Zazueta C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. British journal of pharmacology. 2006;149:829–837. doi: 10.1038/sj.bjp.0706932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlid KD, Beavis AD. Evidence for the existence of an inner membrane anion channel in mitochondria. Biochimica et biophysica acta. 1986;853:187–204. doi: 10.1016/0304-4173(87)90001-2. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ. Use of ruthenium red as an inhibitor of mitochondrial Ca(2+) uptake in single rat cardiomyocytes. FEBS letters. 2000;486:257–260. doi: 10.1016/s0014-5793(00)02268-7. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. Journal of molecular and cellular cardiology. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochimica et biophysica acta. 2009;1787:1324–1333. doi: 10.1016/j.bbabio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Gross GJ, Peart JN. KATP channels and myocardial preconditioning: an update. American journal of physiology. 2003;285:H921–930. doi: 10.1152/ajpheart.00421.2003. [DOI] [PubMed] [Google Scholar]
- Gupta MP, Dixon IM, Zhao D, Dhalla NS. Influence of ruthenium red on rat heart subcellular calcium transport. The Canadian journal of cardiology. 1989;5:55–63. [PubMed] [Google Scholar]
- Gupta MP, Innes IR, Dhalla NS. Responses of contractile function to ruthenium red in rat heart. The American journal of physiology. 1988;255:H1413–1420. doi: 10.1152/ajpheart.1988.255.6.H1413. [DOI] [PubMed] [Google Scholar]
- Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovascular research. 2008;77:334–343. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. What is the mitochondrial permeability transition pore? Journal of molecular and cellular cardiology. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovascular research. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochimica et biophysica acta. 2009;1787:1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- Hanley PJ, Drose S, Brandt U, Lareau RA, Banerjee AL, Srivastava DK, Banaszak LJ, Barycki JJ, Van Veldhoven PP, Daut J. 5-Hydroxydecanoate is metabolised in mitochondria and creates a rate-limiting bottleneck for beta-oxidation of fatty acids. The Journal of physiology. 2005;562:307–318. doi: 10.1113/jphysiol.2004.073932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley PJ, Gopalan KV, Lareau RA, Srivastava DK, von Meltzer M, Daut J. Beta-oxidation of 5-hydroxydecanoate, a putative blocker of mitochondrial ATP-sensitive potassium channels. The Journal of physiology. 2003;547:387–393. doi: 10.1113/jphysiol.2002.037044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovascular research. 2002;55:534–543. doi: 10.1016/s0008-6363(02)00455-8. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. American journal of physiology. 2004;287:H841–849. doi: 10.1152/ajpheart.00678.2003. [DOI] [PubMed] [Google Scholar]
- Headrick JP, Willems L, Ashton KJ, Holmgren K, Peart J, Matherne GP. Ischaemic tolerance in aged mouse myocardium: the role of adenosine and effects of A1 adenosine receptor overexpression. The Journal of physiology. 2003;549:823–833. doi: 10.1113/jphysiol.2003.041541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdiness MR. Clinical pharmacokinetics of N-acetylcysteine. Clinical pharmacokinetics. 1991;20:123–134. doi: 10.2165/00003088-199120020-00004. [DOI] [PubMed] [Google Scholar]
- Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Annals of the New York Academy of Sciences. 2005;1047:248–258. doi: 10.1196/annals.1341.022. [DOI] [PubMed] [Google Scholar]
- Huser J, Blatter LA. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. The Biochemical journal. 1999;343(Pt 2):311–317. [PMC free article] [PubMed] [Google Scholar]
- Imani A, Faghihi M, Sadr SS, Keshavarz M, Niaraki SS. Noradrenaline reduces ischemia-induced arrhythmia in anesthetized rats: involvement of alpha1-adrenoceptors and mitochondrial K ATP channels. Journal of cardiovascular electrophysiology. 2008;19:309–315. doi: 10.1111/j.1540-8167.2007.01031.x. [DOI] [PubMed] [Google Scholar]
- Inoue I, Nagase H, Kishi K, Higuti T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352:244–247. doi: 10.1038/352244a0. [DOI] [PubMed] [Google Scholar]
- Jin H, Nass RD, Joudrey PJ, Lyon AR, Chemaly ER, Rapti K, Akar FG. Altered spatiotemporal dynamics of the mitochondrial membrane potential in the hypertrophied heart. Biophysical journal. 2010;98:2063–2071. doi: 10.1016/j.bpj.2010.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara K, Takase M, Yamauchi Y. Ruthenium red-induced transition from ventricular fibrillation to tachycardia in isolated rat hearts: possible involvement of changes in mitochondrial calcium uptake. Cardiovasc Pathol. 2003;12:311–321. doi: 10.1016/s1054-8807(03)00090-5. [DOI] [PubMed] [Google Scholar]
- Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. American journal of physiology. 2006;290:H2024–2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- Kimura H, Kawahara K, Yamauchi Y, Miyaki J. On the mechanisms for the conversion of ventricular fibrillation to tachycardia by perfusion with ruthenium red. Journal of electrocardiology. 2005;38:364–370. doi: 10.1016/j.jelectrocard.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Kiss A, Juhasz L, Huliak I, Vegh A. Peroxynitrite decreases arrhythmias induced by ischaemia reperfusion in anaesthetized dogs, without involving mitochondrial KATP channels. British journal of pharmacology. 2008;155:1015–1024. doi: 10.1038/bjp.2008.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleber AG. Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfused guinea pig hearts. Circ Res. 1983;52:442–450. doi: 10.1161/01.res.52.4.442. [DOI] [PubMed] [Google Scholar]
- Konya L, Kekesi V, Juhasz-Nagy S, Feher J. The effect of superoxide dismutase in the myocardium during reperfusion in the dog. Free radical biology & medicine. 1992;13:527–532. doi: 10.1016/0891-5849(92)90147-9. [DOI] [PubMed] [Google Scholar]
- Liu M, Liu H, Dudley SC., Jr Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circulation research. 2010;107:967–974. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomuscio A, Vergani D, Marano L, Castagnone M, Fiorentini C. Effects of glibenclamide on ventricular fibrillation in non-insulin-dependent diabetics with acute myocardial infarction. Coronary artery disease. 1994;5:767–771. [PubMed] [Google Scholar]
- Lyon AR, Joudrey PJ, Jin D, Nass RD, Aon MA, O’Rourke B, Akar FG. Optical imaging of mitochondrial function uncovers actively propagating waves of mitochondrial membrane potential collapse across intact heart. J Mol Cell Cardiol. 2010a doi: 10.1016/j.yjmcc.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon AR, Joudrey PJ, Jin D, Nass RD, Aon MA, O’Rourke B, Akar FG. Optical imaging of mitochondrial function uncovers actively propagating waves of mitochondrial membrane potential collapse across intact heart. Journal of molecular and cellular cardiology. 2010b;49:565–575. doi: 10.1016/j.yjmcc.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S. Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. American journal of physiology. 2004;286:H1923–1935. doi: 10.1152/ajpheart.00935.2003. [DOI] [PubMed] [Google Scholar]
- Michelakis ED. Mitochondrial medicine: a new era in medicine opens new windows and brings new challenges. Circulation. 2008;117:2431–2434. doi: 10.1161/CIRCULATIONAHA.108.775163. [DOI] [PubMed] [Google Scholar]
- Minners J, van den Bos EJ, Yellon DM, Schwalb H, Opie LH, Sack MN. Dinitrophenol, cyclosporin A, and trimetazidine modulate preconditioning in the isolated rat heart: support for a mitochondrial role in cardioprotection. Cardiovascular research. 2000;47:68–73. doi: 10.1016/s0008-6363(00)00069-9. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiological reviews. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazareth W, Yafei N, Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. Journal of molecular and cellular cardiology. 1991;23:1351–1354. doi: 10.1016/0022-2828(91)90181-k. [DOI] [PubMed] [Google Scholar]
- Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- O’Rourke B. Myocardial K(ATP) channels in preconditioning. Circulation research. 2000;87:845–855. doi: 10.1161/01.res.87.10.845. [DOI] [PubMed] [Google Scholar]
- O’Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circulation research. 2004;94:420–432. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke B. Mitochondrial ion channels. Annu Rev Physiol. 2007;69:19–49. doi: 10.1146/annurev.physiol.69.031905.163804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke B, Ramza BM, Marban E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science (New York, NY) 1994;265:962–966. doi: 10.1126/science.8052856. [DOI] [PubMed] [Google Scholar]
- O’Rourke B, Ramza BM, Romashko DN, Marban E. Metabolic oscillations in heart cells. Advances in experimental medicine and biology. 1995;382:165–174. doi: 10.1007/978-1-4615-1893-8_17. [DOI] [PubMed] [Google Scholar]
- Ohnuma Y, Miura T, Miki T, Tanno M, Kuno A, Tsuchida A, Shimamoto K. Opening of mitochondrial K(ATP) channel occurs downstream of PKC-epsilon activation in the mechanism of preconditioning. American journal of physiology. 2002;283:H440–447. doi: 10.1152/ajpheart.00434.2001. [DOI] [PubMed] [Google Scholar]
- Oka N, Wang L, Mi W, Caldarone CA. Inhibition of mitochondrial remodeling by cyclosporine A preserves myocardial performance in a neonatal rabbit model of cardioplegic arrest. The Journal of thoracic and cardiovascular surgery. 2008a;135:585–593. doi: 10.1016/j.jtcvs.2007.09.023. [DOI] [PubMed] [Google Scholar]
- Oka N, Wang L, Mi W, Zhu W, Honjo O, Caldarone CA. Cyclosporine A prevents apoptosis-related mitochondrial dysfunction after neonatal cardioplegic arrest. The Journal of thoracic and cardiovascular surgery. 2008b;135:123–130. 130 e121–122. doi: 10.1016/j.jtcvs.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Ozaydin M, Peker O, Erdogan D, Kapan S, Turker Y, Varol E, Ozguner F, Dogan A, Ibrisim E. N-acetylcysteine for the prevention of postoperative atrial fibrillation: a prospective, randomized, placebo-controlled pilot study. European heart journal. 2008;29:625–631. doi: 10.1093/eurheartj/ehn011. [DOI] [PubMed] [Google Scholar]
- Ozcan C, Terzic A, Bienengraeber M. Effective pharmacotherapy against oxidative injury: alternative utility of an ATP-sensitive potassium channel opener. Journal of cardiovascular pharmacology. 2007;50:411–418. doi: 10.1097/FJC.0b013e31812378df. [DOI] [PubMed] [Google Scholar]
- Paucek P, Mironova G, Mahdi F, Beavis AD, Woldegiorgis G, Garlid KD. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. The Journal of biological chemistry. 1992;267:26062–26069. [PubMed] [Google Scholar]
- Peixoto PM, Ryu SY, Kinnally KW. Mitochondrial ion channels as therapeutic targets. FEBS letters. 584:2142–2152. doi: 10.1016/j.febslet.2010.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, Andre-Fouet X, Revel D, Kirkorian G, Monassier JP, Derumeaux G, Ovize M. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. The New England journal of medicine. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- Rajesh KG, Sasaguri S, Suzuki R, Xing Y, Maeda H. Ischemic preconditioning prevents reperfusion heart injury in cardiac hypertrophy by activation of mitochondrial KATP channels. International journal of cardiology. 2004;96:41–49. doi: 10.1016/j.ijcard.2003.06.010. [DOI] [PubMed] [Google Scholar]
- Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, Pozzan T. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. The EMBO journal. 2001;20:4998–5007. doi: 10.1093/emboj/20.17.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romashko DN, Marban E, O’Rourke B. Subcellular metabolic transients and mitochondrial redox waves in heart cells. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:1618–1623. doi: 10.1073/pnas.95.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu SY, Lee SH, Ho WK. Generation of metabolic oscillations by mitoKATP and ATP synthase during simulated ischemia in ventricular myocytes. Journal of molecular and cellular cardiology. 2005;39:874–881. doi: 10.1016/j.yjmcc.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Sasaki N, Sato T, Marban E, O’Rourke B. ATP consumption by uncoupled mitochondria activates sarcolemmal K(ATP) channels in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2001;280:H1882–1888. doi: 10.1152/ajpheart.2001.280.4.H1882. [DOI] [PubMed] [Google Scholar]
- Schwartz LM, Welch TS, Crago MS. Cardioprotection by multiple preconditioning cycles does not require mitochondrial K(ATP) channels in pigs. American journal of physiology. 2002;283:H1538–1544. doi: 10.1152/ajpheart.00040.2002. [DOI] [PubMed] [Google Scholar]
- Slodzinski MK, Aon MA, O’Rourke B. Intracellular and intercellular mitochondrial membrane potential oscillations in the Langendorff perfused heart. Biophys J. 2004;86:461a. [Google Scholar]
- Slodzinski MK, Aon MA, O’Rourke B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. Journal of molecular and cellular cardiology. 2008;45:650–660. doi: 10.1016/j.yjmcc.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Saito T, Sato T, Tamagawa M, Miki T, Seino S, Nakaya H. Cardioprotective effect of diazoxide is mediated by activation of sarcolemmal but not mitochondrial ATP-sensitive potassium channels in mice. Circulation. 2003;107:682–685. doi: 10.1161/01.cir.0000055187.67365.81. [DOI] [PubMed] [Google Scholar]
- Takashi E, Wang Y, Ashraf M. Activation of mitochondrial K(ATP) channel elicits late preconditioning against myocardial infarction via protein kinase C signaling pathway. Circulation research. 1999;85:1146–1153. doi: 10.1161/01.res.85.12.1146. [DOI] [PubMed] [Google Scholar]
- Tokube K, Kiyosue T, Arita M. Openings of cardiac KATP channel by oxygen free radicals produced by xanthine oxidase reaction. The American journal of physiology. 1996;271:H478–489. doi: 10.1152/ajpheart.1996.271.2.H478. [DOI] [PubMed] [Google Scholar]
- Tsai CH, Su SF, Chou TF, Lee TM. Differential effects of sarcolemmal and mitochondrial K(ATP) channels activated by 17 beta-estradiol on reperfusion arrhythmias and infarct sizes in canine hearts. The Journal of pharmacology and experimental therapeutics. 2002;301:234–240. doi: 10.1124/jpet.301.1.234. [DOI] [PubMed] [Google Scholar]
- Vajda S, Baczko I, Lepran I. Selective cardiac plasma-membrane K(ATP) channel inhibition is defibrillatory and improves survival during acute myocardial ischemia and reperfusion. European journal of pharmacology. 2007;577:115–123. doi: 10.1016/j.ejphar.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Kanazirska MP, Tien HT. Ca2+ channels from brain microsomal membranes reconstituted in patch-clamped bilayers. Biochimica et biophysica acta. 1987;897:324–330. doi: 10.1016/0005-2736(87)90428-7. [DOI] [PubMed] [Google Scholar]
- Vegh A, Parratt JR. The role of mitochondrial K(ATP) channels in antiarrhythmic effects of ischaemic preconditioning in dogs. British journal of pharmacology. 2002;137:1107–1115. doi: 10.1038/sj.bjp.0704966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinbrenner C, Liu GS, Downey JM, Cohen MV. Cyclosporine A limits myocardial infarct size even when administered after onset of ischemia. Cardiovascular research. 1998;38:678–684. doi: 10.1016/s0008-6363(98)00064-9. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Yang L, Qu Z. Systems biology approaches to metabolic and cardiovascular disorders: network perspectives of cardiovascular metabolism. Journal of lipid research. 2006;47:2355–2366. doi: 10.1194/jlr.R600023-JLR200. [DOI] [PubMed] [Google Scholar]
- Wilson LD, Wan X, Rosenbaum DS. Cellular alternans: a mechanism linking calcium cycling proteins to cardiac arrhythmogenesis. Annals of the New York Academy of Sciences. 2006;1080:216–234. doi: 10.1196/annals.1380.018. [DOI] [PubMed] [Google Scholar]
- Wirth KJ, Rosenstein B, Uhde J, Englert HC, Busch AE, Scholkens BA. ATP-sensitive potassium channel blocker HMR 1883 reduces mortality and ischemia-associated electrocardiographic changes in pigs with coronary occlusion. The Journal of pharmacology and experimental therapeutics. 1999;291:474–481. [PubMed] [Google Scholar]
- Yang L, Korge P, Weiss JN, Qu Z. Mitochondrial oscillations and waves in cardiac myocytes: insights from computational models. Biophysical journal. 98:1428–1438. doi: 10.1016/j.bpj.2009.12.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Aon MA, Almas T, Cortassa S, Winslow RL, O’Rourke B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput Biol. 6:e1000657. doi: 10.1371/journal.pcbi.1000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. The Journal of experimental medicine. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochimica et biophysica acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]