

Abstract
During long-term potentiation (LTP), synapses undergo stable changes in synaptic strength. The molecular memory processes that maintain strength have not been identified. One hypothesis is that the complex formed by the Ca2+/calmodulin-dependent protein kinase II (CaMKII) and the NMDA-type glutamate receptor (NMDAR) is a molecular memory at the synapse. To establish a molecule as a molecular memory, it must be shown that interfering with the molecule produces a persistent reversal of LTP. We used the CN class of peptides that inhibit CaMKII binding to the NR2B subunit in vitro to test this prediction in rat hippocampal slices. We found that CN peptides can reverse saturated LTP, allowing additional LTP to be induced. The peptide also produced a persistent reduction in basal transmission. We then tested whether CN compounds actually affect CaMKII binding in living cells. Application of CN peptide to slice cultures reduced the amount of CaMKII concentrated in spines, consistent with delocalization of the kinase from a binding partner in the spine. To more specifically assay the binding of CaMKII to the NMDAR, we used coimmunoprecipitation methods. We found that CN peptide decreased synaptic strength only at concentrations necessary to disrupt the CaMKII/NMDAR complex, but not at lower concentrations sufficient to inhibit CaMKII activity. Importantly, both the reduction of the complex and the reduction of synaptic strength persisted after removal of the inhibitor. These results support the hypothesis that the CaMKII/NMDAR complex has switch-like properties that are important in the maintenance of synaptic strength.
Introduction
CaMKII is a critical protein in the induction of LTP (Lisman et al., 2002). The kinase is activated by Ca2+ entry through the NMDA receptor (NMDAR) (Hudmon and Schulman, 2002). If this activation is blocked pharmacologically or by CaMKIIα knock-out, LTP induction is prevented (Malinow et al., 1989; Silva et al., 1992). A necessary aspect of this process is the autophosphorylation of T286 (Giese et al., 1998; Buard et al., 2010), which makes the kinase partially independent of Ca2+ (autonomous) (Miller and Kennedy, 1986). Once activated, CaMKII undergoes translocation to synaptic sites (Shen and Meyer, 1999; Shen et al., 2000; Bayer et al., 2001; Otmakhov et al., 2004; Hudmon et al., 2005; Bayer et al., 2006; Zhang et al., 2008; Lee et al., 2009) and becomes bound in the postsynaptic density (PSD) (Strack et al., 1997; Dosemeci et al., 2001; Otmakhov et al., 2004).
An important binding partner in the PSD is the NMDAR (Strack and Colbran, 1998; Leonard et al., 1999; Strack et al., 2000; Bayer et al., 2001; Leonard et al., 2002). The CaMKII/NMDAR complex exists under basal conditions but is increased by strong synaptic stimulation (Leonard et al., 1999). Preventing the formation of this complex by overexpression of mutated NR2B or NR2B C-tail strongly reduces LTP induction (Barria and Malinow, 2005; Zhou et al., 2007). These results suggest that complex formation is necessary for LTP induction. Whether the complex also has a role in LTP maintenance, i.e., acts as a molecular memory, remains unclear.
A critical test of any model of LTP maintenance is to apply an agent that interferes with the function of the putative memory molecule and determine whether potentiation is persistently reduced. Several inhibitors of CaMKII activity do not reverse LTP (Malinow et al., 1989; Otmakhov et al., 1997; Chen et al., 2001; Wang et al., 2008; Buard et al., 2010). However, it is possible that the CaMKII/NMDAR complex serves as a synaptic memory through a structural rather than enzymatic process. A new class of CaMKII inhibitor derived from the endogenous protein CaMKIIN (Chang et al., 1998, 2001) interferes with the binding of CaMKII to NR2B in vitro (Vest et al., 2007). We have used peptide inhibitors (CN peptides) derived from CaMKIIN to test the hypothesis that synaptic strength is maintained by the CaMKII/NMDAR complex. Previous experiments to test this hypothesis used antCN27 (also known as ant-CaMKIINtide) (Sanhueza et al., 2007). However, it was then shown that the cell-permeabilizing sequence ant directly binds calmodulin (Buard et al., 2010). Here, we use improved CN peptides that contain a tat permeabilizing sequence that does not bind calmodulin (Buard et al., 2010). We find that tatCN peptides are able to reverse LTP maintenance and produce a persistent reduction in basal transmission. In parallel experiments, we directly demonstrate that CN compounds reduce the CaMKII/NMDAR complex in living cells. Importantly, the reduction of both synaptic strength and the CaMKII/NMDAR complex was not an equilibrium effect, but instead had the switch-like properties required of a molecular memory.
Materials and Methods
Acute slice preparation and preincubation.
Animal care was in accordance with institutional guidelines of the University of Chile. Transverse hippocampal slices (400 μm) were prepared from 18- to 23-d-old male Sprague Dawley rats in ice-cold dissection solution containing the following (in mm): 125 NaCl, 2.6 KCl, 10 MgCl2, 0.5 CaCl2, 26 NaHCO3, 1.23 NaH2PO4, and 10 d-glucose (equilibrated with 95% O2 and 5% CO2), pH 7.3. CA3 area was removed, and slices were allowed to recover for a minimum of 1 h at room temperature in inverted interface chambers (tissue inserts, 8 μm; EMS) maintained in an atmosphere saturated with 95% O2 and 5% CO2. Each insert held two slices completely submerged in a drop of 100 μl of artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 26 NaHCO3, 1 NaH2PO4, 2.6 KCl, 2 CaCl2, 1 MgCl2, and 10 d-glucose. In experiments in which slices were preincubated with peptides, the drop of solution bathing slices was carefully removed and replaced by freshly oxygenated ACSF containing 5 or 20 μm peptides. After 1 or 2 h preincubation, drop solution was replaced by regular oxygenated ACSF four times and maintained for at least 1 h before recordings. Slices used for coimmunoprecipitation analyses received identical treatment and were transferred to 2 ml cryotubes (2 slices per tube) in which the ACSF was gently removed. Cryotubes were quickly transferred to a liquid nitrogen tank, where they were stored until biochemical analyses.
Electrophysiological recordings in acute slices.
For recordings, slices were transferred to a submersion-type recording chamber mounted on an upright microscope (Nikon E600FN) and were continuously superfused (2–4 ml/min) with ACSF bubbled with 95% O2 and 5% CO2 in a 20 ml syringe before entering the chamber. A total volume of 10 ml of ACSF was recirculated during each experiment using a two-way pump. Experiments were conducted at 30–31°C. Extracellular field potential recordings were made with borosilicate glass recording electrodes (0.5–1 MΩ) filled with ACSF and placed in stratum radiatum of the CA1 region. Schaffer collaterals were stimulated with monopolar stimulating electrodes (glass pipettes filled with ACSF; 0.5 MΩ) or bipolar electrodes (FHC), positioned 100–150 μm to the recording electrode. Stimulus duration was 0.1 ms, and interstimulus interval was 10 s.
To obtain input–output (I–O) curves, stimuli of increasing intensities were applied (5–100 μA, in 5 or 10 μA steps; 4 records per amplitude) until signal saturation or population spike generation. Construction of I–O curve started only after stable basal transmission was reached (typically, after 10 min). For LTP induction, two independent synaptic pathways were alternately stimulated. LTP was induced in one pathway by four tetani (100 Hz, 1 s each), separated by 20 s. In LTP saturation experiments, 1 additional tetanus was applied 10 min later to confirm that saturation was reached. Stimulus strength was adjusted to have a field EPSP (fEPSP) amplitude of 50–60% of the population spike threshold. Stimulus strength was the same for both test and tetanic stimulation. A stable fEPSP slope baseline of 20 min was taken before LTP induction, and transmission was further recorded for at least 30 min. Data were acquired at 20 kHz by a PC with an ITC-1600 interface (HEKA Instruments), using custom procedures (G.F.-V.) in Igor Pro 6.03A (WaveMetrics).
Field potential data analysis and statistics.
Igor Pro 6.03A and Microsoft Excel were used for analysis. fEPSP initial slope and fiber volley peak amplitude were measured to build an I–O curve for each experiment. I–O curve slope per experiment was determined by linear regression, and a mean slope per animal and drug (2–4 slices for each condition) was calculated and used for paired test analyses (see Fig. 5C,F). Mean I–O test and control curves (see Fig. 5B,D) were calculated by first averaging curves for each drug per animal and then averaging among animals. Plot error bars correspond to SEM. The specific type of statistical analysis used is indicated in legends.
Figure 5.

TatCN21 persistently decreases synaptic strength at 20 μm, but not 5 μm. Acute hippocampal slices were incubated with tatCN21 or tatCN21 SCR peptide and washed with ACSF for 1 h before being transferred to the recording chamber. A–C, Low concentration of TatCN21 (5 μm for 1 h) does not affect synaptic strength. A, Superimposed fEPSP obtained for increasing stimulation after tatCN21 SCR or tatCN21 treatment. B, Average I–O curves. C, Each pair represents mean test and control I–O curve slopes of slices from the same animal [filled symbols: average I–O slope ± SEM: 3.45 ± 0.97 (1/ms), for tatCN21 and 3.07 ± 1.09 (1/ms) for tatCN21 SCR; p = 0.157, paired t test; data from 7 animals, 2–4 slices per treatment]. D–F, Longer incubation with a higher tatCN21 concentration (20 μm for 2 h) persistently decreased synaptic strength. D–F, Same as A–C for the present treatment [average I–O slope ± SEM: 1.81 ± 0.54 (1/ms) for tatCN21, 2.39.0 ± 0.57 (1/ms) for tatCN21 SCR; data from 9 animals, 2–4 slices per treatment]. *p < 0.05. Calibration: 0.5 mV, 5 ms. Error bars, SEM.
Drug application in the recording chamber.
During the experiments, stock peptide solutions were added to the syringe with ACSF to achieve the desired final concentration. We have observed that, during excessive bubbling, there is foam formation in the syringe after peptide addition that could alter the effective peptide concentration in the chamber. Therefore, bubbling was decreased to a level at which foam formation was avoided but enough slice oxygenation was provided (as indicated by stable fEPSP recording and a fEPSP/fiber volley ratio more than fourfold). This method ensured highly reproducible effects of peptide application. Drugs were washed out by changing syringe solution: recirculation was stopped, and the syringe was refilled with fresh, oxygenated ACSF. After changing at least four times the original solution volume, the final 10 ml recirculating volume was reestablished.
Peptides.
Tat-conjugated CaMKIIN-derived peptide, tatCN21 (Vest et al., 2007), and a control peptide that was a fusion of tat to a scrambled CN21 sequence were obtained from Biomatik.
Immunoprecipitation and immunoblotting.
After treatment, hippocampal slices were shock frozen and later extracted with 1% deoxycholate, cleared by ultracentrifugation, and analyzed by immunoprecipitation (IP) and quantitative immunoblotting (IB) with antibodies against CaMKIIα, NR1, NR2B, and α-actinin as described by Leonard et al. (1999).
Slice culture preparation.
Hippocampal slice cultures were prepared from postnatal day 6 (P6)–P7 male Sprague Dawley rats in accordance with the animal care protocol of the Brandeis University. In sterile conditions, the rats were decapitated, the brain was removed, and 260 μm hippocampal slices were prepared using a Leica VT 1000S vibratome. Sterile, cold (4°C) modified ACSF saturated with 95% O2 and 5% CO2 was used during the preparation. Modified ACSF contained the following (in mm): 248 sucrose, 4 KCl, 1 CaCl2, 5 MgCl2, and 26 NaHCO3, pH 7.3, 320 mOsm. Slices were plated on the membrane of six-well plate inserts (Falcon; 0.4 μm pore size) and incubated in a CO2 incubator at 36°C in culture medium containing MEM (Cellgro 50–019-PB) supplemented with the following: 20% horse serum (Sigma), 1 μg/ml insulin, 1 mm glutamine (or GlutaMAX), 30 mm HEPES, 1 mm CaCl2, 2 mm MgSO4, 5 mm NaHCO3, 16.5 mm d-glucose, and 0.5 mm ascorbate. The medium was changed three times per week.
Field potential recordings in slice cultures.
A single cultured slice was cut out from the cell culture insert, the connections between CA1 and CA3 were cut, and the slice was placed in the recording chamber mounted on the microscope stage. Slices were continuously superfused (2.5–3 ml/min) with ACSF containing the following (in mm): 124 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 20 d-glucose. In imaging experiments, ACSF was also supplemented by 200 mm Trolox to decrease fluorescence bleaching and related cell damage. In those experiments, the concentration of dextrose was lowered (10 mm) to maintain an osmolarity of 320 mOsm. Before entering the chamber, the ACSF was continuously bubbled with 95% O2 and 5% CO2. The experiments were conducted at room temperature. For field potential recordings, electrodes (glass pipettes filled with ACSF; 300 kΩ resistance) were positioned in the stratum radiatum of the CA1 region. To stimulate two independent inputs, two monopolar stimulating electrodes were positioned to each side of the recording electrode. For imaging experiments with field potential recordings, only one stimulating electrode was used. Single shock stimuli (a square pulse of 50–100 μA of 70–150 μs duration) were delivered every 30 or 60 s. When two stimulating electrodes were used, test stimulation alternated between the two electrodes.
Single-cell electroporation.
After 7–15 d in vitro, slice cultures were transfected with cDNA containing coding sequences of CaMKIIα fused with that of monomeric green fluorescent protein (GFP-CaMKII) and monomeric red fluorescent protein cherry (RFP). GFP-CN19 plasmid was constructed by inserting CN19 sequence (S. J. Coultrap and K. U. Bayer, unpublished observations) into EGFP-C1 vector (Clontech) between BamHI and XbaI restriction sites. The transfection was performed using single-cell electroporation technique (Haas et al., 2001; Pi et al., 2010). A single cultured slice was cut out from the cell culture insert and placed in electroporation buffer in the chamber (similar to the recording chamber used for imaging) mounted on the microscope stage. The electroporation buffer contained the following (in mm): 160 NaCl, 5.4 KCl, 12 MgCl2, 2 CaCl2, 5 HEPES; pH 7.4; osmolarity 320 mOsm. Glass pipettes (3–4 MΩ resistance) were filled with DNA diluted in electroporation buffer, and pyramidal cells in the CA1 were transfected by approaching the cell membrane and delivering square pulses of 10 V of 1 ms duration at 200 Hz for 500 ms. Slices were then briefly washed in electroporation buffer and placed back onto the insert membrane and incubated in a CO2 incubator in standard culture medium. Slices were used for imaging 1–3 d after the transfection.
Imaging and image analysis.
Imaging experiments were performed using a Leica confocal microscope with Ar–Kr lasers. Green and red images were taken consecutively at each focal plane using 488 and 543 nm excitation wavelengths and 500–550 and 590–650 emission, respectively. Stacks of 10–15 images (256 × 256, 0.093 μm pixel size, 3 frame averages) with 0.5 μm focal steps were collected. Experiments were analyzed using Igor Pro custom software. To eliminate background fluorescence, a binary mask was created for each image in a given stack of z-plane images. The mask was constructed to cover all the pixels below an arbitrarily chosen threshold equal to double of the background fluorescence. Regions of interest (ROIs) were drawn around spines and nearby dendrites. For each ROI, the in-focus z-plane was selected from the z-stack and the fluorescence of pixels within the selected region, and not covered by the mask, was measured. All of the data are presented as mean ± SEM. Statistical significance was calculated using two population t test (independent or paired) or unbalanced two-way ANOVA.
Calculation of bound GFP-CaMKIIα in spines and spine size.
The calculation of the bound amount (BA) and bound fraction (BF) was as used by Otmakhov et al. (2004). Briefly, if all the GFP-CaMKIIα in spines is soluble, then the ratio of green fluorescence in spines to green fluorescence in dendrites [(s/d)G] should be equal to the ratio of red fluorescence in spines to red fluorescence in dendrites [(s/d)R]. A ratio of (s/d)G greater than (s/d)R is an indication of a bound pool of GFP-CaMKIIα in the spine. Therefore, the fluorescence of soluble GFP-CaMKIIα in the spines is estimated as follows: dG · (s/d)R. The amount of GFP-CaMKIIα bound in spines can then be calculated by subtracting the fluorescence of soluble GFP-CaMKIIα in spines from the total green fluorescence in spines: sG − dG · (s/d)R. One limitation in the calculations of bound CaMKII in spines is that it is based on the assumption that all CaMKII in the dendritic region is soluble. Although there are some data supporting this view (Shen and Meyer, 1999), other studies indicate that some CaMKII may also be bound in dendrites. Our unpublished data indicate that the dendritic bound amount of CaMKII is approximately 10-fold smaller than that in spines. Therefore, the method of calculation of bound CaMKII in spines may have an ∼10% error. The fraction of GFP-CaMKIIα bound in the spine is represented by the bound amount of GFP-CaMKIIα divided by the total green fluorescence in the spine: [sG − dG · (s/d)R]/sG or 1 − (d/s)G · (s/d)R. The spine size was measured using the method of Holtmaat et al. (2005): the integral of red fluorescence in the spine ROI was measured and then adjusted to red fluorescence in the dendrite.
Whole-cell recordings.
Whole-cell recordings were performed using standard methods as previously described. Slices were continuously superfused (1.8 ml/min) with ACSF containing the following (in mm): 124 NaCl, 2.5 KCl, 4 CaCl2, 4 MgCl2, 1.25, NaH2PO4, 26 NaHCO3, and 0.05 picrotoxin, balanced with 95% O2 and 5% CO2, pH 7.4. Whole-cell recordings were performed at room temperature (22–24°C). The pipette solution contained (in mm) 120 Cs methane sulfonate, 20 CsCl, 10 HEPES, 4 MgATP, 0.3 Na3GTP, 0.2 EGTA, and 10 Na2-phosphocreatine, pH 7.3; osmolarity, 320 mOsm. EPSCs were evoked by stimulating the Schaffer-collateral pathway via a stimulation electrode placed at 100–200 μm from cell body layer and measured in voltage-clamp mode at −65 mV. For cell pair recordings, two neurons (one transfected with GFP-CN19 plasmid and one nontransfected) within 30 μm were selected. The test stimulation was delivered every 6 s with 0.2 ms duration, and the current intensity was adjusted to induce 50–100 pA EPSC. After adjusting stimulation strength in one neuron, sequential recording was performed in the other neuron with the same intensity and location of stimulation. EPSC magnitudes were calculated by averaging a 5 ms time window centered at the peak of the EPSC, and 20–50 EPSCs were averaged. Series and input resistance were monitored throughout each experiment, and recordings for which series resistance varied by >20% were rejected. In some experiments, 100 μm dl-2-amino-5-phosphonovaleric acid (APV) was added immediately after transfection, and it was present for the following 2 d; after 2 d, the recordings were performed in ACSF without the APV.
Results
The CN compounds are based on the sequence of an endogenous protein, CaMKIIN (Chang et al., 1998, 2001). CN compounds have been progressively optimized to enhance CaMKII inhibition and made shorter (CN27; CN21; CN19) (Vest et al., 2007; Coultrap and Bayer, unpublished observations). Furthermore, the permeabilizing sequence ant used in early versions has been replaced by tat because tat, unlike ant, does not by itself affect calmodulin (Buard et al., 2010). We first evaluated the effect of transient application of tat-fused CN peptides on basal transmission. Figure 1 shows experiments in which tatCN21 (20 μm) or a more potent version, tatCN19 (5 μm), was applied and then removed. During application, there was a reduction of transmission. After removal of peptide there was some recovery of the fEPSP (see Discussion), but there remained a persistent reduction. This persistent reduction was not seen with scrambled peptide (tatCN21 SCR) and was not evident in the fiber volley (Fig. 1A,C,D). These results indicate that a component of synaptic strength can be persistently reduced by transient treatment with CN peptides.
Figure 1.
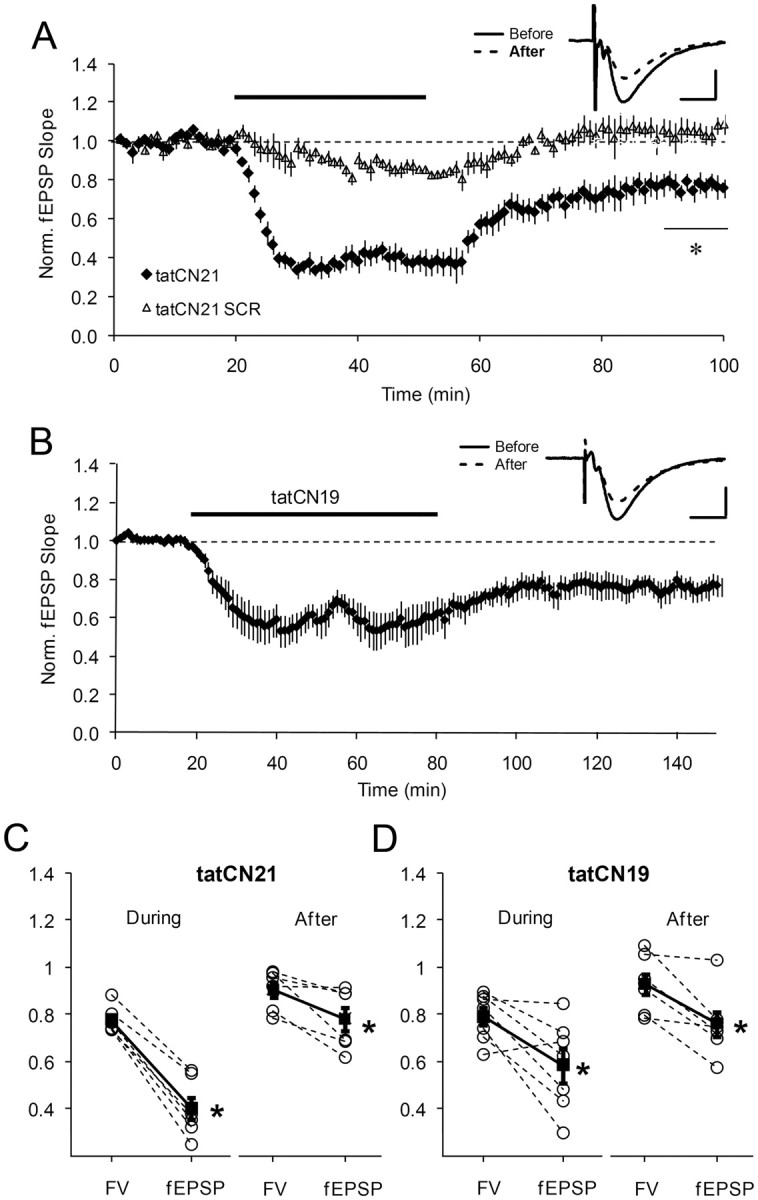
Persistent depression of basal transmission by bath-applied tat-fused CN peptides. A, Basal transmission was monitored before, during, and after transient application (30 min) of 20 μm tatCN21 (n = 6) or tatCN21 scrambled (SCR) peptide (n = 4). t test, *p < 0.005. Inset, Representative averaged basal fEPSP before and 50 min after drug washout. B, Same as A for 1 h application of 5 μm tatCN19 (n = 7). Inset, Averaged fEPSP before and 60 min after drug washout. C, fEPSP and fiber (FV) were reduced during peptide application; after washout, FV recovered, but there was a persistent reduction of fEPSP. Each pair represents data from a single experiment and the data were normalized relative to baseline values before drug application. Filled symbols, Average ± SEM; FV = 0.78 ± 0.02, fEPSP = 0.40 ± 0.05, during treatment; FV = 0.91 ± 0.03, fEPSP = 0.78 ± 0.05, after treatment (Wilcoxon signed test, *p < 0.05). D, Same as C, for tatCN19. FV = 0.79 ± 0.04, fEPSP = 0.58 ± 0.07, during treatment; FV = 0.93 ± 0.04, fEPSP = 0.76 ± 0.05, after treatment (*p < 0.05). Calibration: 5 ms, 0.4 mV. Error bars represent SEM in all figures.
This reduction of basal transmission could be due to an effect on LTP that occurred when the animal was alive (Whitlock et al., 2006). If so, CN peptides should also be able to reverse LTP that was induced experimentally. To examine this issue, we conducted two-pathway experiments and induced saturating LTP in one pathway. Subsequently, tatCN21 or scrambled peptide was transiently applied. If this procedure reversed saturated LTP, it should then be possible to reinduce LTP (Sanhueza et al., 2007). An example of such an experiment is shown in Figure 2A. LTP was initially induced by four tetani; an additional tetanus had no effect, demonstrating saturation. Transient application of peptide produced a persistent reduction of the fEPSP. One hour after washout of tatCN21, LTP could then be reinduced. Summary results for five experiments are presented in Figure 2B. When the same protocol was followed using tat-SCR peptide, LTP could not be reinduced (Fig. 2C). We conclude that tatCN21 can at least partially reverse the LTP maintenance process.
Figure 2.

TatCN21 partially reverses saturated LTP, allowing subsequent potentiation. A, Sample experiment in which saturated LTP was induced (by 4 tetani (4T), and 1 additional tetanus (1T) to show saturation). Twenty micromolar tatCN21 was next applied for 30 min, and drug was washed out for 1 h. Subsequently, LTP reinduction was tested. B, Average plot for five experiments; data were normalized to basal transmission before the first tetanization. Due to slight differences among experiments in the time for LTP reinduction (a few minutes), data were realigned with respect to tetanization time (right). C, Summary plot of mean fEPSP slope for the time intervals a–c, indicated in B; a: 1.50 ± 0.03, b: 1.20 ± 0.08, c: 1.49 ± 0.10; average ± SEM (*p < 0.01; one-way ANOVA for correlated samples, post hoc Tukey test). D, To quantify LTP reinduction, transmission was renormalized to baseline before the last series of tetani. The plot includes average data from similar experiments where SCR peptide was applied instead of tatCN21 (n = 4). After treatment with SCR peptide, tetanization induced no further potentiation, as fEPSP slope was not statistically different as compared to 20 min baseline (average ± SEM = 1.05 ± 0.05; p = 0.156), indicating that LTP saturation was unaffected. LTP reinduction after tatCN21 was 18.5 ± 1.9% (*p = 0.011; Wilcoxon rank test). The averaging interval is indicated by a continuous line.
To determine whether CN compounds reduce transmission by a postsynaptic action, we transfected cells with GFP-CN19. After 2 d, the EPSC in these cells was measured by whole-cell recording and compared to nearby untransfected cells (Hayashi et al., 2000). Figure 3A shows that the EPSC was reduced by postsynaptic CN19. Control experiments show that transfection itself does not reduce the EPSC (Pi et al., 2010). Given that spontaneous activity in cultured slices can induce LTP (Zhu et al., 2002), the smaller EPSP in transfected cells might be due to reduced LTP induction during the 2 d after transfection of CN19, a known CaMKII inhibitor, rather than to a reduction of a maintenance process. We therefore applied APV immediately after transfection with CN19 to block spontaneous LTP during this period. Figure 3B shows that the EPSC was still reduced. This result argues that the reduction is not due to a block of NMDAR-dependent LTP. Furthermore, since the reduction is seen during APV, it is not dependent on NMDAR-dependent LTD. We conclude that CN compounds act postsynaptically to reduce the maintenance of basal transmission.
Figure 3.

CN19 decreases synaptic transmission in an NMDAR-independent way. CA1 neurons from hippocampal slice culture were transfected to overexpress GFP-CN19 for 2 d. Cell-pair recordings were performed from a transfected neuron and a nearby untransfected neuron. A, AMPAR EPSCs were significantly decreased by the overexpression of GFP-CN19 (average ± SEM: 128.7 ± 26.2 vs 50.6 ± 7.8 pA for untransfected and transfected cells, respectively; n = 11, *p = 0.013 with paired Student's t test). B, Slices were incubated in 100 μm APV after transfection for 2 d, and AMPAR EPSCs were recorded with APV removed. CN19 still significantly decreased AMPAR EPSCs after APV treatment (117.0 ± 26.9 vs 28.7 ± 6.6 pA for untransfected and transfected cells, respectively; n = 10, *p = 0.017 with paired Student's t test).
Given the ability of CN compounds to reverse maintenance processes, it is important to establish whether they in fact affect the CaMKII/NMDAR complex in living cells, as suggested by work in cell-free assays (Vest et al., 2007). CaMKII is enriched in spines (Otmakhov et al., 2004; Zhang et al., 2008; Lee et al., 2009) and in the PSD (Otmakhov et al., 2004) even under basal conditions, and this may partly be due to basal CaMKII/NMDAR complex (Leonard et al., 1999). Thus, if the CaMKII/NMDAR complex is reduced by CN compounds, there should be a reduction in spine-bound CaMKII content. To test this prediction, cultured hippocampal slices that had been transfected with GFP-CaMKIIα and RFP as a volume marker were treated with 20 μm tatCN21 or tatCN21 SCR. Peptides were applied for 30 min and the effects monitored by simultaneous electrophysiological recording and imaging. The tatCN21 peptide produced the expected persistent reduction in the fEPSP (Fig. 4A,B). The small reduction at time >2 h observed after SCR peptide was often seen in long recordings when we used the small volume bath recirculation method. Measurements of GFP-CaMKIIα fluorescence showed that the peptide produced a 15–20% reduction in the bound fraction of CaMKII (Fig. 4C,D) (see Materials and Methods). Control peptide had no effect on either the synaptic response or the bound fraction GFP-CaMKIIα. Spine volume did not change significantly (Fig. 4C,D).
Figure 4.

TatCN21 decreases synaptic transmission and bound fraction of CaMKII in dendritic spines in slice cultures. A, The graph shows a significant and persistent (for at least 3 h) decrease of fEPSP amplitude after application of 20 μm tatCN21 (black symbols), while control tatCN21 SCR peptide was ineffective. The gray bar indicates the period of drug application. B, Examples of average waveforms of fEPSP at different times during the experiment. C, Graphs showing that bound fraction of GFP-CaMKII in spines (black symbols) but not spine volume (red symbols) significantly and persistently decreases after application of tatCN21 (black bar). D, SCR peptide did not affect either bound fraction (black symbols) or spine volume (red symbols). Concentrations of both peptides were 20 μm. Thin horizontal line shows a period taken for analysis of statistical significance between control and test experiments (p < 0.001). E, Top, A representative example of CA1 neurons transfected with RFP (left) and GFP-CaMKIIα (right). Images are maximum projection of 3D stack. Bottom, A representative image of a dendritic segment used for analysis in these experiments (this is an overlay of green and red fluorescence).
The above results are consistent with a reduction in the CaMKII/NMDAR complex, but a more specific test of this hypothesis is desirable. We therefore conducted a series of experiments that could directly monitor the effect of CN compound on the complex, as measured by coimmunoprecipitation (Leonard et al., 1999). Because these biochemical experiments required many treated slices, we used a procedure in which multiple slices could be treated with peptide. Most slices were then frozen for subsequent biochemical analysis. Other slices, prepared and treated in parallel, were used to determine whether peptide treatment with this incubation protocol produced a persistent reduction of synaptic strength. The slope of the curve relating fiber volley to fEPSP (I–O curve) gives a measure of average synaptic strength. This measure was compared for slices incubated for equal times with either tatCN21 or tatCN21 SCR. Following a procedure sufficient to inhibit induction, but not maintenance, of LTP (Buard et al., 2010), we used a 1 h incubation with 5 μm tatCN21. Slices were then washed for 1 h. As shown in Figure 5A–C, this procedure had no effect on basal transmission (Fig. 5C) (p > 0.1). As shown in Figure 6A, the procedure also had no effect on the CaMKII/NMDAR complex (p > 0.1). Thus, 5 μm tatCN21 does not affect basal transmission, consistent with previous findings (Buard et al., 2010), but also does not affect the CaMKII/NMDAR complex.
Figure 6.

TatCN21 persistently disrupts the CaMKII/NMDAR complex at 20 μm, but not 5 μm. After the same preincubation procedure described in Figure 5, slices were frozen for biochemical analysis. The NMDA receptor complex was solubilized with 1% deoxycholate before IP with control IgG or CaMKIIα antibody and IB for NR2B, NR1, α-actinin, and CaMKIIα. The immunosignals were quantified by densitometry, NMDAR and α-actinin signals were divided by CaMKIIα signals to correct for any variability in CaMKII IP, and tatCN21 values were normalized to SCR values (100%). Bars, Averages ± SEM. A, Five micromolar tatCN21 for 1 h does not affect basal CaMKII interaction with NMDAR (NR2B: CN21/SCR = 1.177 ± 0.076, p = 0.199, n = 4; NR1: CN21/SCR = 1.399 ± 0.135, p = 0.266, n = 4; averages ± SEM, paired t test) or α-actinin (CN21/SCR = 0.892 ± 0.088, n = 3). B, Twenty micromolar tatCN21 for 2 h produces persistent reduction of basal CaMKII interaction with NMDAR (NR2B: CN21/SCR = 0.386 ± 0.068, p = 0.0008, n = 5; NR1: CN21/SCR = 0.508 ± 0.156, p = 0.021, n = 6), but not α-actinin (CN21/SCR = 1.083 ± 0.189, n = 3).
We then tested 20 μm tatCN21 using the same procedure (Fig. 5D–F). This higher concentration produced a persistent reduction of synaptic strength (Fig. 5F) (p < 0.05). Moreover, there was a persistent reduction in the CaMKII/NMDAR complex, as determined by coprecipitation of either NR2B or NR1 (Fig. 6B) (p < 0.001 for NR2B and p < 0.05 for NR1). Total CaMKII, total NMDAR, and CaMKII binding to α-actinin were unaltered. Thus, tatCN21 concentrations sufficient to produce a persistent reduction of the CaMKII/NMDAR complex produce a persistent reduction of synaptic strength. This reduction was accompanied by the ability to induce enhanced LTP in response to subsequent stimuli (Fig. 7), as expected if the peptide had reduced a LTP maintenance process.
Figure 7.
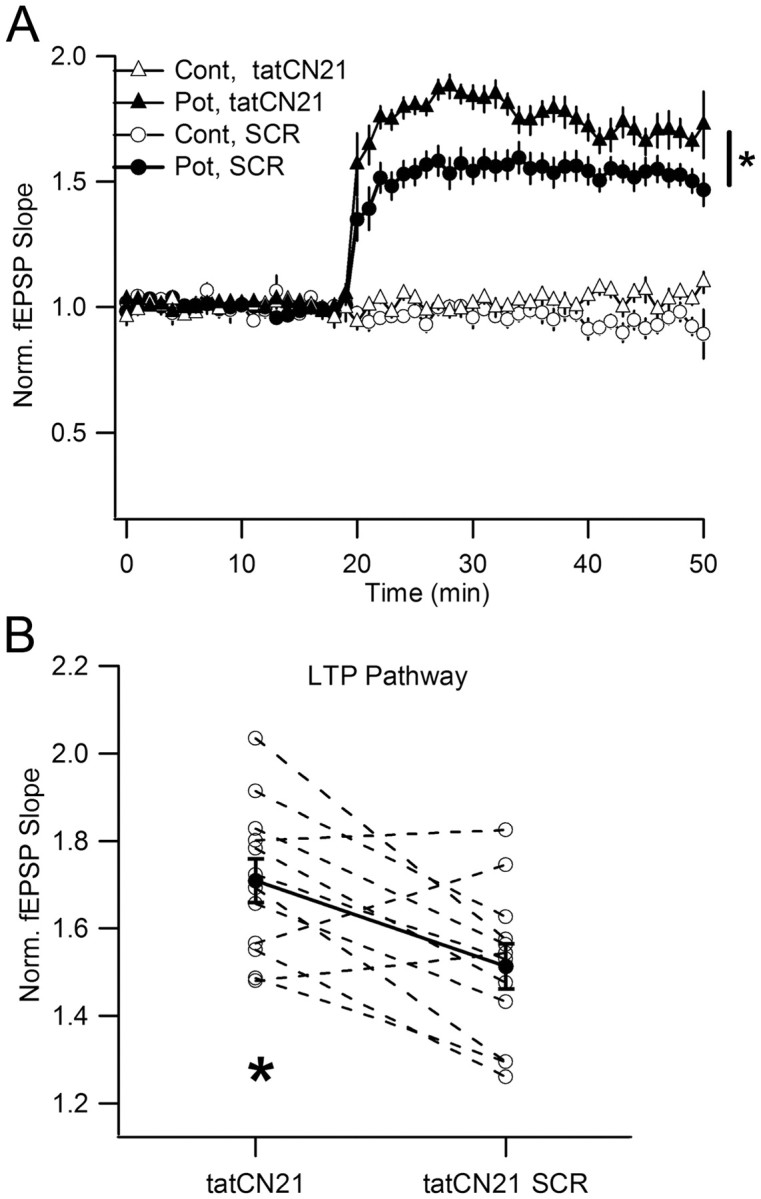
Transient treatment with tatCN21 allows higher subsequent potentiation of basal transmission than in slices similarly treated with scrambled peptide. A, Average plots for LTP induction after preincubation with tatCN21 or SCR peptide (20 μm for 2 h; 1 h drug washout). Cont, Control pathway; Pot, potentiated pathway. B, Each pair represents normalized fEPSP slope in potentiated pathway after tatCN21 or SCR peptide treatment of slices from the same animal (averaging interval: last 10 min). Filled symbols, Average ± SEM: 1.51 ± 0.05 for SCR and 1.71 ± 0.05 for tatCN21 (*p = 0.0045, paired t test). Data from 12 animals.
Discussion
If the CaMKII/NMDAR complex has a role in the maintenance of synaptic strength, reducing this complex should produce a reduction of synaptic strength. Here, we report the first experiments in which both synaptic strength and the amount of CaMKII/NMDAR complex in the slice preparation could be directly measured. We found that the CaMKII inhibitor, tatCN21 (Vest et al., 2007), reduced basal synaptic strength at concentrations that disrupt this complex (20 μm). At lower concentrations (5 μm) sufficient to inhibit CaMKII activity (Buard et al., 2010), but not to reduce the complex, there was no reduction in synaptic strength. We further found that tatCN21 could reverse saturated LTP, a reversal that then allowed additional LTP to be induced.
The effects of tatCN21 persisted after a 1 h washout of the peptide, a persistence that has important implications for the kind of biochemical processes involved. The effectiveness of washout is indicated by the fact that LTP cannot be induced in presence of CN peptide (Sanhueza et al., 2007; Buard et al., 2010) but can be induced after washout. The persistence of the effects of peptide thus argue that the CaMKII/NMDAR complex does not spontaneously reform after removal of the peptide, i.e., that the peptide is not simply interfering with equilibrium binding. These findings suggest that the complex has switch-like properties in which formation of the storage molecule is driven by synaptic activity rather than equilibrium processes.
CaMKII is present both presynaptically and postsynaptically, and both pools have important functions (Lisman, 2003; Hojjati et al., 2007). It has been argued (Sanhueza et al., 2007) that the persistent effects of bath-applied CN are postsynaptic, given that similar effects could be seen in experiments in which the postsynaptic cells were excited by AMPA application. Our results strengthen this conclusion by showing that postsynaptic expression of CN19 reduces basal transmission. Similar results (although not in APV) have been reported previously for CN27 (Goold and Nicoll, 2010). In contrast, Zhou et al. (2007) did not observe a significant effect on basal synaptic transmission by NR2B C-tail expression in transgenic mice. However, the variance of the measurements in Zhou et al. (2007) is on the same order as the effect we observe (∼20%) and so may have been undetected. Bath application of CaMKII inhibitor also produces a reversible decrease in presynaptic function (Waxham et al., 1993; Sanhueza et al., 2007). A component of this is due to a reduction in axon excitability (Fig. 1C,D) and probably arises from a change in Na channel excitability (Carlier et al., 2000; Wagner et al., 2006); an additional component may be due to the effect of CN compounds on presynaptic Ca channels (Jiang et al., 2008).
We found that CN compounds produce a persistent reduction of the bound CaMKII content of spines. This is consistent with the hypothesis that CN reduces the CaMKII/NMDAR complex in the PSD. The magnitude of the reduction (10–15%) is roughly consistent with FRAP, as well as photoactivation experiments showing that only ∼15% of CaMKII in spines is tightly bound (Otmakhov et al., 2007) and protein measurements indicating that ∼10% of spine CaMKII is in the PSD (B. Feng and J. Lisman, unpublished observations). A major fraction of spine CaMKII is weakly bound to actin (Shen and Meyer, 1999; Okamoto et al., 2004).
The persistent reduction in transmission produced by tatCN21 is unlikely to be due to a reduction in catalytic activity of CaMKII. This follows from the observation that 5 μm tatCN21 is sufficient to block LTP induction (Buard et al., 2010), a process that requires CaMKII activity (Malinow et al., 1989; Silva et al., 1992) but does not affect basal synaptic transmission (Buard et al., 2010) or the CaMKII/NMDAR complex (shown here). Other CaMKII inhibitors that are relatively ineffective blockers of the CaMKII/NMDA receptor complex formation (Bayer et al., 2006) can block LTP induction but also do not affect synaptic strength or LTP maintenance (Malinow et al., 1989; Otmakhov et al., 1997; Chen et al., 2001; Wang et al., 2008). Thus, the fact that 5 μm tatCN21 is not sufficient to disrupt the complex provides a satisfactory explanation for why we did not find any effect of that concentration on synaptic strength, as reported previously (Buard et al., 2010). It is unlikely that the effect of CN21 was due to inhibition of other CaM-kinases or PKC, because inhibition of these kinases does not affect basal transmission (Huber et al., 1995; Kasten et al., 2007; Redondo et al., 2010). Together, these results strongly suggest that CN compounds affect synaptic strength through a structural rather than a catalytic process.
What might be the structural processes by which CN affects synaptic strength? There are several known proteins that CaMKII binds in addition to NMDA receptors. These include actin, α-actinin, densin-180, SAP97, MUPP1, and L-type Ca channels (Walikonis et al., 2001; Krapivinsky et al., 2004; Okamoto et al., 2004; Hudmon et al., 2005; Robison et al., 2005; Buard et al., 2010; Nikandrova et al., 2010) [for review, see Colbran and Brown (2004) and Merrill et al. (2005)]. Several of these proteins may bind to the same site on CaMKII as the NR2B. Therefore, we cannot exclude that CN compounds might also interfere with the binding of CaMKII to some of these targets. However, it is likely that the changes in synaptic strength that we observe arise mostly from the demonstrated change in the CaMKII/NMDAR complex because blocking the formation of this complex by other means (overexpression of mutated NR2B or NR2B C-tail) strongly reduces LTP induction (Barria and Malinow, 2005; Zhou et al., 2007). Additionally, expression of NR2B has been described to reconstitute activity-driven translocation of CaMKII in heterologous cells (Strack et al., 2000; Bayer et al., 2006).
The binding of CaMKII to the NMDAR places the kinase in a unique environment where the important T286 site is protected from dephosphorylation (Mullasseril et al., 2007). This may explain why T286 phosphorylation is detected in the PSD under basal conditions (Lengyel et al., 2004; Larsson and Broman, 2005; Dosemeci and Jaffe, 2010). Although phospho-T286 is not required for the initial binding to NR2B (Bayer et al., 2001), it strengthens this binding (Strack and Colbran, 1998; Bayer et al., 2006). In addition, several other proteins (Densin-180, NR1) also preferentially bind to the phosphorylated state of CaMKII through different binding sites on CaMKII (Walikonis et al., 2001; Leonard et al., 2002). A 12 subunit holoenzyme CaMKII can simultaneously bind several different PSD proteins, allowing the formation of macromolecular complexes (Robison et al., 2005). A simple possibility is that one or more of these CaMKII binding proteins serve to anchor AMPARs at the synapse and thereby control synaptic strength through mainly structural mechanisms (Lisman and Zhabotinsky, 2001).
It is noteworthy that CN compounds produce only a partial reduction in transmission and partial reversal of experimentally induced LTP. One possible explanation is that there remains substantial complex, as shown by our immunoprecipitation results. Another possibility is that molecular processes involving PKMζ (Sacktor, 2008) support other components of synaptic strength. Further experiments will be required to gain insight into this issue.
Several criteria must be met to conclude that a molecule serves as a memory mechanism for the synapse. One criterion is that reducing the molecule's function must produce a persistent reduction in the maintenance of synaptic strength. Our results demonstrate such a reduction and provide a direct verification that the putative memory molecule (in this case, the CaMKII/NMDAR complex) has, in fact, been reduced. This satisfies one important criterion for establishing that the CaMKII/NMDAR complex is a synaptic memory. The other required criterion is to show that LTP induction produces a persistent increase in the complex. Strong synaptic action (NMDA application) can produce a persistent increase in this complex. However, it will be important to test whether such an increase occurs during actual LTP induction.
Footnotes
This work was supported by Fondo Nacional de Desarrollo Científico y Tecnológico Grant 1080630 (M.S., G.F-V.), Minesterio de Planificacion y Cooperacion Grant ICMP05-001-F (M.S.), and National Institutes of Health Grants R01 NS052644 (K.U.B.), NS046450 (J.W.H.), and NS027337 (J.L.).
The University of Colorado is currently seeking patent protection for tatCN21 and its uses.
References
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O'Leary H, Schulman H, De Koninck P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci. 2006;26:1164–1174. doi: 10.1523/JNEUROSCI.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buard I, Coultrap SJ, Freund RK, Lee YS, Dell'Acqua ML, Silva AJ, Bayer KU. CaMKII “autonomy” is required for initiating but not for maintaining neuronal long-term information storage. J Neurosci. 2010;30:8214–8220. doi: 10.1523/JNEUROSCI.1469-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier E, Dargent B, De Waard M, Couraud F. Na(+) channel regulation by calmodulin kinase II in rat cerebellar granule cells. Biochem Biophys Res Commun. 2000;274:394–399. doi: 10.1006/bbrc.2000.3145. [DOI] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci U S A. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Calcium/calmodulin-dependent protein kinase II inhibitor protein: localization of isoforms in rat brain. Neuroscience. 2001;102:767–777. doi: 10.1016/s0306-4522(00)00520-0. [DOI] [PubMed] [Google Scholar]
- Chen HX, Otmakhov N, Strack S, Colbran RJ, Lisman JE. Is persistent activity of calcium/calmodulin-dependent kinase required for the maintenance of LTP? J Neurophysiol. 2001;85:1368–1376. doi: 10.1152/jn.2001.85.4.1368. [DOI] [PubMed] [Google Scholar]
- Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol. 2004;14:318–327. doi: 10.1016/j.conb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Dosemeci A, Jaffe H. Regulation of phosphorylation at the postsynaptic density during different activity states of Ca2+/calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 2010;391:78–84. doi: 10.1016/j.bbrc.2009.10.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci A, Tao-Cheng JH, Vinade L, Winters CA, Pozzo-Miller L, Reese TS. Glutamate-induced transient modification of the postsynaptic density. Proc Natl Acad Sci U S A. 2001;98:10428–10432. doi: 10.1073/pnas.181336998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68:512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas K, Sin WC, Javaherian A, Li Z, Cline HT. Single-cell electroporation for gene transfer in vivo. Neuron. 2001;29:583–591. doi: 10.1016/s0896-6273(01)00235-5. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hojjati MR, van Woerden GM, Tyler WJ, Giese KP, Silva AJ, Pozzo-Miller L, Elgersma Y. Kinase activity is not required for alphaCaMKII-dependent presynaptic plasticity at CA3-CA1 synapses. Nat Neurosci. 2007;10:1125–1127. doi: 10.1038/nn1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Huber KM, Mauk MD, Thompson C, Kelly PT. A critical period of protein kinase activity after tetanic stimulation is required for the induction of long-term potentiation. Learn Mem. 1995;2:81–100. doi: 10.1101/lm.2.2.81. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Structure/function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Lebel E, Roy H, Sik A, Schulman H, Waxham MN, De Koninck P. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J Neurosci. 2005;25:6971–6983. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Lautermilch NJ, Watari H, Westenbroek RE, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc Natl Acad Sci U S A. 2008;105:341–346. doi: 10.1073/pnas.0710213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasten MR, Fan Y, Schulz PE. Activation of silent synapses with sustained but not decremental long-term potentiation. Neurosci Lett. 2007;417:84–89. doi: 10.1016/j.neulet.2007.02.035. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE. SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron. 2004;43:563–574. doi: 10.1016/j.neuron.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Larsson M, Broman J. Different basal levels of CaMKII phosphorylated at Thr286/287 at nociceptive and low-threshold primary afferent synapses. Eur J Neurosci. 2005;21:2445–2458. doi: 10.1111/j.1460-9568.2005.04081.x. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel I, Voss K, Cammarota M, Bradshaw K, Brent V, Murphy KP, Giese KP, Rostas JA, Bliss TV. Autonomous activity of CaMKII is only transiently increased following the induction of long-term potentiation in the rat hippocampus. Eur J Neurosci. 2004;20:3063–3072. doi: 10.1111/j.1460-9568.2004.03748.x. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Bayer KU, Merrill MA, Lim IA, Shea MA, Schulman H, Hell JW. Regulation of calcium/calmodulin-dependent protein kinase II docking to N-methyl-D-aspartate receptors by calcium/calmodulin and alpha-actinin. J Biol Chem. 2002;277:48441–48448. doi: 10.1074/jbc.M205164200. [DOI] [PubMed] [Google Scholar]
- Lisman J. Long-term potentiation: outstanding questions and attempted synthesis. Philos Trans R Soc Lond B Biol Sci. 2003;358:829–842. doi: 10.1098/rstb.2002.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Zhabotinsky AM. A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron. 2001;31:191–201. doi: 10.1016/s0896-6273(01)00364-6. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Merrill MA, Chen Y, Strack S, Hell JW. Activity-driven postsynaptic translocation of CaMKII. Trends Pharmacol Sci. 2005;26:645–653. doi: 10.1016/j.tips.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- Mullasseril P, Dosemeci A, Lisman JE, Griffith LC. A structural mechanism for maintaining the ‘on-state’ of the CaMKII memory switch in the post-synaptic density. J Neurochem. 2007;103:357–364. doi: 10.1111/j.1471-4159.2007.04744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikandrova YA, Jiao Y, Baucum AJ, Tavalin SJ, Colbran RJ. Ca2+/calmodulin-dependent protein kinase II binds to and phosphorylates a specific SAP97 splice variant to disrupt association with AKAP79/150 and modulate alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor (AMPAR) activity. J Biol Chem. 2010;285:923–934. doi: 10.1074/jbc.M109.033985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Griffith LC, Lisman JE. Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. J Neurosci. 1997;17:5357–5365. doi: 10.1523/JNEUROSCI.17-14-05357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhov N, Tao-Cheng JH, Carpenter S, Asrican B, Dosemeci A, Reese TS, Lisman J. Persistent accumulation of calcium/calmodulin-dependent protein kinase II in dendritic spines after induction of NMDA receptor-dependent chemical long-term potentiation. J Neurosci. 2004;24:9324–9331. doi: 10.1523/JNEUROSCI.2350-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhov N, Asrican B, Lisman J. Measuring dynamic exchange of bound CaMKIIa in spines and dendrites. Soc Neurosci Abstr. 2007;33:788–8. [Google Scholar]
- Pi HJ, Otmakhov N, Lemelin D, De Koninck P, Lisman J. Autonomous CaMKII can promote either long-term potentiation or long-term depression, depending on the state of T305/T306 phosphorylation. J Neurosci. 2010;30:8704–8709. doi: 10.1523/JNEUROSCI.0133-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo RL, Okuno H, Spooner PA, Frenguelli BG, Bito H, Morris RG. Synaptic tagging and capture: differential role of distinct calcium/calmodulin kinases in protein synthesis-dependent long-term potentiation. J Neurosci. 2010;30:4981–4989. doi: 10.1523/JNEUROSCI.3140-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Bass MA, Jiao Y, MacMillan LB, Carmody LC, Bartlett RK, Colbran RJ. Multivalent interactions of calcium/calmodulin-dependent protein kinase II with the postsynaptic density proteins NR2B, densin-180 and alpha-actinin-2. J Biol Chem. 2005;280:35329–35336. doi: 10.1074/jbc.M502191200. [DOI] [PubMed] [Google Scholar]
- Sacktor TC. PKMzeta, LTP maintenance, and the dynamic molecular biology of memory storage. Prog Brain Res. 2008;169:27–40. doi: 10.1016/S0079-6123(07)00002-7. [DOI] [PubMed] [Google Scholar]
- Sanhueza M, McIntyre CC, Lisman JE. Reversal of synaptic memory by Ca2+/calmodulin-dependent protein kinase II inhibitor. J Neurosci. 2007;27:5190–5199. doi: 10.1523/JNEUROSCI.5049-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Connor JH, Shenolikar S, Meyer T. Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase II. Nat Neurosci. 2000;3:881–886. doi: 10.1038/78783. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- Strack S, Choi S, Lovinger DM, Colbran RJ. Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. J Biol Chem. 1997;272:13467–13470. doi: 10.1074/jbc.272.21.13467. [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- Vest RS, Davies KD, O'Leary H, Port JD, Bayer KU. Dual mechanism of a natural CaMKII inhibitor. Mol Biol Cell. 2007;18:5024–5033. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walikonis RS, Oguni A, Khorosheva EM, Jeng CJ, Asuncion FJ, Kennedy MB. Densin-180 forms a ternary complex with the α-subunit of Ca2+/calmodulin-dependent protein kinase II and α-actinin. J Neurosci. 2001;21:423–433. doi: 10.1523/JNEUROSCI.21-02-00423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Feng R, Phillip Wang L, Li F, Cao X, Tsien JZ. CaMKII activation state underlies synaptic labile phase of LTP and short-term memory formation. Curr Biol. 2008;18:1546–1554. doi: 10.1016/j.cub.2008.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxham MN, Malenka RC, Kelly PT, Mauk MD. Calcium/calmodulin-dependent protein kinase II regulates hippocampal synaptic transmission. Brain Res. 1993;609:1–8. doi: 10.1016/0006-8993(93)90847-g. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Zhang YP, Holbro N, Oertner TG. Optical induction of plasticity at single synapses reveals input-specific accumulation of alphaCaMKII. Proc Natl Acad Sci U S A. 2008;105:12039–12044. doi: 10.1073/pnas.0802940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Takahashi E, Li W, Halt A, Wiltgen B, Ehninger D, Li GD, Hell JW, Kennedy MB, Silva AJ. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J Neurosci. 2007;27:13843–13853. doi: 10.1523/JNEUROSCI.4486-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]