

Abstract
Notch signaling is pivotal for the morphogenesis and homeostasis of many tissues. We found that aberrant Notch activation in mouse neural-crest-derived periocular mesenchymal cells (POMCs), which contribute to the formation of corneal and eyelid stroma, results in blepharophimosis. Compound transgenic mice overexpressing the Notch1 intracellular domain (N1-ICD) in POMCs (POMCN1-ICD) showed relatively minor effects on the cornea, but increased cell apoptosis and decreased cell proliferation during eyelid morphogenesis. Eyelid closure at E15.5 and eyelid formation at birth were incomplete. In further analyses, overexpression of N1-ICD impaired eyelid levator smooth muscle formation by downregulating the transcription factor FoxL2. This is similar to the effect of haploinsufficiency of FOXL2 in humans, which results in type II BPES (blepharophimosis, ptosis and epicanthus inversus syndrome). In vitro studies showed that FoxL2 expression is augmented by a low dose of N1-ICD but was downregulated by a high dose, depending on the extent of Hes-1 and Hey-1 activation. Moreover, transfection of CMV-FoxL2 enhanced α-SMA promoter activity. These data strongly imply that a physiologically low level of Notch1 is crucial for proper FoxL2 expression in POMCs, which is, in turn, essential for Müeller muscle formation and normal eyelid development.
Key words: BPES, FoxL2, Neural crest, Notch signaling, Eyelid, Morphogenesis
Introduction
In mammals, eyelids are of paramount importance, not only for forming a functional visual system, but also for protecting the ocular surface from environmental insults. Eyelid morphogenesis is a dynamic process involving active interactions between the epidermis and the dermis (Findlater et al., 1993). To achieve eyelid closure in the fetus, a coordinated movement of the neural-crest-derived peri-ocular mesenchymal cells (POMCs) has a pivotal role in forming the lid-specific structures, including levator smooth muscle, tarsus and Meibomian glands (Nien et al., 2010). Developmental defects in POMCs during eyelid closure can lead to congenital disorders such as blepharophimosis, ptosis and epicanthus inversus syndrome (BPES).
Human BPES is an autosomal dominant genetic disorder characterized by craniofacial defects that mainly affect eyelid development, often in association with premature ovarian failure (POF). Genetic and epidemiological studies have shown that mutations of the gene encoding the transcription factor forkhead box L2 (FoxL2) are responsible for BPES, among which, 70% have intragenic mutations, but the remaining 30% have mutations found upstream or downstream of the coding region (De Baere et al., 2003). Mice lacking FoxL2 exhibit eyelid open at birth (EOB) and ovarian malformations (Uda et al., 2004; Schmidt et al., 2004). These observations suggest that mutations that lead to qualitative or quantitative changes of FoxL2 are involved in the pathogenesis of eyelids and ovary in BPES. In the ovary, reactive oxidative stress is the major inducer for upregulating FoxL2, which triggers the modulation of stress-related target genes such as manganese superoxide dismutase (MnSOD) (Benayoun et al., 2009). However, little is known about the effects of mutations upstream and downstream of FoxL2 during eyelid morphogenesis.
Notch signaling has been shown to have a pivotal role in various cellular processes, including cell fate determination, differentiation, proliferation, apoptosis, cell–cell adhesion and migration events through local cell-cell interactions (reviewed by Bolós et al., 2007; Fiúza and Arias, 2007; Gridley, 2007; Borggrefe and Oswald, 2009; Kopan and Ilagan, 2009). The Notch receptor exists at the cell surface as a proteolytically cleaved heterodimer consisting of a large ectodomain and a membrane-tethered intracellular domain. Ligands of the Delta-like (DLL1, DLL3, DLL4) and Jagged (JAG1 and JAG2) families interact with receptors of Notch family (NOTCH1–NOTCH4) on an adjacent cell. The binding between ligand and receptor induces further proteolytic cleavages of Notch that release the Notch intracellular domain (NICD) from the cell membrane. The NICD translocates into the nucleus, where it forms a complex with the recombination signal binding protein for immunoglobulin kappa J region (RBP-Jκ) protein, displacing a histone deacetylase (HDAc)-co-repressor (CoR) complex from the RBP-Jκ protein. Components of an activation complex, mastermind-like protein 1 (MAML1) and histone acetyltransferases (HAc), are recruited to the NICD–RBP-Jκ complex, leading to the transcriptional activation of Notch target genes.
Notch signaling has been shown to have pivotal roles in corneal homeostasis (Ma et al., 2007; Vauclair et al., 2007; Djalilian et al., 2008; Nakamura et al., 2008), but its function in other ocular surface tissues such as the eyelid has not been explored. In the present study, we took a gain-of-function approach in transgenic mice conditionally misexpressing the Notch1 intracellular domain (N1-ICD) in POMCs during eyelid morphogenesis. As a consequence, eyelid closure was delayed at embryonic day (E) 15.5, resulting in poor lid closure at birth as a result of downregulation of FoxL2, the absence of levator muscle, tarsus and Meibomian glands, which together, resembled BPES in humans. We investigated how activation of Notch1 might serve as the upstream controller for expression of FoxL2 in periocular mesenchyma cells, which are destined to become levator smooth muscle cells in the eyelids.
Results
Generation and characterization of KR/TC transgenic driver for POMC gene manipulation in vivo
To manipulate expression of loss-of-function and/or gain-of-function genes at the desired time to study their roles in POMCs during embryonic development, we first generated a novel transgenic mouse line called KR which harbors a 1.1 kb mutant reverse tetracycline transactivator (rtTA2S-M2) minigene (Clontech) driven by a 4.8 kb keratocan gene regulatory cassette (Liu et al., 2000; Holmberg et al., 2004; Hayashi et al., 2005). The KR mice were then crossed with a TetO-Cre (TC) mouse strain (Perl et al., 2002), carrying a Dox-inducible TetO-CMVmin promoter-driven Cre recombinase minigene, to obtain the KR/TC double transgenic mouse strain, which served as a Dox-inducible driver (Fig. 1A). The functionality of the KR/TC strain was tested by crossing with Z/EG, a Cre reporter mouse line (Novak et al., 2000). The resulting KR/TC/Z/EG triple transgenic mouse (Fig. 1, bottom left) were induced with Dox chow in the pregnant mother from gestation day 12.5 (E12.5) and examined at birth. We found that strong green fluorescent signals were readily detected in specific regions such as eyelids, snout, ears and limbs, using dissecting epi-fluorescent microscopy (Fig. 1B,D). Such a pattern is consistent with our previously published results in Kerapr3.2-β-Geo-BpA transgenic mice (Liu et al., 2000). At the cellular level, EGFP was expressed in the stromal but not in the epithelial cells, as shown in a section of the eye region. Eyelids and corneal stromal cells displayed a strong EGFP-positive signal, but no positive signal was observed in epithelial or endothelial cells, or in other ocular tissues such as the sclera, lens and retina (Fig. 1E,F). Thus, the KR/TC mouse strain is a novel transgenic driver to manipulate expression of loss-of-function and/or gain-of-function genes at desired time to study their roles in aforementioned tissues including peri-ocular mesenchymal cells during embryonic development.
Fig. 1.

Functional analysis of the KR/TC double transgenic mice. (A) KR/TC/Z/EG triple transgenic mice were generated by natural crossing between KR, TC and Z/EG mouse lines. EGFP reporter gene expression can be induced by feeding the mouse with Dox. Images from stereomicroscope with (B) and without (C) green fluorescence show EGFP (green) expression pattern in Dox-treated KR/TC/Z/EG transgenic eye at P0. (D) The eye region. (E) A section across the P0 eye showing EGFP (green) expression patterns in the ocular tissues. (F) Enlargement of boxed region in E. Co, cornea; Er, ear; Ey, eye; Le, lens; LL, lower lid; Re, retina; s, snout; UL, upper lid.
Aberrant expression of N1-ICD in POMCs interrupts fetal eyelid closure
Notch signaling is central to vertebrate development, and analysis of Notch has provided important insights into pathogenic mechanisms in many tissues (Gridley, 2007). However, little is known about the role of Notch in the development and pathology of ocular surfaces. To test whether there is potential function of Notch1 in the specification and differentiation of POMCs for ocular surface morphogenesis, we first examined the Notch1 expression pattern during eyelid morphogenesis. Immunostaining with anti-Notch1 antibody clearly showed that Notch1 was predominantly expressed by the epidermis of the eyelid and moderately in corneal epithelium (Fig. 2A,B). The expression level of Notch1 during eyelid closure decreased and the expression pattern was gradually shifted to the migrating edges at E15.5 and became restricted to the eyelid epithelial fusion junction at E17.5 (Fig. 2, compare C with B). Likewise, the corneal epithelium downregulated Notch1 between E15.5 and E17.5 when the lids were merged (Fig. 2, compare C with B). However, we noticed that there was very little Notch1 expression in dermis of the eyelid and corneal stroma during eyelid closure from E13.5–E17.5 (Fig. 2A–C). We then asked whether aberrantly expression of N1-ICD in POMCs would have dramatic impact on cell specification and differentiation during eyelid morphogenesis. We used triple transgenic mice KR/TC/R26fN1-ICD in which the rtTA was constitutively expressed in the Kera+ POMCs. Upon administration of Dox, transcriptional activation of TetO-CMVmin promoter yielded the production of Cre recombinase, which functioned as a molecular scissor to delete the loxP flanking sequence and thus activate the expression of N1-ICD driven by the R26 promoter (Murtaugh et al., 2003) (Fig. 2D).
Fig. 2.

Notch1 expression pattern during embryonic eyelid closure and genetic overexpression of N1-ICD in POMC. (A–C) Immunoflurescence staining with anti-Notch1 antibody on sections of mouse eyelids at E13.5~E17.5. Note that Notch1-positive signals (red) are detected predominantly in eyelid epidermis, with very few in the dermis. Nuclear counter staining with DAPI is shown in blue. (D) Schematic representation of N1-ICD expression via Cre-loxP system in POMC. The rtTA is constitutively expressed in Kera-positive POMCs. In the presence of Dox, Cre recombinase is induced to delete the Neo/Stop cassette, permitting transcription of Notch1 ICD. Co, cornea; Le, lens; LL, lower lid; Re, retina; UL, upper lid.
To examine phenotypic changes caused by the overexpression of N1-ICD during development, time-mated embryos were obtained by crossing KR/R26fN1-ICD male and TC/R26fN1-ICD female mouse and Dox chow was administered to the pregnant mouse in the dam from E12.5 to E15.5 (Fig. 3A–D), day of birth (Fig. 3E–H) and P42 (Fig. 3I–N), respectively. Normally, eyelid closure begins at E13.5–E14 and is complete by E15.5–E16. As expected, eyelid closure took place and nearly covered the corneal surface at E15.5 in control TC/R26fN1-ICD double transgenic mouse embryo (Fig. 3A,C); however, it was impaired in Dox-treated KR/TC/R26fN1-ICD embryos (Fig. 3B,D). At birth, the mouse pups had closed eyelids with an obvious fusion line between upper and lower eyelids, as seen in control TC/R26fN1-ICD (Fig. 3E,G) and KR/R26fN1-ICD double transgenic mice (data not shown). However, there was no obvious eyelid fusion line in Dox-treated KR/TC/R26fN1-ICD mice (Fig. 3F,H). Histological examinations revealed that the upper and lower eyelids of POMCN1-ICD did not come close enough to support the formation of this fusion line (Fig. 3H). The mouse eyelid is normally closed at birth and begins to re-open at P12–14 (Findlater et al., 1993). We have noticed that Dox-induced POMCN1-ICD in the KR/TC/R26fN1-ICD mice failed to open their eyelid (blepharophimosis) at P15 and P60, and never fully opened (ptosis-like) (data not shown) as compared with their TC/R26fN1-ICD or KR/R26fN1-ICD control littermates. Histological and immunohistochemical analyses of a Dox-treated KR/TC/R26fN1-ICD mouse at P42 showed that the eyelid fissures were obviously narrower (Fig. 3N) than that of the control TC/R26fN1-ICD littermate (Fig. 3M). This is probably due to the lack of Müeller lavator muscle in POMCN1-ICD eyelid (Fig. 3, compare N with M). In addition to eyelid malformations, these Dox-treated triple transgenic mice also had other defects, including craniofacial malformation, shortened outer ears and forelimb, but they did not have gait problems (supplementary material Movie 1) and both genders were fertile. Altogether, our data demonstrate that POMCN1-ICD delayed embryonic eyelid closure and impaired eyelid re-opening around P12–P14. The phenotypic manifestations resemble PBES type II in humans (Crisponi et al., 2001).
Fig. 3.

POMCN1-ICD impairs eyelid development. (A–D) Photographs and H&E-stained paraffin sections of mouse eyes at E15.5. Arrowheads in A and C indicate the formation of fusion line between upper and lower eyelids. The eyelid of POMCN1-ICD remained open (arrows in B). (E–H) Photographs and H&E staining of mouse eyes at birth (P0). POMCN1-ICD causes incomplete eyelid closure. Arrowheads in E and G indicate the formation of a fusion line between upper and lower eyelids. Arrows in F and H indicate an eyelid closure defect. (I–N) Photographs and H&E staining of mouse eyes at P42. POMCN1-ICD exhibits an abnormality in the palpebral fissure. Arrowheads in M indicate the eyelid lavatory muscle which was not found in the POMCN1-ICD in N. Co, cornea; Le, lens; LL, lower eyelid; Re, retina; UL, upper eyelid.
Immunofluorescent staining showed that in addition to signals in eyelid epidermis, strong nuclear Notch1 immunofluorescent signals were detected in eyelids and corneal stromal cells of the Dox-treated KR/TC/R26fN1-ICD triple transgenic embryos (Fig. 4Ba–c). However, Notch1 expression was detected mainly in the eyelid epidermis, but at relatively low levels in the eyelid stroma of the TC/R26fN1-ICD littermates treated with Dox (Fig. 4Aa) or other age-matched control embryos such as uninduced KR/TC/R26fN1-ICD triple transgenic embryos (data not shown). These results demonstrate that the transgenic N1-ICD mutant protein was aberrantly expressed in the POMC by Dox induction.
Fig. 4.

Nuclear N1-ICD misexpression in Dox-treated KR/TC/R26fN1-ICD/WT (POMCN1-ICD) mice at E15.5. Immunofluorescent staining with anti-Notch1 antibody. Red: Notch positive signal. Blue: DAPI nuclear counterstain. Note that Notch1-positive signals are rare in POMCwild-type eye (A,Aa), but abundant in POMCN1-ICD eyelid and corneal stroma (B,Ba,Bb,Bc). Co, cornea; Le, lens; LL, lower eyelid; Re, retina; UL, upper eyelid.
To investigate whether POMCN1-ICD had any impact to cell proliferation and death, immunostaining of 5-bromo-2-deoxyuridine (BrdU) incorporation and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay were performed using embryos at E14.5, one day before exhibiting prominent eyelid outgrowth phenotypes in POMCN1-ICD. Fig. 5 clearly shows that overexpression of N1-ICD caused a 4.1-fold reduction of BrdU incorporation (Fig. 5A–H), but triggered a 3.7-fold elevation of apoptosis in POMCs (Fig. 5I–O). These data demonstrate that misexpression of N1-ICD in the POMCs had an impact on eyelid dermis cell proliferation and survival during eyelid morphogenesis. It should be noted that the differences in BrdU incorporation were statistically significant only in the eyelid stroma where keratocan was expressed, not in the epidermis where keratocan is not expressed (Fig. 5G,H). This suggests that the effect of NICD on proliferation on the eyelid dermis might be a cell-autonomous effect.
Fig. 5.

Overexpression of N1-ICD decreases BrdU uptake but triggers apoptosis in POMCs during eyelid morphogenesis at E14.5. (A–F) Immunohistochemistry of anti-BrdU antibody. Sections of the eyelids of TC/R26fN1-ICD (POMCWT) (A, BrdU; B, BrdU/DAPI merged; C, close-up view from B) and KR/TC/R26fN1-ICD (POMCN1-ICD) embryos (D, BrdU; E, BrdU and DAPI merged; F, close-up view from E) labeled with BrdU (red) and nuclear counterstained with DAPI. (I–N) TUNEL assay. Sections of the eyelids of TC/R26fN1-ICD (POMCWT) (I, TUNEL; J, DAPI; K, TUNEL and DAPI merged) and KR/TC/R26fN1-ICD (POMCN1-ICD) embryos (L, TUNEL; M, DAPI; N, TUNEL and DAPI merged) labeled with Click-iT® TUNEL Assay kit (green) and nuclear counterstained with DAPI. (G) Quantitative analysis of the percentage of BrdU-labeled cells in eyelid stroma. KR/TC/R26fN1-ICD (POMCN1-ICD, 4.48±0.45%) embryos showed significant reduction in the percentage of BrdU-labeled eyelid stromal cells, compared with the TC/R26fN1-ICD (POMCWT, 18.55±4.34%) littermates in the eyelid mesenchyme. (H) No significant difference of BrdU-labeled eyelid epithelium between POMCN1-ICD (47.54± 7.16%) and POMCWT (46.84±2.84%). (O) Quantitative analysis of TUNEL-labeled cells per section of eyelid margin. KR/TC/R26fN1-ICD (POMCN1-ICD, 18.31± 2.49%) embryos showed significant elevation in TUNEL-labeled cells, compared with the TC/R26fN1-ICD (POMCWT, 4.94±0.51%) littermates in the eyelid stromal mesenchyme. Data are represented as mean ± s.d. (n=5). *P<0.05. Dashed lines in C, F, K, and N demarcate eyelid epithelium from stromal mesenchyme. Co, cornea; epi, eyelid epithelium; der, eyelid dermis; Le, lens; LL, lower lid; Re, retina; UL, upper lid.
Aberrant expression of N1-ICD in POMCs failed to form Müeller smooth muscle and interrupted post-natal eyelid re-opening
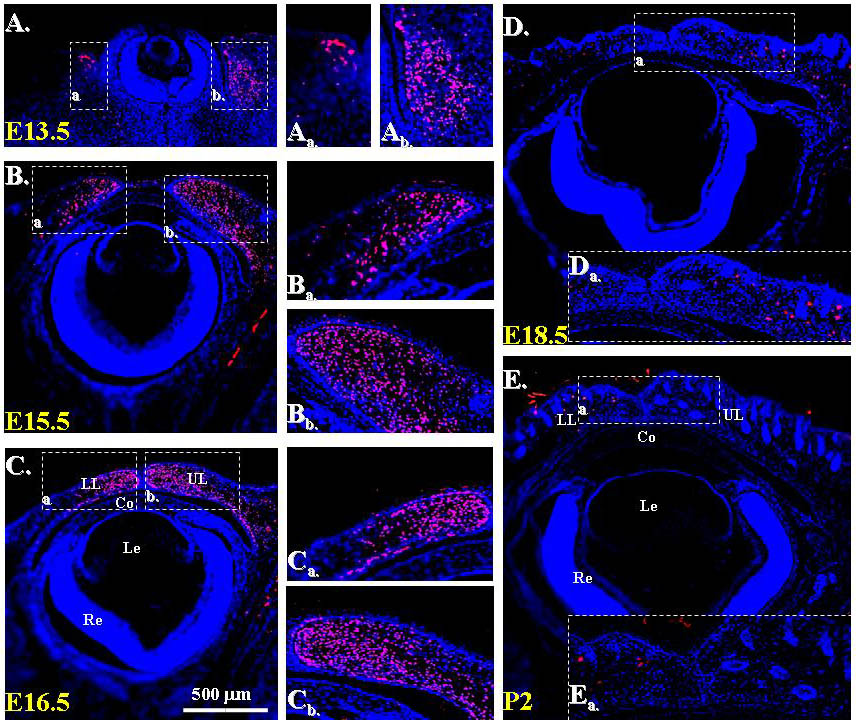
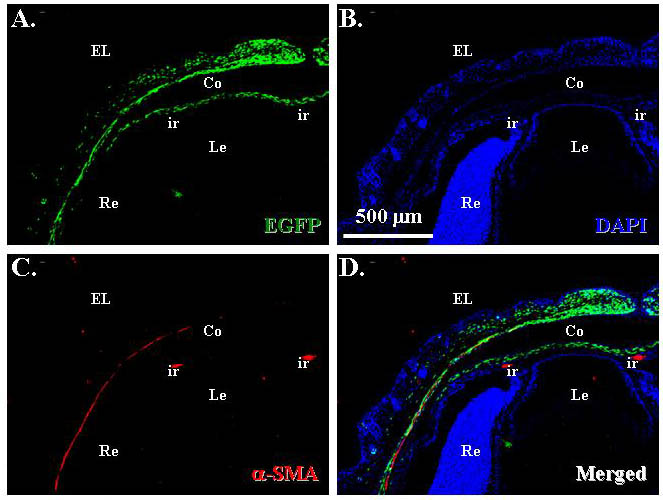
Because the craniofacial and palpebral phenotypes observed in Dox-treated KR/TC/R26fN1-ICD mice vividly resembled human BPES type II, and it has been well documented that FOXL2 gene mutation is directly associated with this disorder, it prompted us to compare the FoxL2 expression level in POMCwild-type and POMCN1-ICD during eyelid development. Immunofluorescent staining revealed that FoxL2 indeed was abundantly and transiently expressed in POMCs during eyelid closure. The FoxL2 protein level gradually increased from E13.5 to E16.5 (supplementary material Fig. S1A,B,C), was dramatically downregulated at E18.5 (supplementary material Fig. S1D) and completely diminished at P2 (supplementary material Fig. S1E). More interestingly, FoxL2 was robustly expressed in POMCwild-type of the Dox-treated TC/R26fN1-ICD (Fig. 6A) but dramatically decreased in those of KR/TC/R26fN1-ICD (Fig. 6B) at E15.5. Similarly, a strong α-SMA-positive smooth muscle sheet was detected in POMCwild-type of the Dox-treated TC/R26fN1-ICD (Fig. 6C), but only a small α-SMA-positive cell mass was present in POMCN1-ICD of the Dox-treated KR/TC/R26fN1-ICD mice (Fig. 6D). Indeed, most of these α-SMA-positive signals overlapped with EGFP-positive cells in the eyelid stroma of the KR/TC/Z/EG mice treated with Dox from E12.5 to P0 (supplementary material Fig. S2), suggesting that eyelid levator muscle was derived from a Kera-positive cell lineage during eyelid morphogenesis. Moreover, when using anti-α-SMA antibody to examine the smooth muscle formation in the eyelid of different ages (P1, P15 and P60), our data showed that unlike in the POMCwild-type, in which there was a continuous α-SMA-positive smooth muscle sheet detected in eyelids (Fig. 7A–C), the anti-α-SMA immunoreactivity was either fragmented or missed in the POMCN1-ICD (Fig. 7D–I), suggesting that they failed to differentiate into α-SMA-positive smooth muscle sheet. Taken together, these results suggested that mis-expression of N1-ICD in POMCs resulted in a dramatic downregulation of FoxL2 and α-SMA expression in the developing eyelids. These findings prompt us to ask whether the N1-ICD–RBP-Jκ–MAML-1 complex directly binds and regulates FoxL2 promoter activity, or whether it has to go through activation of Hes/Hey genes, which in turn regulate FoxL2 protein expression.
Fig. 6.

POMCN1-ICD downregulates FoxL2 and α-SMA expression during eyelid morphogenesis at E15.5. Immunofluorescent staining with anti-FoxL2 (red, A,B) or anti-skeletal muscle myosin (red, C,D) and anti-α-SMA (green, C,D). Note that FoxL2-positive signals (red) are downregulated in the POMCN1-ICD (B) compared with POMCwt (A). Arrows in C indicate α-SMA-positive smooth muscle sheet. Arrowhead in D indicates that POMCN1-ICD failed to form α-SMA-positive muscle sheet. Other green signals are smooth muscle of the blood vessels. DAPI nuclear counterstain is shown in blue. The skeletal muscle expression pattern changed very little. Co, cornea; Le, Lens; LL, lower lid; Re, retina; UL, upper lid.
Fig. 7.

Downregulation of α-SMA expression and eyelid levator smooth muscle malformation in Dox-treated KR/TC/R26fN1-ICD mice at different stages. (A–I) Immunofluorescent staining with anti-skeletal muscle myosin (red), α-SMA (green) and counterstaining with DAPI (blue) at P1 (A,D,G), P15 (B,E,H) and P60 (C,F,I). Arrows indicate α-SMA-positive eyelid smooth muscle sheet. Other green signals are smooth muscle of the blood vessels. Co, cornea; Le, Lens; LL, lower lid; Re, retina; UL, upper lid.
N1-ICD overexpression downregulates FoxL2 promoter activity
To understand the molecular mechanism(s) by which aberrant Notch activation resulted in downregulation of FoxL2 in POMCs, we performed chromatin immunoprecipitation (ChIP) assay in an endogenous FoxL2-positive KK1 mouse ovarian granulosa cell line transfected with mN1-ICD–Myc plasmid DNA. Our data showed that both anti-Myc eptitope tag (for precipitating N1-ICD) and anti-RBP-Jκ antibodies were able to pull-down DNA sequences corresponding to the RBP-Jκ binding site of the mouse FoxL2 promoter region (Fig. 8A,B), suggesting that binding of the N1-ICD–RBP-Jκ complex to the RBP-Jκ site takes place in vivo. Likewise, ChIP assay in KK1 mouse ovarian granulose-derived cells transfected with HES-1 plasmid showed that anti-Hes-1 antibody could pull-down three of four N-Box Hes-1 binding sites (Fig. 8E). These data suggested that FoxL2 promoter activity is regulated by Notch signaling in vivo. Next, we performed a luciferase assay using the mouse 3.1 kb FoxL2 promoter (mFoxL2pr3.1) by transient transfection of COS-7 cells. Our data showed that FoxL2 promoter activity was regulated by N1-ICD in a dosage-dependent manner. The mFoxL2pr3.1 activity was enhanced twofold when co-transfected with low dose (20 ng) N1-ICD; however, it was decreased 3- and 7-fold when the N1-ICD concentration was elevated to 2 μg and 4 μg, respectively (Fig. 9B). Interestingly, the upregulation of mFoxL2pr3.1 activity by co-transfection of low dosage (20 ng) N1-ICD plasmid was attenuated by addition of Hes-1 plasmid (Fig. 9C). Western blotting analysis showed that endogenous expression of Hes-1 and Hey-1 was upregulated in a dose-dependent manner by transfection of increasing amounts of N1-ICD expression vector DNA (Fig. 9D). Indeed, immunofluorescent staining also demonstrated that Hes-1 production was increased concomitantly with downregulation of FoxL2 in POMCN1-ICD at both E14.5 (Fig. 10) and E15.5 (data not shown). However, the pattern of Notch1 expression remained unchanged in eyelids lacking FoxL2 (supplementary material Fig. S3). Thus, our data argued that Notch signaling activation could serve as upstream regulator of FoxL2 and aberrant overexpression of N1-ICD upregulated Hes-1, which, in turn, inhibited FoxL2 expression and impaired levator Müeller smooth muscle formation, leading to a BPES-like phenotype.
Fig. 8.

Binding of RBP-Jκ and Hes-1 to the mFoxL2 promoter. (A) Predicted RBP-Jκ binding site and Hes-1 binding sites within mFoxL2 5′-flanking regulatory region (~3.1 kb). Arrows are DNA primers used in ChIP assay for RBP-Jκ binding site and Hes-1 binding sites. (B–D) ChIP analysis of RBP-Jκ binding to mouse FoxL2 promoter region. The binding region of RBP-Jκ to Hes-1 promoter served as ChIP positive control (C) and its non-binding region as negative control (D). Notice that both anti-Myc tag and anti-RBP-Jκ antibodies pull down RBP-Jκ binding region of the mFoxL2 promoter, indicating the binding of the N1-ICD–RBP-Jκ complex to the RBP-Jκ site of the mFoxL2 promoter. (E) ChIP analysis of Hes-1 binding to mouse FoxL2 promoter region. N-box sites 1, 2 and 4 show evident Hes-1 binding, but site 3 does not bind to the mouse FoxL2 promoter region.
Fig. 9.

Dosage-dependent N1-ICD regulation of FoxL2 promoter activity in vitro. (A) Schematic drawing of mFoxL2pr3.1-Luc vector, which contains one RBP-Jκ site and four potential Hes-1 binding sites in relation to mouse FoxL2 transcription start site (+1). (B) N1-ICD regulates FoxL2 promoter activity. Note that low dose (20 ng) N1-ICD enhances (lane 3) but N1-ICD greater than 2 μg downregulates (lanes 4,5) pGL3.0-mFoxL2pr3.1 promoter activity. (C) Hes1 transcriptional inhibition of pGL3.0-mFoxL2pr3.1 promoter activity (compare lane 3 with lane 2). Note that pGL3.0-mFoxL2pr3.1 promoter activity enhanced by low dose (20ng) N1-ICD is attenuated by co-transfection of CMV-Hes-1 (compare lane 5 with lane 4). (D) Western blotting analysis of Hes-1 and Hey-1 expression in NIH3T3 cells transfected with various amounts of pLIA-mNIC-Myc plasmids. Exogenous N1-ICD upregulates Hes-1 and Hey-1 expression in a dose-dependent manner. Expression level of β-actin serves as a protein loading control.
Fig. 10.

POMCN1-ICD enhances Hes1 expression concurrent with FoxL2 downregulation during eyelid closure at E14.5. Immunofluorescent staining of paraffin sections of TC/R26N1-ICD (POMCWT) (A,C,E) and KR/TC/R26N1-ICD (POMCN1-ICD) (B,D,F) with anti-Notch1 (A,B), anti-Hes-1 (C,D) and anti-FoxL2 (E,F) antibodies. Red color indicates Alexa-Fluor-555-conjugated secondary antibody. Nuclear counterstaining with DAPI is blue. Misexpression of N1-ICD (B) results in an increase of Hes-1 (compare D with C), but downregulation of FoxL2 (compare F with E) expression. Co, cornea; Le, lens; Ll, lower lid; Re, retina; Ul, upper lid.
FoxL2 positively regulates mouse Acta2 promoter activity
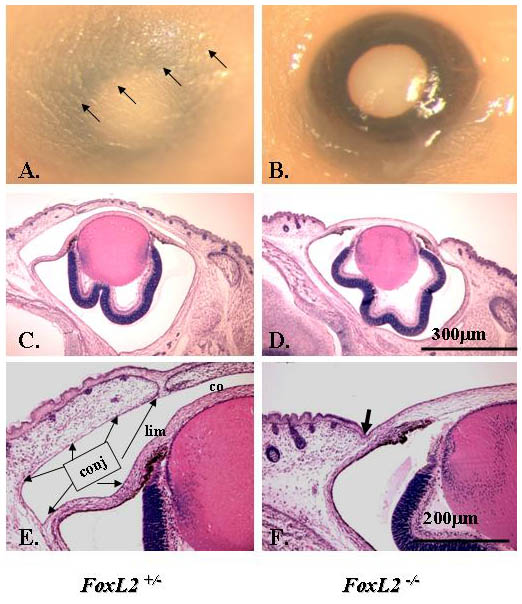
Magnetic resonance imaging (MRI) study of human BPES eyelids showed the absence or hypotrophy of the eyelid superior levator muscle, resulting in ptosis and suggesting a possible role of FOXL2 in the development of this eyelid muscle (Dollfus et al., 2003). To investigate whether FoxL2 can impact on α-SMA gene (Acta2) regulation in POMC during eyelid morphogenesis, we first checked whether α-SMA expression is dependent on the presence of FoxL2. Morphological examinations showed that FoxL2-knockout (FoxL2−/−) mice failed to form eyelids, as revealed by a lack of conjunctiva located in the inner side of the eyelid, and they exhibited an EOB phenotype (Uda et al., 2004) (supplementary material Fig. S4B,D,F). Immunofluorescent staining showed that FoxL2 was dispensable for corneal epithelial (K12-positive) and stromal (keratocan-positive) differentiation (data not shown). By contrast, α-SMA-positive Müeller muscle was absent in FoxL2−/− mice (Fig. 11B,D). These data strongly suggest that FoxL2 is required for expression of α-SMA and formation of Müeller muscle during embryonic eyelid closure. This argument was further strengthened by ChIP analysis in which anti-FoxL2 antibody clearly brought down FoxL2 binding motif in the mouse Acta2 promoter region (Fig. 12A). Moreover, in a cell culture experiment, transfection of CMV–FoxL2 enhanced mouse Acta2 promoter activity (Fig. 12B).
Fig. 11.

Mice lacking FoxL2 do not form levator (Müeller) smooth muscle at E18.5. Immunofluorescent staining clearly shows an α-SMA-positive stripe (green color) indicating the formation of muscle in FoxL2+/− (A,C) but not in the FoxL2−/− (B,D) mouse. Asterisks indicate α-SMA positive blood vessel. Nuclear counterstaining with DAPI is blue. UL, upper eyelid; LL, lower eyelid; Co, cornea; Le, lens; Re, retina.
Fig. 12.

FoxL2 binds to and upregulates mouse Acta2 promoter activity. (A) FoxL2 binds to mActa2 promoter in ChIP assay. Predicted FoxL2 binding sites within the mActa2 5′-flanking regulatory region (~1.1 kb). Arrows indicate DNA primers used in ChIP assay for FoxL2 binding site. A 235 bp PCR-amplified product was generated from ChIP with anti-Myc tag antibody (lane 3) but not from mouse IgG (lane 4). (B) FoxL2 enhances mActa2 promoter luciferase activity in vitro. Diagram of mActa2 promoter-luciferase construct. Co-transfection of pcDNA3.1-mFoxL2 plasmid enhances luciferase activity threefold.
Discussion
In this study, we developed a novel Dox-inducible mouse driver strain, KR/TC, to manipulate expression of loss-of-function and/or gain-of-function genes at the desired time to study their roles in keratocan-expressing cells, which represent a subset of neural-crest-derived cell lineage destined to become eyelid and ear dermis, corneal stroma and limbs during embryonic development. The KR/TC driver mouse can complement other neural crest cell lineage drivers such as Wnt1-Cre (Danielian et al., 1998) and P0-Cre (Feltri et al., 1999) mice because of two facts. First, in the KR/TC system, Cre activity is tightly controlled by Dox; however, Wnt1-Cre and P0-Cre are not inducible. Second, in the KR/TC system, the Cre activity is not turned on earlier than E13.5 owing to Kera promoter activity (Liu et al., 2000). However, the Cre activity derived from the Wnt1-Cre and P0-Cre systems can ‘flox-out’ the gene of interest as early as E8.5, which often causes embryonic lethality before most organogenesis has taken place. Therefore, the KR/TC system is a valuable tool to study the role of signaling molecules during and following development in those aforementioned tissues (Fig. 1).
We investigated the role of Notch signaling activation in POMCs during embryonic eyelid development. Our results showed that KR/TC/R26fN1-ICD triple transgenic mice administered with Dox exhibited eyelid defects with poor eyelid closure at birth. Interestingly, they did not develop a secondary exposure keratitis, but had a relatively healthy ocular surface. POMCs also contribute to the corneal stroma and are the major source of keratocytes, although there were some quantitative differences in K12 and keratocan expression but, to our surprise, mis-expression of N1-ICD did not have adverse impact on corneal keratocyte and stromal morphogenesis (data not shown). Interestingly, however, the POMCN1-ICD effects caused malformation of Meibomian gland and dysplasia of the Muc5A/C-positive goblet cells in the conjunctival region (data not shown). The phenotypes of POMCN1-ICD in cornea and conjunctiva will be reported elsewhere.
In the course of investigating the eyelid development of POMCN1-ICD mice, we found that eyelid re-opening was delayed compared with that in control littermates at P12–P14 and never reached complete opening, resembling congenital type II BPES (Fig. 3). Therefore, the KR/TC/R26fN1-ICD triple transgenic mouse strain can serve as a novel transgenic mouse model for studying eyelid morphogenesis and the pathogenesis of human type II BPES. We found that POMCN1-ICD inhibited cell proliferation, whereas it triggered apoptosis. Although not fully documented in the present study, other cell behavior, such as cell migration and cell fate changes, might also be involved in the pathogenic progression of POMCN1-ICD in vivo. More intriguing was the fact that POMCN1-ICD downregulated FoxL2 expression, and thus impaired smooth muscle formation during embryonic eyelid closure. Genetic and epidemiological studies have shown that mutations in FOXL2 cause 70% of BPES, but ~30% of people with BPES did not have an identified FOXL2 gene mutation. The cause of BPES in these people is unknown, but regulation of FOXL2 might be altered. Our studies strongly support the idea that those cases of the disease not due to mutation of FOXL2 result from activation of aberrant Notch signaling and changes in expression of FOXL2.
This result was further strengthened by the ChIP and promoter-luciferase assays concerning FoxL2 regulation by N1-ICD. Sequence analysis showed one RBP-Jκ and four Hes-1 putative binding (N-Box) sites in the mouse FoxL2 3.1 kb promoter region. Endogenous Notch signaling was not able to reveal the presence of N1-ICD–RBP-Jκ complex binding to RBP-Jκ in ChIP assay of mouse KK1 cells, but overexpressed N1-ICD clearly formed complexes with endogenous RBP-Jκ within the FoxL2 promoter. Similarly, three out of four N-Box sites were able to bind to Hes-1 in cells transiently transfected with Hes-1 expression vector. Moreover, the mFoxL2pr3.1-luc exhibited a fourfold increase in luciferase activity compared with that in the control vector pGL3-basic. Co-transfection with N1-ICD plasmid vector significantly downregulated mFoxL2 promoter activity in a dose-dependent manner (Fig. 9B). At low doses, N1-ICD enhanced FoxL2 promoter activity, whereas N1-ICD in high dosage negatively regulated mFoxL2pr3.1 promoter activity, probably through activation of the transcriptional repressor Hes/Hey. Indeed, transfection of N1-ICD upregulated Hes-1 and Hey-1 protein production (Fig. 9D). We could not detect Hes-5 protein by either western blotting analysis using NIH-3T3 cells, or by immunohistochemistry in mouse eyelid (data not shown), so it was hard to judge whether Hes-5 had any role in this regard. Furthermore, co-transfection of the Hes-1 expression vector could attenuate low-dosage N1-ICD-enhanced FoxL2 promoter activity (Fig. 9C). This result is consistent with the in vivo data that N1-ICD overexpression caused upregulation of Hes-1 (Fig. 10D) and downregulation of FoxL2 (Fig. 10F) in POMCs during eyelid morphogenesis. Our data indicated that, depending on its concentration, N1-ICD might execute dual functions in the regulation of FoxL2 gene expression. Interestingly, it has also been documented that the transcription factor forkhead box O3a (FoxO3a) is a key negative transcriptional target of canonical Notch1 signaling, exerting a protective function in the UVB-induced apoptosis in skin keratinocytes (Mandinova et al., 2008). Similarly, N1-ICD regulates the transcription factor forkhead box P3 (FOXP3) promoter through RBP-Jκ- and Hes-1-dependent mechanisms in FOXP3+CD4+CD25+ regulatory T cells, or ‘Tregs’ (Ou-Yang et al., 2009; Radtke et al., 2010). These data suggest that FOX-related genes might be a common downstream target of the canonical Notch signaling in different cell types. However, during embryonic development, Notch1 expression in the mouse eyelid stroma was not altered in the FoxL2−/− mutant, suggesting that Notch signaling is not regulated by FoxL2 (supplementary material Fig. S3).
In eyelid morphogenesis, FoxL2 expression is specific in POMCs and closely associated with embryonic eyelid closure. Previous published data (Crisponi et al., 2001; Uda et al., 2004; Uhlenhaut and Treier, 2006) and our results (supplementary material Fig. S1) showed that FoxL2 expression first appears in POMCs at E12.5~E14.5 and is restricted to the eyelid dermis at E15.5~E16.5. More interestingly, after morphogenetic eyelid closure is complete, FoxL2 expression was dramatically downregulated at E18.5 and eventually diminished at P2. FoxL2 expression was never detected in the corneal stroma, although it shared the common ascendant of POMC. These data implicated that FoxL2+Kera+ cells mainly contribute to eyelid stroma, but FoxL2-Kera+ cells become corneal keratocytes. Most of these FoxL2+Kera+ POMCs should normally differentiate into levator Müeller smooth muscle, but fail to do so if the expression of N1-ICD is aberrantly high. We found that FoxL2 was able to directly bind to and activate mouse Acta2 promoter activity (Fig. 12). In addition, expression of Acta-2 was absent in the FoxL2-knockout mouse. Taken together, our transgenic mice and cell culture data argue that a physiologically low level of notch activation might be very critical for the control of FoxL2 expression, which in turn ensures levator muscle differentiation and normal eyelid morphogenesis. By contrast, aberrantly sustained Notch activation inhibits FoxL2 expression and impairs levator Müeller smooth muscle formation, leading to eyelid malformation and BPES-like phenotypes (see model in Fig. 13).
Fig. 13.

A proposed model of POMCN1-ICD effects that impair levator muscle formation during eyelid development, leading to BPES-like phenotypes. Aberrantly high expression of N1-ICD causes activation of transcriptional repressor Hes-1 expression, which suppresses FoxL2 expression, which, in turn, downregulates α-SMA and thus leads to impaired levator muscle formation.
It remains unknown whether the effects of POMCN1-ICD on levator Müeller smooth muscle formation during eyelid morphogenesis depend on RBP-Jκ and MAML-1 (canonical pathway) or go through non-canonical pathways (Perumalsamy et al., 2009; Sanalkumar et al., 2010). Notch loss-of-function approaches by crossing RBP-Jκfloxed/floxed or RosadnMAML−1 mouse lines with POMCwild-type and POMCN1-ICD, respectively, will allow elucidation of the molecular pathway(s) of Notch activation in POMCs, which lead to eyelid malformation and BPES-like phenotypes. Moreover, endogenous factors that fine-tune the appropriate Notch signaling during eyelid morphogenesis remain unknown. It has been recently reported that Notch can be phosphorylated on multiple sites by Nemo-like kinase (NLK), and the density of NLK phosphorylation sites in Notch serves as a molecular rheostat to fine-tune Notch activity. Knockdown of NLK leads to hyperactivation of Notch signalling, and consequently decreases neurogenesis in zebrafish (Ishitani et al., 2010). It would be interesting to know whether this mode of regulation in the Notch signaling pathway also exists in mammalian eyelid morphogenesis.
Materials and Methods
Construction and generation of Kerapr3.2-rtTA2S-M2 (KR) transgenic mice
To generate the transgenic mice of C57BL/6 background, a rtTA2S-M2 DNA fragment (1.1 kb) was excised from pTet-ON-advanced plasmid vector (Clontech) with EcoRI and HindIII digestion and subcloned into the EcoRI and HindIII sites of the pKerapr3.2-int-BpA plasmid vector (Liu et al., 2000). The resulting plasmid was designated pKerapr3.2-int-rtTA2S-M2-BpA from which a NotI and SalI digested DNA fragment containing Kerapr3.2-rtTA2S-M2-SV40 poly A (~6.2 kb) was purified and microinjected into fertilized eggs to generate transgenic mice. Thirty-four transgenic pups were obtained in which 16 transgenic founders carrying the transgene were identified by tail DNA polymerase chain reaction (PCR) genotyping with primers: Forward, 5′-TCAGCCATCGCTATGACTCAGTTC-3′ and Reverse, 5′-TTGTTCTTCACGTGCCAGTACAGG-3′. The initial identification of functional KR mice was carried out by outbreeding each founder line with a rtTA transgenic reporter mouse line called PTR which harbors a bi-directional TetO-EGFP and TetO-TβRII (Frugier et al., 2005). The resulting KR/PTR double transgenic pups were induced with 1 g per kg doxycycline (dox) chow (Bio-Serve, Laurel, MD) through pregnant females from E12.5 to the date of birth. Newborn mice were examined under a Zeiss stereomicroscope with epi-fluorescence. The founder lines displaying either weak or no enhanced green fluorescent protein (EGFP) signal were no longer studied. Two founder lines designated KR4 and KR5 exhibited identical strong EGFP signals in eyelid, snout and limb (data not shown) and were used in the present study. Other mouse strains such as TetO-Cre (Perl et al., 2002), Z/EG (Novak et al., 2000), R26fN1-ICD (Murtaugh et al., 2003) are commercially available from the Jackson laboratory. FoxL2-knockout mice have been described previously (Crisponi et al., 2001). All the mice except FoxL2 knockouts were bred at the Animal Facility of the University of Cincinnati Medical Center. They were housed in a room maintained on a 12 hour/12 hour light/dark cycle (light on at 7 a.m.) with a continuous supply of food and water. Experimental procedures for handling the mice were approved by the Institutional Animal Care and Utilization Committee, University of Cincinnati College of Medicine.
Cell cultures
NIH3T3 mouse fibroblast cell line and COS-7 monkey kidney cell line were purchased from American Type Culture Collection (ATCC). KK1 mouse granulosa tumour cell line was obtained from Kenneth W. Escudero (Escudero et al., 2010). 3T3 cells were maintained in DMEM supplemented with 10% FBS and 100 U/ml penicillin-streptomycin solution (Hyclone). KK1 and COS-7 cells were maintained in DMEM/F12 (1:1) advanced medium supplemented with 5% FBS and 100 U/ml penicillin-streptomycin solution (Hyclone). All cells were cultured at 37°C in a 5% CO2 incubator.
Antibodies
See supplementary material Table S1 for the antibodies used for immunofluorescence, western blotting and chromatin immunoprecipitation (ChIP).
Plasmid construction
PCR was used to generate mouse FoxL2 full-length cDNA from one BAC clone (RP24-225E14, BACPAC) which was subcloned into the CMV-driven eukaryotic expression vector pcDNA3.1/Myc-His(−) (Invitrogen). PCR condition was as follows: 95°C for 2 minutes, 35 cycles of 94°C for 30 seconds, 58°C for 30 seconds, 72°C for 1 minute, followed by 72°C for 10 minutes as a final extension. Mouse FoxL2 promoter region (−2887 to +244) generated by PCR from the same BAC clone was subcloned into pGL3.0 luciferase basic vector (Promega). Mouse α-smooth muscle actin promoter (mActa-2pr) region (−1146 to −1) was generated by PCR from one BAC clone (RP24-329P13; BACPAC) and cloned into pGL4.10 vector (Promega). PCR conditions were same as above except longer extension time at 72°C. All PCR-amplified DNA were verified by DNA sequencing. Supplementary material Table S2 lists the primers used to generate the constructs.
Histological analysis
Mouse samples were fixed overnight in 4% paraformaldehyde (PFA) in 1× phosphate-buffered saline (PBS; pH 7.2), followed by paraffin embedding. De-paraffinized sections (5 μm) were stained with hematoxylin and eosin (H&E) or periodic acid Schiff reagent (PAS).
Immunofluorescence and immunohistochemistry
Tissue sections (5 μm) were de-paraffinized, rehydrated and subjected to antigen retrieval in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween, pH 6.0) at boiling temperature for 30 minutes. Eyelid sections were then blocked with 3% BSA in PBS containing 0.025% Nonidet P-40 for 1 h at room temperature, then incubated overnight at 4°C with the primary antibodies diluted in the same buffer. After three washes with PBST (PBS, 0.1% Tween 20), slides were incubated at room temperature for 1 hour with Alexa-Fluor-488- or Alexa-Fluor-555-conjugated secondary antibodies (Invitrogen) and 1 μg/ml DAPI (4′,6-diamidino-2-phenylindole) as a nuclear counterstain, washed with PBST again, and mounted with Mowiol (Sanofi-Aventis). Sections were examined and photographed using a Zeiss microscope equipped with a camera (Axiocam Mrm; Carl Zeiss). For data acquisition, we used the Axiovision 4.6 software (Carl Zeiss).
Detection of cell proliferation
For detection of cell proliferation in the developing eyelids, timed pregnant female mice were injected intraperitoneally on gestational day 14.5 with BrdU (Sigma) (80 μg/g body weight). Two hours after injection, embryos were dissected, fixed in 4% PFA in PBS, dehydrated through graded alcohols, embedded in paraffin and sectioned in the coronal plane at 5 μm thickness. De-paraffinized and rehydrated sections were treated with 3N HCl in double distilled water for 15 minutes at room temperature followed by three washes with PBS. Immunodetection of BrdU was performed using mouse anti-BrdU monoclonal antibody followed by Alexa-Fluor-555-labeled rabbit anti-mouse IgG and the sections were counterstained with DAPI. The total number of cell nuclei (DAPI-positive) as well as the number of BrdU-labeled nuclei on sections through the middle of the eyelids were counted and recorded for the mesenchyme and epidermis, respectively, from five adjacent sections. The cell proliferation index was calculated as the percentage of cell nuclei with BrdU labeling. Student's t-test was used to analyze the significance of difference and a P value less than 0.05 was considered statistically significant.
Detection of apoptosis in situ
Embryos at E14.5 were obtained through time-mated females. Cell apoptosis status in Dox-treated and non-treated groups were subjected to TUNEL assay using Click-iT® TUNEL Assay kit (Invitrogen) according the manufacturer's protocol.
Western blot analysis
Cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% deoxycholate). Frozen tissue was homogenized in a buffer containing phosphatase and protease inhibitors. Protein lysates (20 μg) from each sample were separated on a 4–20% linear gradient Tris-HCl denaturing polyacrylamide Ready Gel® (Bio-Rad) and transferred to nitrocellulose membranes (Whatman). Antibody incubations were performed in 5% non-fat dry milk in TBS-T (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 0.05% Tween-20) or in 5% BSA in TBS-T, depending on the requirement of the antibody.
Chromatin immunoprecipitation (ChIP) assay
KK1 cells were transfected with 20 μg pLIA-mNIC-myc plasmid (Addgene CC#341) (Bao and Cepko, 1997) or pCMV-SPORT6-mouse Hes-1 cDNA clone (Openbiosystems MMMM1013-9200434 using GeneJammer transfection reagent (Invitrogen). Forty-eight hours post transfection, cells were crosslinked with 1% formaldehyde at 37°C for 10 minutes and subjected to ChIP assay with antibodies against Myc epitope tag (5 μg/ml; RDI) and RBP-Jκ (6 μg/ml; Santa Cruz) using the ChIP assay Kit (Millipore) following the manufacturer's instructions. Purified DNAs after ChIP were used as templates for PCR to verify the interaction between DNA promoter and protein. ChIP assay for FoxL2 binding in mouse Acta2 promoter, NIH3T3 cells were transfected with 20 μg pcDNA3.1-mFoxL2. PCR primers are listed in supplementary material Table S3.
Promoter luciferase assay
COS-7 cells seeded in six-well plates at 80% confluence were transiently transfected with a mixture of three different types of plasmids: (1) prhL-TK served as transfection efficiency control vector (Promega); (2) empty vector (pGL3.0 or pGL4.10) or recombinant plasmids harboring mouse FoxL2 promoter (pGL3.0-mFoxL2pr3.1) or α-smooth muscle actin promoter (pGL4.0-Acta2pr) and (3) cDNA expression plasmids (pLIA mNIC myc, or mouse Hes-1 cDNA clone or pcDNA3.1-mFoxL2) at different dosages as indicated. 36 hours after transfection, cells were collected with 1× PassiveLysis buffer (200 μl), luciferase assay was conducted using Dual Luciferase® Reporter (DLR) Assay System (Promega) according to the manufacturer's recommendations. Luminescence was measured using a Synergy2 plate reader luminescence module (BioTek).
Statistical analysis
Two-tailed Student's t-test (Excel, Microsoft, Redmond, WA) was used to analyze the percentage of BrdU-positive and the number of TUNEL-positive cells. All quantification data are presented as mean ± s.d.
Supplementary Material
Acknowledgments
We thank the reviewers for their instructive comments and suggestions. We thank Nadean Brown (Cincinnati Childrens Hospital) for providing the anti-Hes-1 antibody. This work was supported by grants from NIH/NEI RO1 # EY12486 (C.Y.L.) and EY13755 (W.W.K.), Research Prevent Blindness, Ohio Lions Foundation for Eye Research, and the Intramural Research Program of the NIH, National Institute on Aging. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/15/2561/DC1
References
- Bao Z. Z., Cepko C. L. (1997). The expression and function of Notch pathway genes in the developing rat eye. J. Neurosci. 17, 1425-1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun B. A., Batista F., Auer J., Dipietromaria A., L'Hôte D., De Baere E., Veitia R. A. (2009). Positive and negative feedback regulates the transcription factor FOXL2 in response to cell stress: evidence for a regulatory imbalance induced by disease-causing mutations. Hum. Mol. Genet. 18, 632-644 [DOI] [PubMed] [Google Scholar]
- Bolós V., Grego-Bessa J., de la Pompa J. L. (2007). Notch signaling in development and cancer. Endocr. Rev. 28, 339-363 [DOI] [PubMed] [Google Scholar]
- Borggrefe T., Oswald F. (2009). The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell. Mol. Life Sci. 66, 1631-1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisponi L., Deiana M., Loi A., Chiappe F., Uda M., Amati P., Bisceglia L., Zelante L., Nagaraja R., Porcu S., et al. (2001). The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat. Genet. 27, 159-166 [DOI] [PubMed] [Google Scholar]
- Danielian P. S., Muccino D., Rowitch D. H., Michael S. K., McMahon A. P. (1998). Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323-1326 [DOI] [PubMed] [Google Scholar]
- De Baere E., Beysen D., Oley C., Lorenz B., Cocquet J., De Sutter P., Devriendt K., Dixon M., Fellous M., Fryns J. P., et al. (2003). FOXL2 and BPES: mutational hotspots, phenotypic variability, and revision of the genotype-phenotype correlation. Am. J. Hum. Genet. 72, 478-487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djalilian A. R., Namavari A., Ito A., Balali S., Afshar A., Lavker R. M., Yue B. Y. (2008). Down-regulation of Notch signaling during corneal epithelial proliferation. Mol. Vis. 14, 1041-1049 [PMC free article] [PubMed] [Google Scholar]
- Dollfus H., Stoetzel C., Riehm S., Lahlou Boukoffa W., Bediard Boulaneb F., Quillet R., Abu-Eid M., Speeg-Schatz C., Francfort J. J., Flament J., et al. (2003). Sporadic and familial blepharophimosis-ptosis-epicanthus inversus syndrome: FOXL2 mutation screen and MRI study of the superior levator eyelid muscle. Clin. Genet. 63, 117-120 [DOI] [PubMed] [Google Scholar]
- Escudero J. M., Haller J. L., Clay C. M., Escudero K. W. (2010). Microarray analysis of Foxl2 mediated gene regulation in the mouse ovary derived KK1 granulosa cell line: over-expression of Foxl2 leads to activation of the gonadotropin releasing hormone receptor gene promoter. J. Ovarian Res. 3, 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri M. L., D'Antonio M., Previtali S., Fasolini M., Messing A., Wrabetz L. (1999). P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann. N. Y. Acad. Sci. 883, 116-123 [PubMed] [Google Scholar]
- Findlater G., McDougall R., Kaufman M. (1993). Eyelid development, fusion and subsequent reopening in the mouse. J. Anat. 183, 121-129 [PMC free article] [PubMed] [Google Scholar]
- Fiúza U. M., Arias A. M. (2007). Cell and molecular biology of Notch. J. Endocrinol. 194, 459-474 [DOI] [PubMed] [Google Scholar]
- Frugier T., Koishi K., Matthaei K. I., McLennan I. S. (2005). Transgenic mice carrying a tetracycline-inducible, truncated transforming growth factor beta receptor (TbetaRII). Genesis 42, 1-5 [DOI] [PubMed] [Google Scholar]
- Gridley T. (2007). Notch signaling in vascular development and physiology. Development 134, 2709-2718 [DOI] [PubMed] [Google Scholar]
- Hayashi Y., Liu C. Y., Jester J. J., Hayashi M., Wang I. J., Funderburgh J. L., Saika S., Roughley P. J., Kao C. W., Kao W. W. (2005). Excess biglycan causes eyelid malformation by perturbing muscle development and TGF-alpha signaling. Dev. Biol. 277, 222-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg J., Liu C. Y., Hjalt T. A. (2004). PITX2 gain-of-function in Rieger syndrome eye model. Am. J. Pathol. 165, 1633-1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishitani T., Hirao T., Suzuki M., Isoda M., Ishitani S., Harigaya K., Kitagawa M., Matsumoto K., Itoh M. (2010). Nemo-like kinase suppresses Notch signalling by interfering with formation of the Notch active transcriptional complex. Nat. Cell Biol. 12, 278-285 [DOI] [PubMed] [Google Scholar]
- Kopan R., Ilagan M. X. (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. Y., Arar H., Kao C., Kao W. W. (2000). Identification of a 3.2 kb 5′-flanking region of the murine keratocan gene that directs beta-galactosidase expression in the adult corneal stroma of transgenic mice. Gene 250, 85-96 [DOI] [PubMed] [Google Scholar]
- Ma A., Boulton M., Zhao B., Connon C., Cai J., Albon J. (2007). A role for notch signaling in human corneal epithelial cell differentiation and proliferation. Invest. Ophthalmol. Vis. Sci. 48, 3576-3585 [DOI] [PubMed] [Google Scholar]
- Mandinova A., Lefort K., Tommasi di Vignano A., Stonely W., Ostano P., Chiorino G., Iwaki H., Nakanishi J., Dotto G. P. (2008). The FoxO3a gene is a key negative target of canonical Notch signalling in the keratinocyte UVB response. EMBO J. 27, 1243-1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh L. C., Stanger B. Z., Kwan K. M., Melton D. A. (2003). Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl. Acad. Sci. USA 100, 14920-14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T., Ohtsuka T., Sekiyama E., Cooper L. J., Kokubu H., Fullwood N. J., Barrandon Y., Kageyama R., Kinoshita S. (2008). Hes1 regulates corneal development and the function of corneal epithelial stem/progenitor cells. Stem Cells 26, 1265-1274 [DOI] [PubMed] [Google Scholar]
- Nien C. J., Massei S., Lin G., Liu H., Paugh J. R., Liu C. Y., Kao W. W., Brown D. J., Jester J. V. (2010). The development of meibomian glands in mice. Mol. Vis. 16, 1132-1140 [PMC free article] [PubMed] [Google Scholar]
- Novak A., Guo C., Yang W., Nagy A., Lobe C. G. (2000). Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis 28, 147-155 [PubMed] [Google Scholar]
- Ou-Yang H. F., Zhang H. W., Wu C. G., Zhang P., Zhang J., Li J.-C., Hou L.-H., He F., Ti X.-Y., Song L.-Q., et al. (2009). Notch signaling regulates the FOXP3 promoter through RBP-J- and Hes1-dependent mechanisms. Mol. Cell. Biochem. 320, 109-114 [DOI] [PubMed] [Google Scholar]
- Perl A. K., Wert S. E., Nagy A., Lobe C. G., Whitsett J. A. (2002). Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc. Natl. Acad. Sci. USA 99, 10482-10487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perumalsamy L. R., Nagala M., Banerjee P., Sarin A. (2009). A hierarchical cascade activated by non-canonical Notch signaling and the mTOR-Rictor complex regulates neglect-induced death in mammalian cells. Cell Death Differ. 16, 879-889 [DOI] [PubMed] [Google Scholar]
- Radtke F., Fasnacht N., Macdonald H. R. (2010). Notch signaling in the immune system. Immunity 32, 14-27 [DOI] [PubMed] [Google Scholar]
- Sanalkumar R., Dhanesh S. B., James J. (2010). Non-canonical activation of Notch signaling/target genes in vertebrates. Cell. Mol. Life Sci. 67, 2957-2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D., Ovitt C. E., Anlag K., Fehsenfeld S., Gredsted L., Treier A.-C., Treier M. (2004). The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development 131, 933-942 [DOI] [PubMed] [Google Scholar]
- Uda M., Ottolenghi C., Crisponi L., Garcia J. E., Deiana M., Kimber W., Forabosco A., Cao A., Schlessinger D., Pilia G. (2004). Foxl2 disruption causes mouse ovarian failure by pervasive blockage of follicle development. Hum. Mol. Genet. 13, 1171-1181 [DOI] [PubMed] [Google Scholar]
- Uhlenhaut N. H., Treier M. (2006). Foxl2 function in ovarian development. Mol. Genet. Metab. 88, 225-234 [DOI] [PubMed] [Google Scholar]
- Vauclair S., Majo F., Durham A. D., Ghyselinck N. B., Barrandon Y., Radtke F. (2007). Corneal epithelial cell fate is maintained during repair by Notch1 signaling via the regulation of vitamin A metabolism. Dev. Cell 13, 242-253 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}