

Abstract
We have previously shown that PIP5KIβ and PIP5KIγ generate functionally distinct pools of phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] important for antigen-stimulated Ca2+ entry in mast cells. In the present study, we find that association of the endoplasmic reticulum (ER) Ca2+ sensor, STIM1, and the store-operated Ca2+ channel, Orai1, stimulated by thapsigargin-mediated ER store depletion, is enhanced by overexpression of PIP5KIβ and inhibited by overexpression of PIP5KIγ. These different PIP5KI isoforms cause differential enhancement of PtdIns(4,5)P2 in detergent-resistant membrane (DRM) fractions, which comprise ordered lipid regions, and detergent-solubilized membrane (DSM) fractions, which comprise disordered lipid regions. Consistent with these results, the inositol 5-phosphatase L10-Inp54p, which is targeted to ordered lipids, decreases PtdIns(4,5)P2 in the DRM fraction and inhibits thapsigargin-stimulated STIM1–Orai1 association and store-operated Ca2+ entry, whereas the inositol 5-phosphatase S15-Inp54p, which is targeted to disordered lipids, decreases PtdIns(4,5)P2 in the DSM fraction and enhances STIM1–Orai1 association. Removal of either the STIM1 C-terminal polylysine sequence (amino acids 677–685) or an N-terminal polyarginine sequence in Orai1 (amino acids 28–33) eliminates this differential sensitivity of STIM1–Orai1 association to PtdIns(4,5)P2 in the distinctive membrane domains. Our results are consistent with a model of PtdIns(4,5)P2 balance, in which store-depletion-stimulated STIM1–Orai1 association is positively regulated by the ordered lipid pool of PtdIns(4,5)P2 and negatively regulated by PtdIns(4,5)P2 in disordered lipid domains.
Key words: Store-operated Ca2+ entry, Lipid raft, Phosphoinositide, STIM1, Orai1
Introduction
Store-operated Ca2+ entry (SOCE) is a ubiquitous process that regulates intracellular Ca2+ as a secondary messenger in nonexcitable cells (Hoth and Penner, 1993; Putney and Bird, 1993). The two proteins that are necessary for this process are the endoplasmic reticulum (ER) transmembrane Ca2+ sensor STIM1 (Liou et al., 2005; Zhang et al., 2005) and the plasma membrane tetraspan Ca2+ channel Orai1, also known as CRACM1 (Feske et al., 2006; Vig et al., 2006). Oligomerized Orai1 forms the Ca2+-release-activated Ca2+ (CRAC) channel that is activated by association with STIM1 at ER–plasma membrane (ER–PM) junctions following depletion of Ca2+ from the ER. Coupling of these proteins results in Orai1-mediated influx of Ca2+ from the extracellular medium. We previously identified an acidic coiled-coil on the C-terminus of Orai1 as being important for the functional interaction between STIM1 and Orai1 (Calloway et al., 2009), and in a subsequent study we showed that a short sequence of lysine residues within the CRAC-activating domain (CAD) (Park et al., 2009) of STIM1 must interact with the acidic coiled-coil of Orai1 for effective coupling (Calloway et al., 2010).
In addition to these sequences, the lysine-rich C-terminus of STIM1 (amino acids 677–685 in humans) has been a region of much interest for its hypothesized capacity to bind phosphatidylinositol phosphates (PtdIns-Ps) at the plasma membrane as part of the coupling mechanism that is enhanced by STIM1 oligomerization (Liou et al., 2007). Korzeniowski and colleagues (Korzeniowski et al., 2009) demonstrated that activation of SOCE is sensitive to inhibition of phosphoinositide 4-kinase (PI4K) but is not prevented by depletion of PtdIns(4,5)P2 at the plasma membrane, suggesting a role for PtdIns(4)P. Walsh and colleagues (Walsh et al., 2010) showed that inhibition of multiple pathways of PtdIns(4,5)P2 generation is necessary to prevent thapsigargin-mediated translocation of STIM1 to the plasma membrane, but that expression of Orai1 permits STIM1 to concentrate at ER–PM junctions even in the absence of PtdIns(4,5)P2. Furthermore, they showed that PtdIns(4,5)P2 plays an inhibitory role in the interaction of Orai1 with STIM1. Additionally, several studies have provided evidence for involvement of the C-terminal polylysine sequence of STIM1 in other interactions. This polylysine sequence has been identified as a direct binding partner for canonical transient receptor potential (TRP) channels (Zeng et al., 2008), as a structural determinant of the inwardly rectifying character of ICRAC (Yuan et al., 2009) and as a binding site for calmodulin (Bauer et al., 2008).
We previously demonstrated that different isoforms of type I phosphatidylinositol 4-phosphate 5-kinase (PIP5KI), namely PIP5KIβ and PIP5KIγ, synthesize functionally distinguishable pools of PtdIns(4,5)P2 that have distinct roles in SOCE and inositol trisphosphate (Ins(1,4,5)P3) generation, respectively (Vasudevan et al., 2009). In the present study we characterize a membrane structural basis for distinctive pools of PtdIns(4,5)P2. Previous studies provided evidence for participation of ordered lipid membrane domains, sometimes called ‘lipid rafts’, in the interaction between STIM1 and TRPC1 (Pani et al., 2008; Alicia et al., 2008). Our present results provide evidence for a positive role for PtdIns(4,5)P2 in ordered lipid domains and a negative role for PtdIns(4,5)P2 in disordered lipid domains in thapsigargin-stimulated STIM1–Orai1 association, such that the balance appears to be important. Furthermore, we find that the C-terminal polylysine sequence of STIM1, as well as a previously uncharacterized polyarginine sequence in Orai1 (amino acids 28–33 in humans) modulate STIM1–Orai1 association during SOCE, and this might be explained by their differential association with PtdIns(4,5)P2 pools in ordered and disordered membrane domains.
Results
Cholesterol and phosphoinositides contribute to stimulated association of STIM1 and Orai1
Consistent with previous evidence that cholesterol-dependent membrane domains participate in activation of SOCE (Pani et al., 2008; Galan et al., 2010), we found that relatively mild reduction of cholesterol in RBL mast cells by 4 mM methyl β-cyclodextrin (MβCD) for 20 minutes at 37°C caused substantial inhibition of thapsigargin-stimulated SOCE (Fig. 1A). Under these conditions, cholesterol is decreased by ~30% in these cells (Surviladze et al., 2001), and, in multiple experiments, we found that SOCE measured 5 minutes after addition of thapsigargin was inhibited by 69±6% (s.d., n=3). Sensitivity of this Ca2+ influx to 1 μM Gd3+ (Fig. 1A) indicates that it is largely due to ICRAC (Broad et al., 1999). A recent study indicated that inhibition of SOCE by cholesterol depletion can be caused by depolarization of the plasma membrane (DeHaven et al., 2009). To check this possibility in our cells, we evaluated the effect of high K+-mediated depolarization on the Ca2+ response to thapsigargin in RBL cells treated with MβCD and in control cells. We found that the MβCD-inhibited Ca2+ response remained sensitive to depolarization to a similar extent to the untreated control cells, indicating that cholesterol depletion does not inhibit the Ca2+ response due to plasma membrane depolarization in the these conditions (supplementary material Fig. S1).
Fig. 1.

Thapsigargin-stimulated SOCE and STIM1–Orai1 FRET following cholesterol depletion and PI4K inhibition. (A) Representative Ca2+ responses to thapsigargin (TG) in control RBL cells (black circles) and cells treated with 4 mM MβCD for 20 minutes (red circles). Sensitivity to 1 μM Gd3+ is shown by addition at the indicated time points. (B) Effect of cholesterol reduction on thapsigargin-stimulated FRET between AcGFP–Orai1 and STIM1–mRFP. Cells were untreated (black) or incubated with 5 mM MβCD for 10 minutes (red) before stimulation by thapsigargin. Error bars show s.e.m. (C) Representative Ca2+ responses in control RBL cells (black), and cells treated with 10 μM wortmannin for 10 minutes (green) before stimulation by thapsigargin. (D) Thapsigargin-stimulated FRET between AcGFP–Orai1 and STIM1–mRFP in unperturbed cells (black) and cells treated with 10 μM wortmannin (green). Error bars show s.e.m.
We previously established a microscopy method to monitor stimulated association of STIM1 and Orai1 by fluorescence resonance energy transfer (FRET) in individual cells (Calloway et al., 2009). Using this method for RBL cells transiently transfected with donor AcGFP–Orai1 and acceptor STIM1–mRFP, we found that similar mild cholesterol reduction by MβCD inhibits thapsigargin-stimulated association of STIM1 with Orai1 by 73±8% after 10 minutes (similar at 5 minutes; Fig. 1B), consistent with a role for cholesterol-dependent plasma membrane structures in this process.
Broad and colleagues (Broad et al., 2001) reported that inhibition of PI4P synthesis by 10–20 μM wortmannin causes substantial inhibition of ICRAC in RBL mast cells, indicating that phosphoinositides are involved in SOCE. Consistent with these results, we found that treatment of RBL cells with 10 μM wortmannin for 10 minutes at 37°C inhibits thapsigargin-stimulated Ca2+ entry by ~40% when assessed 5 minutes after the addition of thapsigargin (Fig. 1C). We also found that thapsigargin-stimulated FRET between AcGFP–Orai1 and STIM1–mRFP was inhibited 52±8% by 10 μM wortmannin under similar conditions (Fig. 1D). This finding is consistent with reports that wortmannin and other PI4K inhibitors prevent colocalization of STIM1 and Orai1 in ER–plasma membrane puncta (Korzeniowski et al., 2009; Walsh et al., 2010). These results indicate that functional coupling between STIM1 and Orai1 depends on PtdIns(4)P, either directly or because it is a precursor for other phosphoinositide species such as PtdIns(4,5)P2.
PIP5KIβ and PIP5KIγ differentially modulate stimulated association of STIM1 with Orai1
We recently showed that PIP5KIβ and PIP5KIγ synthesize distinguishable pools of PtdIns(4,5)P2 that differentially affect antigen-stimulated SOCE (Vasudevan et al., 2009). To investigate the significance of PtdIns(4,5)P2 synthesis by these PIP5K isoforms in STIM1–Orai1 interactions, we monitored stimulated FRET between AcGFP–Orai1 and STIM1–mRFP in RBL cells overexpressing either PIP5KIβ or PIP5KIγ. As shown in Fig. 2A, we found that overexpression of the Iβ isoform consistently enhanced thapsigargin-stimulated association between STIM1–mRFP and AcGFP–Orai1, whereas overexpression of the Iγ isoform consistently inhibited this interaction.
Fig. 2.

Differential effects of PIP5KI isoforms and targeted inositol 5-phosphatases on thapsigargin-stimulated association between AcGFP–Orai1 and STIM1–mRFP and PtdIns(4,5)P2 concentrations. (A) Thapsigargin (TG)-stimulated FRET between STIM1 and Orai1 in control cells (black), cells coexpressing PIP5KIβ (cyan) and cells coexpressing PIP5KIγ (red). Error bars show s.e.m. (B) Stimulated FRET between STIM1 and Orai1 in control cells (black), cells coexpressing the DRM-targeted inositol 5-phosphatase L10-Inp54p (green) and cells coexpressing the DSM-targeted inositol 5-phosphatase S15-Inp54p (pink). Error bars show s.e.m. (C) PIP5KI isoforms differentially enhance PtdIns(4,5)P2 levels in DRMs and DSMs. Values are normalized to the total PtdIns(4,5)P2 level in control cells from each experiment. Error bars show s.d. Unpaired, one-tailed Student's t-test between indicated populations are: *P=0.03, **P=0.05, ***P=0.05 (n=3 for each type of sample). (D) Targeted inositol 5-phosphatases selectively hydrolyze PtdIns(4,5)P2. Values are represented as in C; *P=0.001, **P=0.007 (n=6).
This differential modulation of STIM1–Orai1 association by the two isoforms of PIP5KI is consistent with our previous finding that they produce distinguishable membrane pools of PtdIns(4,5)P2. The cholesterol dependence of SOCE that we and others have observed suggests that spatial segregation of PtdIns(4,5)P2 can arise from membrane heterogeneity and underlying lipid order, which is modulated by the presence of cholesterol. As established previously, sucrose gradients can be used to separate detergent-resistant membrane (DRM) from membrane proteins and lipids that are solubilized by Triton X-100 (detergent-solubilized membrane; DSM). Although simply correlative, this biochemical approach has consistently proven useful for separating membrane subregions that are distinguished by their composition of lipids with ordered (DRM) compared with disordered (DSM) acyl chains (Brown and Rose, 1992; Sheets et al., 1999; Simons and Toomre, 2000; Brown, 2006). This biochemical approach previously provided evidence for ordered and disordered lipid pools of PtdIns(4,5)P2 that are differentially altered by EGF-stimulated phospholipase C (PLC) activation (Pike and Casey, 1996) and contribute differentially to T cell activation (Johnson et al., 2008). To test the possibility that PIP5KI isoforms synthesize pools of PtdIns(4,5)P2 that preferentially associate with distinctive lipid pools, we compared the relative concentrations of PtdIns(4,5)P2 that fractionate with DRM and DSM from cells with and without overexpression of PIP5KIβ and PIP5KIγ. As summarized in Fig. 2C, overexpression of each of the enzymes enhanced total PtdIns(4,5)P2 in RBL cells, but PIP5KIγ selectively enhanced PtdIns(4,5)P2 in the DSM fraction, whereas PIP5KIβ enhanced PtdIns(4,5)P2 in both DRM and DSM pools. The enhancing effect of PIP5KIβ overexpression on thapsigargin-stimulated STIM1–Orai1 association that we observed with FRET correlates most strongly with an increase in PtdIns(4,5)P2 in ordered lipid subregions, whereas the negative effect of PIP5KIγ on stimulated STIM1–Orai1 interactions correlates with a selective increase in PtdIns(4,5)P2 in disordered lipid subregions. The dot-blot method that we used for these measurements, previously developed by Johnson et al. (Johnson et al., 2008), is laborious but reproducible in multiple experiments.
Reduction of PtdIns(4,5)P2 in the ordered lipid pool inhibits thapsigargin-stimulated association of STIM1 and Orai1 and SOCE
We next measured thapsigargin-stimulated FRET between STIM1 and Orai1 in RBL cells expressing inositol 5-phosphatases that are selectively targeted to membrane subregions. Phosphatase L10-Inp54p targets ordered lipid regions, and phosphatase S15-Inp54p targets disordered regions, as characterized previously (Johnson et al., 2008). As shown in Fig. 2B, coexpression of L10-Inp54p with AcGFP–Orai1 and STIM1–mRFP resulted in substantial inhibition of thapsigargin-stimulated association of these proteins as detected by FRET. By contrast, coexpression of S15-Inp54p with these reporter constructs resulted in an increase in thapsigargin-stimulated FRET. As expected from previous results in HEK-293 cells (Johnson et al., 2008), we found that L10-Inp54p significantly reduced the levels of PtdIns(4,5)P2 in the DRM fraction, but not the DSM fraction, in RBL cells (Fig. 2D). Also consistent with previous results, S15-Inp54p caused some reduction in the pool of DSM-associated PtdIns(4,5)P2, but it did not significantly reduce the pool of DRM-associated PtdIns(4,5)P2. These are similar to trends observed for overexpression of PIP5KIβ and PIP5KIγ: they indicate that a higher concentration of PtdIns(4,5)P2 in ordered lipid regions promotes thapsigargin-stimulated Orai1–STIM1 association, whereas a higher concentration of PtdIns(4,5)P2 in disordered lipid regions inhibits this association.
To evaluate whether reductions in PtdIns(4,5)P2 pools alter thapsigargin-stimulated SOCE, we used confocal microscopy and Fluo-4 to monitor Ca2+ responses in individual cells under conditions similar to the FRET measurements. Either L10-Inp54p or S15-Inp54p was co-transfected with mRFP to identify positive tranfectants in RBL cells, and nontransfected, control cells were monitored in the same fields. Fig. 3 shows results from a representative experiment that averages Ca2+ changes from multiple cells of each type. Consistent with the indo-1 fluorimetry results in Fig. 1, traces of Fluo-4 from individual cells, as averaged over multiple cells, showed an initial fast rise in Ca2+ in response to thapsigargin that typically peaked in <1 minute, followed by a more sustained phase that was maximal between 1 and 3 minutes, then gradually declined. From previous thapsigargin experiments, carried out with RBL cells in the presence and absence of extracellular Ca2+ (Vasudevan et al., 2009), we assign the initial fast phase of this response to the passive release of Ca2+ from the ER store and the more sustained phase (>1 minute) to SOCE. In these and other experiments (Fig. 1), stimulated FRET develops somewhat more slowly than SOCE, and it is often more sustained than the Ca2+ response. These differences in kinetics are due to several factors, one of which is that robust SOCE can occur with only limited coupling between STIM1 and Orai1, as we previously found in the case of antigen stimulation (Calloway et al., 2009). Additionally, processes that inactivate the CRAC channel, such as Ca2+-dependent inactivation (Mullins et al., 2009), do not necessarily result in rapid dissociation of STIM1 and Orai1.
Fig. 3.
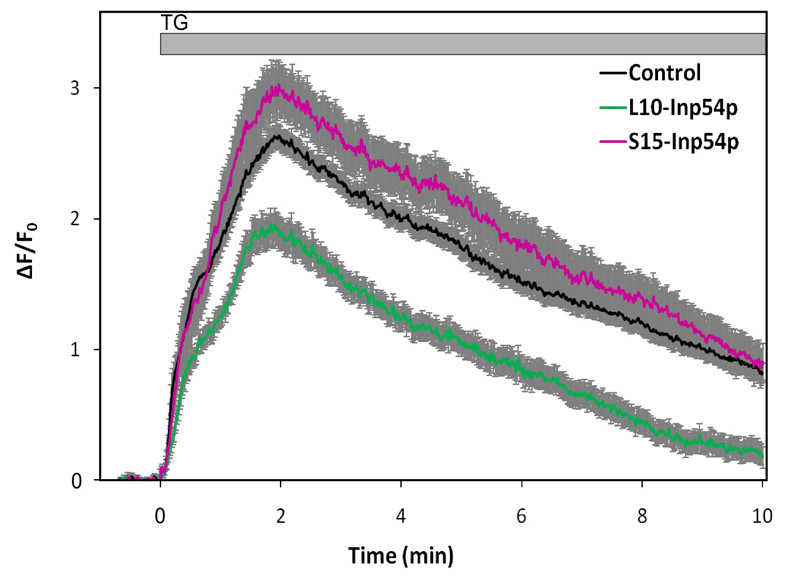
Effects of targeted inositol 5-phosphatases on thapsigargin-stimulated Ca2+ responses. Cells were transfected with L10-Inp54p–mRFP or S15-Inp54p–mRFP and loaded with Fluo-4 before analysis of individual cells with confocal microscopy. Thapsigargin (TG) was added at t=0. Each trace corresponds to Ca2+ responses as averaged over at least 12 cells of each type; the control is the untransfected cells in the same fields. Error bars show s.e.m.
Fig. 3 shows that for cells expressing S15-Inp54p, there was a small increase in the average SOCE response to thapsigargin compared with non-transfected cells in the same fields, although this difference might not be statistically significant (P>0.05 as calculated for t=3, 5 or 10 minutes after thapsigargin addition). Significantly reduced responses were observed for cells expressing L10-Inp54p, with the greatest reductions observed at the longer time points (P<10−5 as calculated for t=3, 5 or 10 minutes). In control experiments, we found that the Ca2+ responses in cells transfected with mRFP but not L10-Inp54p or S15-Inp54p were not significantly different from non-transfected cells (data not shown). From these results, we conclude that L10-Inp54p and S15-Inp54p alter thapsigargin-stimulated Ca2+ responses in adherent RBL cells in a manner that is consistent with their effects on stimulated FRET.
Polybasic sequences in STIM1 and Orai1 influence the dependence of STIM1–Orai1 coupling on PtdIns(4,5)P2 in membrane subregions
The polylysine sequence at the C-terminus of STIM1 (amino acids 677–685) has been shown to mediate puncta formation by STIM1 at the plasma membrane in the absence of Orai1 overexpression, but it is not necessary for puncta formation when Orai1 is coexpressed (Park et al., 2009; Walsh et al., 2009). We deleted this basic sequence from STIM1–mRFP (STIM1ΔK–mRFP), and we observed a modest enhancement in the kinetics of thapsigargin-stimulated FRET between this mutant protein and AcGFP–Orai1 compared with that for wild-type STIM1–mRFP, but the extent of this interaction was similar at longer time points (Fig. 4A, black curve, compare with Fig. 1B, black curve). Coexpression of S15-Inp54p with STIM1ΔK–mRFP and AcGFP–Orai1 yielded slightly more FRET (Fig. 4A, pink curve compared with black curve) compared with a greater enhancing effect with the wild-type proteins (Fig. 2B, magenta curve compared with black curve). By contrast, although the interaction between wild-type STIM1–mRFP and AcGFP–Orai1 was substantially inhibited by coexpression of L10-Inp54p (Fig. 2B, green curve compared with black curve), the interaction between STIM1ΔK–mRFP and AcGFP–Orai1 was enhanced under these conditions (Fig. 4A, light green curve compared with black curve).
Fig. 4.

Removal of either the polybasic sequences on STIM1 (STIM1ΔK) or Orai1 (Orai1ΔR) alters PtdIns(4,5)P2 pool selectivity. (A,B) Thapsigargin (TG)-stimulated FRET between AcGFP–Orai1 and STIM1ΔK–mRFP in control cells (black, A,B), cells expressing L10-Inp54p (light green, A) and cells expressing S15-Inp54p (pink, A), cells expressing PI5KIβ (cyan, B), and cells expressing PI5KIγ (bright red, B). FRET between wild-type (wt) STIM1 and Orai1 in the presence of L10-Inp54p (dark green, A; reproduced from Fig. 2B) or in the presence of PIP5KIγ (dark red, B; reproduced from Fig. 2A). (C,D) Thapsigargin-stimulated FRET between AcGFP–Orai1ΔR and STIM1–mRFP in control cells (black, C,D), cells expressing L10-Inp54p (light green, C) and cells expressing S15-Inp54p (pink, C), and cells expressing PIP5KIβ (cyan, D) and cells expressing PIP5KIγ (red, D). FRET between STIM1 and wild-type Orai1 in the presence of S15-Inp54p (purple, B; reproduced from Fig. 2B) or in the presence of PIP5KIβ (blue, D; reproduced from Fig. 2A). Error bars show s.e.m.
The effect of overexpressing PIP5KIβ on thapsigargin-stimulated association of STIM1ΔK–mRFP and AcGFP–Orai1 is consistent with that of L10-Inp54p: the enhancing effect of PIP5KIβ on the association of wild-type proteins (Fig. 2A, cyan curve compared with black curve) was not observed with STIM1ΔK–mRFP (Fig. 4B, cyan curve compared with black curve). Overexpression of PIP5KIγ, which inhibits wild-type STIM1–Orai1 interactions (Fig. 2A, red curve compared with black curve), did not inhibit the association between STIM1ΔK–mRFP and AcGFP–Orai1 (Fig. 4B, bright red curve compared with black curve). Changes caused by the STIM1ΔK mutation on the effects mediated by PIP5KIβ (Fig. 2A) and L10-Inp54p (Fig. 2B) on stimulated STIM1–Orai1 association are consistent with a mechanism in which the C-terminal polybasic sequence of STIM1 interacts with the ordered lipid pool of PtdIns(4,5)P2 in the plasma membrane during the normally regulated process of coupling with Orai1.
Orai1 has a polybasic sequence composed primarily of arginine residues near its N-terminus (amino acids 28–33). When this sequence is removed, the mutant (AcGFP–Orai1ΔR) localized normally to the plasma membrane (supplementary material Fig. S2), and stimulated FRET between AcGFP–Orai1ΔR and STIM1–mRFP was similar to that for the wild-type proteins (Fig. 4C, black curve compared with Fig. 1B, black curve). Also similar to results with the wild-type proteins, stimulated FRET between AcGFP–Orai1ΔR and STIM1–mRFP was decreased in cells expressing L10-Inp54p (Fig. 4C, green curve compared with black curve). Stimulated association of STIM1–mRFP and AcGFP–Orai1ΔR was also decreased after reduction of PtdIns(4,5)P2 in the disordered lipid pool by S15-Inp54p (Fig. 4C pink curve compared with black curve), and this contrasts with enhanced association of wild-type STIM1 and Orai1 caused by S15-Inp54p (Fig. 2B, magenta curve compared with black curve).
Deletion of the polyarginine sequence near the N-terminus of Orai1 also alters the effect of PIP5KIβ overexpression on the STIM1–Orai1 association. For the wild-type proteins, this stimulated association was enhanced by PIP5KIβ (Fig. 2A, cyan curve compared with black curve), but for STIM1–mRFP and AcGFP–Orai1ΔR this association was slightly reduced (Fig. 4D, cyan curve compared with black curve). By contrast, PIP5KIγ overexpression similarly affected stimulated FRET between STIM1–mRFP and either AcGFP–Orai1ΔR (Fig. 4D, red curve compared with black curve) or wild-type AcGFP–Orai1 (Fig. 2A, red curve compared with black curve), causing some inhibition in both cases. These results indicate that the basic polyarginine sequence at the N-terminus of Orai1 is involved in the enhancing effect of PI5KIβ but is relatively insensitive to the inhibitory effect of PIP5KIγ. This would be expected if the overall structure of Orai1 preferentially partitions in the disordered membrane lipid pool and interaction of the N-terminal basic sequence with PtdIns(4,5)P2 present in that pool provides little additional stabilization. However, this disposition of Orai1 could be modulated if the polybasic sequence was driven towards interactions with ordered lipid domains when the PtdIns(4,5)P2 content in that pool is sufficiently high. In this manner the coupling of Orai1 with STIM1 localized to the ordered lipid regions would be facilitated.
Discussion
Thapsigargin-stimulated STIM1–Orai1 association is regulated by the balance of PtdIns(4,5)P2 in ordered and disordered lipid subregions of the plasma membrane
Our results provide substantial evidence for the importance of cholesterol-dependent membrane heterogeneity and segregated pools of phosphoinositides in regulating the association of STIM1 with Orai1 during stimulated SOCE. We found that reduction of cholesterol inhibits thapsigargin-stimulated physical coupling between Orai1 and STIM1, as detected by FRET, in parallel with the inhibition of SOCE (Fig. 1A,B). Similarly, inhibition of PtdIns(4)P synthesis and SOCE by 10 μM wortmannin inhibits this stimulated protein–protein interaction (Fig. 1C,D). These and our previous results with PIP5KI isoforms (Vasudevan et al., 2009) led us to hypothesize that PtdIns(4,5)P2 synthesized by PIP5KIβ and PIP5KIγ is segregated in different membrane domains and that STIM1 and Orai1 coupling is influenced by the PtdIns(4,5)P2 content of these two pools. Numerous studies have shown that plasma membrane domains arise in part by cholesterol-based ordering of lipids, and in an approximate manner these can be characterized as ordered lipid and disordered lipid subregions. Proteins are integrally involved in membrane structure in multiple ways, and greater complexity comes from the variety of lipid and protein interactions. Modulation of these interactions can cause functional redistributions in stimulated signaling events (Lingwood and Simons, 2010).
We found that overexpression of PIP5KIβ results in an increase in stimulated association between STIM1 and Orai1 as measured by FRET, whereas overexpression of PIP5KIγ causes a decrease in this association. The distinctive effects might be explained by differences in membrane localization of the PtdIns(4,5)P2 generated by these two isoforms, and we found that overexpression of PIP5KIβ causes an increase in PtdIns(4,5)P2 in both the DRM (ordered lipids) and DSM (disordered lipids) fractions from resting cells, whereas overexpression of PIP5KIγ causes a selective increase in PtdIns(4,5)P2 in the DSM fraction (Fig. 2A,C). These results suggest that PtdIns(4,5)P2 in ordered lipid regions, which increases with PIP5KIβ overexpression, positively regulates STIM1–Orai1 coupling. We tested this possibility directly by selectively reducing PtdIns(4,5)P2 levels in either ordered lipid or disordered lipid subregions with respectively targeted inositol 5-phosphatases. We found that these phosphatases cause changes in stimulated FRET that are consistent with PIP5KIβ and PIP5KIγ overexpression (i.e. reduction in PtdIns(4,5)P2 in ordered lipid pools inhibits stimulated FRET, whereas reduction in PtdIns(4,5)P2 in disordered lipid pools enhances stimulated FRET).
A consistent result from our data is that thapsigargin-stimulated coupling of STIM1 and Orai1 is inhibited under conditions where there is a greater proportion of PtdIns(4,5)P2 in disordered lipid subregions of the membrane than in ordered lipid subregions. This disproportionality can be generated by either hydrolysis of PtdIns(4,5)P2 in ordered lipid subregions, by targeted L10-Inp54p, or when the pool of PtdIns(4,5)P2 in disordered lipid subregions is enhanced by PIP5KIγ. Distinctive effects based on the distribution of PtdIns(4,5)P2 in ordered lipid compared with disordered lipid subregions contrasts with an apparent insensitivity to the total amount of PtdIns(4,5)P2 present in the membrane. This pattern is consistent with previous reports that found only small effects on Ca2+ mobilization or puncta formation with changes in PtdIns(4,5)P2 content unless it was completely removed from the plasma membrane (Korzeniowski et al., 2009; Walsh et al., 2010). These previous reports did not consider a differential effect of multiple pools of PtdIns(4,5)P2 and did not directly measure STIM1–Orai1 association.
Confocal microscopy measurements of thapsigargin-stimulated Ca2+ responses in adherent RBL cells expressing L10-Inp54p and S15-Inp54p (Fig. 3) show reduced and enhanced SOCE responses, respectively, that correlate with the reduced and enhanced FRET we measured under these conditions (Fig. 2B). Thus, differential alterations of PtdIns(4,5)P2 pools associated with ordered and disordered lipid regions, mediated by these inositol 5-phosphatases, confer similar effects on FRET and SOCE on functional STIM1–Orai1 coupling that are consistent with an enhancing effect of PtdIns(4,5)P2 in ordered lipid domains and an inhibitory effect of PtdIns(4,5)P2 in disordered lipid domains. We note that the enhancing effect of PIP5KIβ overexpression on thapsigargin-stimulated FRET that we observe (Fig. 2A) does not correlate with its inhibitory effect on SOCE that we previously characterized in RBL cells stably expressing this construct (Vasudevan et al., 2009). One difference is that those previous Ca2+ measurements were carried out on suspended RBL cells, and it might be that this physical state has an altered capacity for PtdIns(4,5)P2 modulation due to a different cytoskeletal arrangement or some other change. Consistent with this possibility, we do not observe clear effects of transiently transfected L10-Inp54p or S15-Inp54p expression on thapsigargin-stimulated SOCE in suspended RBL cells, as monitored with a co-transfected Ca2+ indicator (GCaMP2) (Cohen et al., 2009) (data not shown). In the study by Johnson et al. (Johnson et al., 2008), targeted modulation of PtdIns(4,5)P2 pools by these inositol 5-phosphatases altered T cell morphology, suggesting that processes such as cell adhesion affect the distribution of phosphoinositides in different membrane pools. In addition, in our previous experiments on suspended cells (Vasudevan et al., 2008), stable expression of PIP5KIβ was generally low, and this too might affect the balance of PtdIns(4,5)P2 in ordered compared with disordered lipid pools, which could result in an altered phenotype. Taken together, these observations suggest that experimental conditions such as the physical state of the cells or different PIP5K expression levels influence the delicate balance of PtdIns(4,5)P2 in ordered and disordered membrane domains or the functional consequences of this balance. Regulation of PtdIns(4,5)P2 distribution is probably subtle, and further studies are needed to define the crucial features that determine functional outcomes under different conditions.
The polybasic sequences on STIM1 and Orai1 confer selectivity for ordered and disordered lipid pools that facilitate their stimulated association
Eliminating either the C-terminal polylysine sequence from STIM1 (STIM1ΔK) or the N-terminal polyarginine sequence from Orai1 (Orai1ΔR) largely eliminated the effects on thapsigargin-stimulated STIM1–Orai1 coupling that are caused by altering the distribution of PtdIns(4,5)P2 in membrane subregions. Unlike with wild-type proteins (Fig. 2A,B), stimulated FRET with STIM1ΔK or with Orai1ΔR is similar for cells expressing L10-Inp54p and S15-Inp54p or overexpressing PIP5KIβ and PIP5KIγ (Fig. 4), although small differences for the two mutated proteins are seen. FRET between STIM1ΔK–mRFP and AcGFP–Orai1 is approximately the same or higher than FRET between the wild-type proteins in every case (Fig. 4A,B compared with Fig. 2A,B). By contrast, FRET between STIM1–mRFP and AcGFP–Orai1ΔR is approximately the same or lower than between the wild-type proteins in every case (Fig. 4C,D compared with Fig. 2A,B). Overall, our results are consistent with the model described below, in which wild-type proteins utilize these polybasic sequences for PtdIns(4,5)P2-mediated translocation of Orai1 from disordered to ordered lipid pools, where it couples with STIM1 associated with PtdIns(4,5)P2 in this subregion.
We note that differences in the degree of association observed following removal of the polybasic sequences from STIM1 and Orai1 might be complicated by competition with the endogenous wild-type counterparts that are present in RBL cells. If the polylysine sequence in STIM1 guides this ER protein to interact with PtdIns(4,5)P2 in ordered lipid subregions, then its removal might allow free engagement of STIM1 with Orai1 that is associated with either ordered or disordered lipid pools. This scenario would be consistent with previous results showing that, although the polylysine sequence on STIM1 is important for translocation to the plasma membrane in the absence of Orai1, interaction with Orai1 is sufficient to concentrate STIM1 into ER–PM puncta (Park et al., 2009).
A model for PtdIns(4,5)P2-regulated STIM1–Orai1 association
A simple model that accounts for our results on the association between STIM1 and Orai1 that is stimulated by thapsigargin is summarized in Fig. 5. Before stimulation, STIM1 in the ER membrane is not functionally coupled to the plasma membrane, and the transmembrane protein Orai1 preferentially partitions into disordered lipid subregions of the plasma membrane, where its N-terminal polyarginine sequence provides some stabilization by interacting with PtdIns(4,5)P2 in those subregions. Upon stimulation by thapsigargin, the ER membrane protein oligomerizes, the C-terminal polylysine sequence of STIM1 targets this oligomerized protein to PtdIns(4,5)P2 in ordered lipid subregions of the plasma membrane and Orai1 translocates to these same subregions to associate with STIM1. STIM1–Orai1 association is stabilized by protein–protein binding, as well as by the polybasic sequences in both proteins interacting with PtdIns(4,5)P2. Thus the relative distributions of PtdIns(4,5)P2 in the ordered lipid and disordered lipid subregions influence the targeting of STIM1 and the propensity of Orai1 to redistribute into the ordered lipid subregions where it can engage STIM1. Consistent with this model, STIM1 was previously shown to undergo increased fractionation with DRMs in sucrose gradients in a thapsigargin-dependent manner (Pani et al., 2008).
Fig. 5.

Proposed scheme for PtdIns(4,5)P2 regulation of thapsigargin-stimulated association of STIM1 and Orai1. (A) In cells coexpressing the wild-type proteins STIM1 and Orai1, SOCE is initiated by the translocation of STIM1 to PtdIns(4,5)P2 associated with ordered lipid subregions of the plasma membrane (LO, blue; ER membrane omitted for clarity). This is followed by PtdIns(4,5)P2-dependent redistribution of Orai1 from disordered to ordered lipid subregions (LD, red) to facilitate the binding interaction with STIM1. (B) In cells expressing STIM1ΔK and wild-type Orai1, STIM1 lacks the polylysine sequence directing it to interact with PtdIns(4,5)P2, allowing STIM1 to directly engage Orai1 that is primarily localized to disordered lipid subregions of the PM. This results in somewhat slower timecourses of association, which are larger in magnitude at longer times. (C) In cells expressing wild-type STIM1 and Orai1ΔR, Orai1 lacks the capacity for PtdIns(4,5)P2-mediated redistribution between membrane pools. In this case, stimulated association of these proteins is more limited in magnitude.
Also consistent with our model is the finding that removal of polybasic sequences in the N-terminus of Orai1 or the C-terminus of STIM1 alters the mode of interaction between these two proteins, as revealed by expression of L10-Inp54p and S15-Inp54p or overexpression of PIP5KIβ and PIP5KIγ. Removal of the polylysine sequence on STIM1 prevents the initial association of STIM1ΔK with the ordered lipid pool of PtdIns(4,5)P2, but does not prevent the direct association of STIM1ΔK with Orai1 as measured by FRET (Fig. 4A,B; Fig. 5B). This scheme is also consistent with a previous report that characterized Orai1-dependent and -independent modes of STIM1 association with the PM following thapsigargin-mediated store depletion and identified the polylysine sequence on STIM1 as important for the Orai1-independent mode of association (Park et al., 2009). The loss of PtdIns(4,5)P2 selectivity in STIM1ΔK is due to its loss of the stimulated association with ordered lipid subregions in the membrane, thereby allowing STIM1ΔK to engage with Orai1 in either the ordered or disordered lipid subregions. The STIM1ΔK deletion generally increases the extent of its association with Orai1, and this can be explained by the capacity of STIM1ΔK to engage Orai1 without restrictions imposed by PtdIns(4,5)P2 in membrane subregions.
Removal of the polyarginine sequence in the N-terminus of Orai1 also results in the loss of PtdIns(4,5)P2 sensitivity in FRET measurements, but in this case the extent of stimulated FRET observed is consistently reduced (Fig. 4C,D), as represented in our model (Fig. 5C). This mutation does not affect the capacity of STIM1 to bind to PtdIns(4,5)P2 in ordered lipid subregions, but it affects the tendency of Orai1 to partition into this subregion to bind to STIM1. In this case, the redistribution of Orai1 between distinctive lipid subregions would depend more on Orai1 partitioning that is based on its transmembrane protein structure, rather than on stabilization provided by interactions with PtdIns(4,5)P2. It will be informative to evaluate these predictions regarding mutant and wild-type Orai1 partitioning between DRM and DSM in future experiments.
The present model does not explain why STIM1 should preferentially associate with PtdIns(4,5)P2 in ordered domains rather than with PtdIns(4,5)P2 in disordered domains, unless the relative PtdIns(4,5)P2 content changes markedly with thapsigargin stimulation. It is possible that a structural property of ordered domains, such as enhanced association with the cytoskeleton (Holowka et al., 2000) could influence this, but such issues remain to be investigated. In addition, this model cannot fully account for more subtle quantitative relationships between PtdIns(4,5)P2 localized to ordered lipid subregions compared with disordered lipid subregions of the membrane. For example, there is a significant increase in the stimulated interaction between STIM1–mRFP and AcGFP–Orai1 following overexpression of PI5KIβ, in comparison with an increase in PtdIns(4,5)P2 that fractionates with both DRM and DSM in resting cells with overexpressed PI5KIβ. It might be that the enhancement of the STIM1–Orai1 interaction by PtdIns(4,5)P2 in ordered lipid subregions is more potent than the inhibition by the PtdIns(4,5)P2 in disordered subregions, but current data are insufficient to evaluate this possibility.
Roles for PtdIns(4,5)P2 pools in antigen-stimulated STIM1–Orai1 association
Our experiments, as described above, were performed systematically with thapsigargin as the stimulus. Our initial experiments with antigen stimulation showed clear effects caused by the PI5K isoforms and the selectively targeted inositol 5-phosphatases, but these results are not yet readily interpreted in terms of a simple model. Antigen-stimulated coupling of STIM1 and Orai1 differs from thapsigargin stimulation in two principal respects: first, as shown previously, antigen stimulation does not prevent store refilling, resulting in substantially attenuated STIM1–Orai1 association detected by FRET (Calloway et al., 2009); and, second, antigen stimulation alters PtdIns(4,5)P2 levels at the plasma membrane by hydrolysis of PtdIns(4,5)P2 into diacylglycerol and Ins(1,4,5)P3, and possibly by other stimulated enzyme activities. Our previous study provided evidence that PIP5KIγ contributes to the pool of PtdIns(4,5)P2 that is hydrolyzed by antigen-activated PLCγ to mediate Ca2+ release from ER stores, but that PIP5KIβ does not (Vasudevan et al., 2009). Measurements of antigen-stimulated FRET between labeled Orai1 and STIM1 under these and other conditions that differentially perturb PtdIns(4,5)P2 pools might help to illuminate the molecular mechanisms for these findings. However, we expect the results might be complicated by the multiple roles for PtdIns(4,5)P2 and possibly by dynamic alterations in ordered and disordered PtdIns(4,5)P2 pools during this more complex physiological process. Although the evidence from a large number of studies for participation of membrane lipid heterogeneity in targeting of cell signaling is quite strong, delineating the subtle and dynamic components remains a daunting challenge.
Conclusions
Our results provide direct evidence that PtdIns(4,5)P2 regulates the stimulated association of STIM1 with Orai1 during activation of SOCE. This regulation is determined by the balance of PtdIns(4,5)P2 in ordered lipid subregions compared with that in disordered lipid subregions. Polybasic sequences at the C-terminus of STIM1 (amino acids 677–685) and in the N-terminal region of Orai1 (amino acids 28–33) are crucial for this regulation by PtdIns(4,5)P2. An outstanding question is the biophysical or biochemical basis for the segregation of the functionally distinguishable pools of PtdIns(4,5)P2 that fractionate with DRM and DSM domains and can be modulated by expression of L10-Inp54p or S15-Inp54p, or overexpression of PIP5KIβ or PIP5KIγ. Mass spectrometry analysis of PtdIns(4,5)P2 acyl chain composition provides evidence for pools of monounsaturated and polyunsaturated PtdIns(4,5)P2 (Wenk et al., 2001), which should preferentially partition into ordered lipid and disordered lipid membrane domains, respectively (Fridriksson et al., 1999). Simple lateral diffusion of newly synthesized PtdIns(4,5)P2 would be expected to result in rapid mixing, such that segregation of order-preferring and disorder-preferring PtdIns(4,5)P2 probably requires dynamic turnover and possibly structural ‘fences’ to maintain such segregated pools or gradients. Selective targeting of PI5K isoforms to ordered or disordered membrane domains could provide the driving force for this segregation on a nanometer scale. Previous evidence for micron-scale segregation of PtdIns(4,5)P2 and PtdIns(3,4,5)P3 in polarized epithelial cells (Martin-Belmonte et al., 2007) might be relevant to this issue, and ongoing studies in this cell type might provide mechanistic clues for nanometer-scale PtdIns(4,5)P2 segregation in non-polarized cells.
Materials and Methods
Constructs and cloning
STIM1ΔK–mRFP was constructed from our previously described STIM1–mRFP vector (Calloway, 2009) using the Stratagene QuikChange site-directed mutagenesis kit. The primers used were 5′-GGAAACAGACTCCAGCCCAGGCCGGGCGGCCGCATCAGGCATGGCCTC-3′ and its reverse complement. AcGFP–Orai1ΔR was constructed using site-directed mutagenesis on our previously described AcGFP–Orai1 vector (Calloway et al., 2009). The mutagenic primers used were 5′-GCAGCACCACCAGCGGCAGCAGCGGGGACGGGGAGCCC-3′ and its reverse complement. Additionally, flanking primers up and downstream of the multiple cloning site on AcGFP–Orai1 were used.
L10-Inp54p and S15-Inp54p were derived from the L10-GFP-Inp54p and S15-GFP-Inp54p vectors (Johnson et al., 2008). GFP was removed from each vector by recloning and ligating the targeting sequence (L10 or S15) with that for Inp54p. PIP5KIβ, PIP5KIγ87, STIM1–mRFP and AcGFP–Orai1 vectors were as previously described (Vasudevan et al., 2009; Calloway et al., 2009).
Cell culture
RBL-2H3 mast cells were cultured in minimal essential medium supplemented with 1 μg/ml gentamicin and 20% (v/v) fetal bovine serum (FBS). In preparation for transfection and imaging, cells were plated at 25% confluence into 35-mm MatTek wells. After approximately 20 hours, cells were transfected with either mutant or wild-type versions of STIM1–mRFP and AcGFP–Orai1 as previously described (Calloway et al., 2009). These constructs were transfected using either Geneporter (Genlantis) or Fugene HD (Roche) according to the manufacturers' instructions, with modifications to enhance transfection efficiency in the RBL cells previously described (Gosse et al., 2005). For Ca2+ imaging experiments using Fluo-4, L10-Inp54p and S15-Inp54p constructs were co-transfected with an mRFP-containing vector at a ratio of 4:1 w/w to identify transfected cells. For FRET experiments, modifications were made to the transfection protocol to achieve higher degrees of expression for both the L10-Inp54p and S15-Inp54p constructs: cells were transfected with 8 μg DNA (7 μg Inp54p construct, 0.5 μg each of STIM1 and Orai1 constructs) and 20 μl Geneporter in 100 μl OptiMem (Invitrogen) added to each MatTek well. These conditions of transfection result in 1–2% of the cells co-expressing fluorescent STIM1 and Orai1. Cells were imaged at 37°C on the day after transfection. For steady-state fluorimetry measurements of Ca2+ changes in RBL cells, indo-1 was loaded into the cells, and these were monitored in a stirred cuvette as previously described (Pierini et al., 1997).
Confocal microscopy of FRET and Ca2+ measurements
RBL cells were imaged for FRET as previously described (Calloway et al., 2009). Prior to imaging, cells were washed and incubated for 5 min at 37°C in 2.5 ml buffered salt solution (BSS: 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 5.6 mM glucose, 1 mg/ml BSA, 20 mM HEPES pH 7.4). Images of cells were collected on a Leica TCS SP2 confocal microscope with a Leica APO 63× dipping objective. Cells were excited at specified wavelengths, with laser intensity and phototube sensitivity adjusted to maximize the signal-to-noise ratio. For FRET measurements, cells were imaged at 10-second intervals with excitation at 476 nm to minimize spectral bleed-through. All FRET measurements were performed at 37°C. After observing the resting state of the cells, they were stimulated for the time specified by the addition of 0.5 ml of BSS containing thapsigargin (150 nM final concentration). An automated mask-drawing algorithm in MATLAB was used to select the pixels of interest at the plasma membrane for every time point, using fluorescence from AcGFP as the template (Calloway et al., 2009). The integrated red and green fluorescence intensities under the mask were adjusted by background subtraction and correction for spectral bleed-through. We report the ratio of the corrected red fluorescence to corrected green fluorescence as FRET. For all of the FRET results reported, data were collected in at least four separate experiments, and a minimum of 12 individual cells were analyzed for each condition shown.
For Ca2+ measurements with Fluo-4, transfected cells were dye-loaded and monitored as previously described for COS-7 cells (Calloway et al., 2009). Individual cells were excited at 488 nm (for Fluo-4) and 547 nm (for mRFP) and imaged at 1-second intervals on a Zeiss LSM 510 META confocal microscope with a 40× oil-immersion objective lens heated at 37°C. Cells were stimulated with thapsigargin as described above. ImageJ (NIH) was used to analyze the changes with time of Fluo-4 fluorescence that was integrated over a representative region of interest (ROI).
Membrane fractionation and dot-blots
At 24 hours before fractionation, cells were electroporated with the indicated construct at a concentration of 32 μg DNA per ml at 280 V and 950 μF using Gene Pulser X (Bio-Rad). Sucrose-gradient fractionation was performed as previously described (Field et al., 1999), with small modifications. Briefly, cells were harvested, resuspended in BSS and solublized with Triton X-100 (Pierce) at a ratio of 0.013% (v/v) Triton X-100 per 1×106 cells. Gradients were constructed by pipetting in order: 250 μl 80% sucrose, 500 μl 50% sucrose, 1.5 ml 40% sucrose containing cell lysate, 750 μl 30% sucrose, 500 μl 20% sucrose and 1 ml 10% sucrose (all concentrations are w/v). After 16–18 hours of centrifugation at 49,000 rpm with a SW60.1 (Beckman) rotor, each gradient was divided into two fractions at the top interface of the 40% sucrose band. Each of these fractions was subsequently extracted for lipids according to the method described by Johnson et al. (Johnson et al., 2008). After extraction, the dried lipid film was resuspended in water at a concentration normalized to 800 cell equivalents per μl. Dot-blotting was performed by first sealing the ventral holes in the dot-blot apparatus (BD Biosciences) with aluminum foil. Then 10 μl of resuspended extract was added to the nitrocellulose membrane in each well and allowed to air dry in the apparatus for 30 minutes, followed by removal of the membrane from the apparatus and further drying for 1.5 hours at room temperature. Blots were blocked in either 3% BSA (w/v) or 20% FBS (v/v), developed with anti-PtdIns(4,5)P2 monoclonal antibody (Assay Designs) and anti-mouse-IgG antibody conjugated to horseradish peroxidase (GE Healthcare). Blots were visualized using Supersignal West Pico chemiluminescent dye (Thermo Scientific). Quantification was performed with ImageJ.
Supplementary Material
Acknowledgments
We thank Jean-Pierre Kinet, Andreas Jeromin and Pietro De Camilli for useful constructs. This work was supported by an American Chemical Society Division of Medicinal Chemistry Predoctoral Fellowship (N.C.), an American Heart Association Predoctoral Fellowship (N.C.) and National Institutes of Health Grants T32 GM008500 (N.C.) and AI022449. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/15/2602/DC1
References
- Alicia S., Angélica Z., Carlos S., Alfonso S., Vaca L. (2008). STIM1 converts TRPC1 from a receptor-operated to a store-operated channel: moving TRPC1 in and out of lipid rafts. Cell Calcium 44, 479-491 [DOI] [PubMed] [Google Scholar]
- Bauer M. C., O'Connell D., Cahill D. J., Linse S. (2008). Calmodulin binding to the polybasic C-termini of STIM proteins involved in store-operated calcium entry. Biochemistry 47, 6089-6091 [DOI] [PubMed] [Google Scholar]
- Broad L. M., Cannon T. R., Taylor C. W. (1999). A non-capacitative pathway activated by arachidonic acid is the major Ca2+ entry mechanism in rat A7r5 smooth muscle cells stimulated with low concentrations of vasopressin. J. Physiol. 517, 121-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad L. M., Braun F. J., Lievremont J. P., Bird G. S., Kurosaki T., Putney J. W. (2001). Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. J. Biol. Chem. 276, 15945-15952 [DOI] [PubMed] [Google Scholar]
- Brown D. A. (2006). Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology 21, 430-439 [DOI] [PubMed] [Google Scholar]
- Brown D. A., Rose J. K. (1992). Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68, 533-544 [DOI] [PubMed] [Google Scholar]
- Calloway N., Vig M., Kinet J. P., Holowka D., Baird B. (2009). Molecular clustering of STIM1 with Orai1/CRACM1 at the plasma membrane depends dynamically on depletion of Ca2+ stores and on electrostatic interactions. Mol. Biol. Cell 20, 389-399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloway N., Holowka D., Baird B. (2010). A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry 49, 1067-1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen R., Torres A., Ma H.-T., Holowka D., Baird B. (2009). Ca2+ waves initiate antigen-stimulated Ca2+ responses in mast cells. J. Immunol. 183, 6478-6488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven W. I., Jones B. F., Petranka J. G., Smyth J. T., Tomita T., Bird G. S., Putney J. W. (2009). TRPC channels function independently of STIM1 and Orai1. J. Physiol. 587, 2275-2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S. H., Tanasa B., Hogan P. G., Lewis R. S., Daly M., Rao A. (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179-185 [DOI] [PubMed] [Google Scholar]
- Field K. A., Holowka D., Baird B. (1999). Structural aspects of the association of FcepsilonRI with detergent-resistant membranes. J. Biol. Chem. 274, 1753-1758 [DOI] [PubMed] [Google Scholar]
- Fridriksson E. K., Shipkova P. A., Sheets E. D., Holowka D., Baird B., McLafferty F. W. (1999). Quantitative analysis of phospholipids in functionally important membrane domains from RBL-2H3 mast cells using tandem high-resolution mass spectrometry. Biochemistry 38, 8056-8063 [DOI] [PubMed] [Google Scholar]
- Galan C., Woodard G. E., Dionisio N., Salido G. M., Rosado J. A. (2010). Lipid rafts modulate the activation but not the maintenance of store-operated Ca2+ entry. Biochim. Biophys. Acta 1803, 1083 [DOI] [PubMed] [Google Scholar]
- Gosse J. A., Wagenknecht-Wiesner A., Holowka D., Baird B. (2005).Transmembrane sequences are determinants of immunoreceptor signaling. J. Immunol. 175, 2123-2131 [DOI] [PubMed] [Google Scholar]
- Holowka D., Sheets E. D., Baird B. (2000). Interactions between Fc(epsilon)RI and lipid raft components are regulated by the actin cytoskeleton. J. Cell Sci. 113, 1009-1019 [DOI] [PubMed] [Google Scholar]
- Hoth M., Penner R. (1993). Calcium release-activated calcium current in rat mast cells. J. Physiol. 465, 359-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G. N., Zeng W., Kim J. Y., Yuan J. P., Han L., Muallem S., Worley P. F. (2006). STIM1 carboxyl-terminus activates native SOC, ICRAC and TRPC1 channels. Nat. Cell Biol. 8, 1003-1010 [DOI] [PubMed] [Google Scholar]
- Hull J. J., Lee J. M., Kajigaya R., Matsumoto S. (2009). Bombyx mori homologs of STIM1 and Orai1 are essential components of the signal transduction cascade that regulates sex pheromone production. J. Biol. Chem. 284, 31200-31213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. M., Chichili G. R., Rodgers W. (2008). Compartmentalization of phosphatidylinositol 4,5-bisphosphate signaling evidenced using targeted phosphatases. J. Biol. Chem. 283, 29920-29928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniowski M. K., Popovic M. A., Szentpetery Z., Varnai P., Stojilkovic S. S., Balla T. (2009). Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J. Biol. Chem. 284, 21027-21035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D., Simons K. (2010). Lipid rafts as a membrane organizing principle. Science 327, 46-50 [DOI] [PubMed] [Google Scholar]
- Liou J., Kim M. L., Heo W. D., Jones J. T., Myers J. W., Ferrell J. E., Jr, Meyer T. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J., Fivaz M., Inoue T., Meyer T. (2007). Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 104, 9301-9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Casey L., Pike L. J. (1998). Compartmentalization of phosphatidylinositol 4,5-bisphosphate in low-density membrane domains in the absence of caveolin. Biochem. Biophys. Res. Commun. 245, 684-690 [DOI] [PubMed] [Google Scholar]
- Martin-Belmonte F., Gassama A., Datta A., Yu W., Rescher U., Gerke V., Mostov K. (2007). PTEN-mediated apical segregation of phophoinositides controls epithelial morphogenesis through Cdc42. Cell 128, 383-397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins F. M., Park C. Y., Dolmetsch R. E., Lewis R. S. (2009). STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc. Natl. Acad. Sci. USA 106, 15495-15500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani B., Ong H. L., Liu X., Rauser K., Ambudkar I. S., Singh B. B. (2008). Lipid rafts determine clustering of STIM1 in endoplasmic reticulum-plasma membrane junctions and regulation of store-operated Ca2+ entry (SOCE). J. Biol. Chem. 283, 17333-17340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C. Y., Hoover P. J., Mullins F. M., Bachhawat P., Covington E. D., Raunser S., Walz T., Garcia K. C., Dolmetsch R. E., Lewis R. S. (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876-890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierini L. M., Harris N. T., Holowka D., Baird B. (1997). Evidence supporting a role for microfilaments in regulating the coupling between poorly dissociable IgE-FceRI aggregates and downstream signaling pathways. Biochemistry 36, 7447-7456 [DOI] [PubMed] [Google Scholar]
- Pike L. J., Casey L. (1996). Localization and turnover of phosphatidylinositol 4,5-bisphosphate in caveolin-enriched membrane domains. J. Biol. Chem. 271, 26453-26458 [DOI] [PubMed] [Google Scholar]
- Putney J. W., Bird G. S. (1993). The inositol phosphate-calcium signaling system in nonexcitable cells. Endocr. Rev. 14, 610-631 [DOI] [PubMed] [Google Scholar]
- Sengupta P., Holowka D., Baird B. (2007). Fluorescence resonance energy transfer between lipid probes detects nanoscopic heterogeneity in the plasma membrane of live cells. Biophys. J. 92, 3564-3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets E. D., Holowka D., Baird B. (1999). Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcepsilonRI and their association with detergent-resistant membranes. J. Cell Biol. 145, 877-887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K., Toomre D. (2000). Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31-39 [DOI] [PubMed] [Google Scholar]
- Surviladze Z., Dráberová L., Kovárová M., Boubelík M., Dráber P. (2001). Differential sensitivity to acute cholesterol lowering of activation mediated via the high-affinity IgE receptor and Thy-1 glycoprotein. Eur. J. Immunol. 31, 1-10 [DOI] [PubMed] [Google Scholar]
- Vasudevan L., Jeromin A., Volpicelli-Daley L., De Camilli P., Holowka D., Baird B. (2009). The β- and γ-isoforms of type I PIP5K regulate distinct stages of Ca2+ signaling in mast cells. J. Cell Sci. 122, 2567-2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M., Peinelt C., Beck A., Koomoa D. L., Rabah D., Koblan-Huberson M., Kraft S., Turner H., Fleig A., Penner R., et al. (2006). CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220-1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. M., Chvanov M., Haynes L. P., Petersen O. H., Tepikin A. V., Burgoyne R. D. (2010). Role of phosphoinositides in STIM1 dynamics and store-operated calcium entry. Biochem. J. 425, 159-168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk M. R., Lucast L., Di Paolo G., Romanelli A. J., Suchy S. F., Nussbaum R. L., Cline G. W., McMurray W., De Camilli P. (2001). Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat. Biotechnol. 21, 813-817 [DOI] [PubMed] [Google Scholar]
- Yuan J. P., Zeng W., Dorwart M. R., Choi Y. J., Worley P. F., Muallem S. (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337-343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W., Yuan J. P., Kim M. S., Choi Y. J., Huang G. N., Worley P. F., Muallem S. (2008). STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell 7, 439-448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., Cahalan M. D. (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902-905 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}