

Abstract
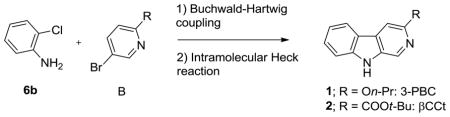
In order to gain access to 3-propoxy-β-carboline hydrochloride (3-PBC·HCl) (1·HCl), and β-carboline-3-carboxylate-t-butyl ester (βCCt) (2), potential clinical agents active against alcohol self-administration, a two-step route was developed. This process involves a palladium catalyzed Buchwald-Hartwig coupling and an intramolecular Heck reaction. This two-step route provides rapid access to multigram quantities of 3-PBC (1) and βCCt (2), as well as analogs for studies of alcohol self-administration. The overall yield of 3-PBC (1) was improved from 8 % to 50 % in this route.
The GABAA receptor is the major inhibitory neurotransmitter receptor of the central nervous system (CNS) and the site of action of a variety of pharmacologically and clinically important drugs, such as benzodiazepines, barbiturates, neuroactive steroids, anesthetics and convulsants.1 There are several disease states thought to be associated with the improper functioning of this system, including anxiety, epilepsy, insomnia, depression, bipolar disorder, and schizophrenia, as well as mild cognitive impairment and Alzheimer’s disease.2
Alcohol addiction and dependence remain significant public health concerns, impacting physical and mental well-being, family structure and occupational stability. The design of clinically safe and effective drugs that reduce alcohol addiction and dependence remains a high priority.3,4 While the precise neuromechanisms regulating alcohol-seeking behaviors remain unknown, there is now compelling evidence that the GABAA receptors within the striatopallidal and extended amygdala system are involved in the “acute” reinforcing actions of alcohol.5–9
The beta-carbolines 3-propoxy-β-carboline hydrochloride 1 · HCl (3-PBC) · HCl and β-carboline-3-carboxylate-t-butyl ester 2 (βCCt) are mixed benzodiazepine agonist-antagonist ligands with binding selectivity at α1 receptors (see Figure 1).10–12 In studies which involve the α1 subtype, the orally active 1·HCl and 2 were observed to selectively reduce alcohol-motivated behaviors in a variety of experiments.10,13 Moreover, both 1·HCl and 2 displayed mixed weak agonist-antagonist profiles in vivo in alcohol P and HAD rats, and may be capable of reducing alcohol intake while eliminating or greatly reducing the anxiety associated with habitual alcohol, abstinence or detoxification.10,13 These types of ligands, subsequently, may be ideal clinical agents for the treatment of alcohol dependent individuals.
Figure 1.

Structures of 3-PBC (1) and βCCt (2)
The synthesis of both 1 and 2 have been accomplished previously.14–17 The overall yield of 1 (via 6 steps) as reported previously was 8 %; while that of 2 (5 steps) was 35% from DL-tryptophan. The syntheses involved a number of steps, some of which occurred in low yields.
In 2005 Weerts et al. reported that 3-PBC·HCl significantly reduced alcohol self-administration in baboons.18 More importantly, it reduced craving in the subjects as well.18 This important result led to the interest in a short and concise synthesis of 3-PBC (1), capable of scale up to multigram levels, as well as a similar route to βCCt (2). Retrosynthetically, both of these compounds can be envisioned to arise from a substituted aniline A and a substituted pyridine derivative B (Scheme 1).
Scheme 1.

Retrosynthetic analysis of 1 and 2
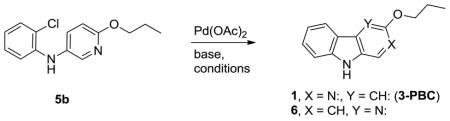
As shown in Scheme 2, bromopyridine 319 was reacted with aniline 4a in toluene at 100°C in the presence of 5 mol % Pd(OAc)2 and 7.5 mol% X-Phos to obtain diarylamine 5a in 93 % yield. Various conditions were screened to effect a coupling reaction between 3 and aniline 4a in high yield.20–23 Other combinations of conditions (lower temperature, different ligands, different bases, copper-based methods) resulted either in inferior yields and/or debromination of 3. In similar fashion, 5b was obtained from 3 and 2′-chloroaniline 4b in 91 % yield.
Scheme 2.

Synthesis of intermediates 5a and 5b
Reagents and conditions: (a) 5 mol% Pd(OAc)2, 7.5 mol% X-Phos, 1.5 eq. Cs2CO3, toluene, 100°C, 15 h
Once diarylamines 5a and 5b were in hand as potential precursors to 1, various methods to carry out the required intramolecular cyclization were attempted. A Heck-type coupling reaction and CH activation/oxidative coupling-cyclization reaction were more appealing because of atom economy.23, 24 In recent years, such direct arylation reactions have been developed extensively.25–30 One advantage is the preactivated, functionalized arenes can be replaced with a simple arene, consequently reducing the number of steps and overall cost of the process.25
Various approaches for the construction of substituted carbazoles via coupling processes have been described in the literature.29–33 Bedford et al. have carried out a microwave-mediated one-pot synthesis of carbazoles from 2-chloroanilines via consecutive amination and CH activation.30 Budén et al. have used photostimulated reactions of chloro diarylamines for the synthesis of substituted carbazoles.31 However, the issue of regioselectivity was not addressed in those reports since the authors dealt with reactions leading to a symmetrical product or one of the possible positions for cyclization was blocked. Hostyn and co-workers have described palladium catalyzed regioselective synthesis of α-carbolines by using 2,3-dichloropyridines and substituted anilines.32 Recently Sridharan et al. have reported a Pd(OAc)2 promoted cyclodehydrogenation of diphenylamines to carbazole using Cu(OAc)2 as an oxidant.33 In addition Ohno et al. have carried out the synthesis of substituted carbazoles by intramolecular oxidative direct arylation in an aerobic atmosphere.29 Interestingly the extension of these methods for the synthesis of β-carbolines had not been demonstrated to these authors knowledge.
With this history in mind, initial attempts were made to cyclize 5a using catalytic Pd (II)-mediated oxidative coupling. Diarylamine 5a failed to cyclize using Pd(OAc)2 as a Pd (II) source and Cu(OAc)2 as the co-oxidant for palladium even after heating in refluxing toluene or in acetic acid.29 Interestingly, when the reaction was carried out at 120°C in acetic acid as a solvent with an oxygen atmosphere, 5a appeared to react, and the β-carboline 1 was obtained as an equimolar mixture with its regiosiomer, a δ-carboline, 6. However, the reaction did not go completion in 15 hours and only 30 % combined yield was obtained. Further modifications of these conditions with 5a (longer duration, increased catalytic loading) did not result in any improvement in the yield, nor ratio of 1 to 6.
At this point it was decided to explore a Heck-type cyclization of model compound 5b utilizing Pd(0) as a catalyst. Initial attempts, which included previously reported Buchwald-Hartwig amination conditions [Pd(OAc)2, X-Phos, Cs2CO3 or NaOt-Bu, toluene, 100°C] did not provide β-carboline 1 even after heating for 24 hours (Table 1, entries 1 and 2). Use of polar solvents such as DMF or DMA led to dechlorination of the starting material (entries 3 and 4). Air-stable mono-dentate ligand (t-Bu)3P·HBF4 had been used previously for similar types of transformations in a different system.30 Interestingly, Pd(OAc)2 with (t-Bu)3P·HBF4 as a catalyst in the presence of NaOt-Bu in DMA gave 32 % yield of the mixture of regioisomers 1 and 6. A large amount of decomposition products were observed on TLC (entry 5). When K2CO3 was employed as the base, the reaction went smoothly within 16 hours at 120°C and afforded a readily separable mixture of regioisomers 1 and 6 in a ratio of 1.75:1 (entries 6 and 7). Substitution of (t-Bu)3P·HBF4 with ligand Cy3P·HBF4 afforded comparable results (entry 8). The desired β-carboline 1 and its regioisomer 6 (δ-carboline) can be differentiated from each other by examination of their corresponding 1H NMR spectra. In addition, X-ray crystal structures (Supporting Information) of both regioisomers 1 and 6 were obtained to further confirm the structures.
Table 1.
Optimization of conditions for the cyclization of 5b to 1a
 | |||||
|---|---|---|---|---|---|
| entry | ligand | base | solvent | temp (time) | results (yield)b |
| 1 | X-Phos | Cs2CO3 | toluene | 100°C (24 h) | no reaction |
| 2 | X-Phos | NaOt-Bu | toluene | 100°C (24 h) | no reaction |
| 3 | X-Phos | NaOt-Bu | DMF | 120°C (24 h) | dechlorination |
| 4 | X-Phos | NaOt-Bu | DMA | 120°C (24 h) | dechlorination |
| 5 | (t-Bu)3P ·HBF4 | NaOt-Bu | DMA | 120°C (24 h) | 32 % yield (mixture of 1 and 6) + decomposed material |
| 6 | (t-Bu)3P ·HBF4 | K2CO3 | DMA | 120°C (24 h) | 52 % 1 + 30 % 6 |
| 7 | (t-Bu)3P ·HBF4 | K2CO3 | DMA | 120°C (16 h) | 56 % 1 + 32 % 6 |
| 8 | Cy3P ·HBF4 | K2CO3 | DMA | 120°C (16 h) | 55 % 1 + 30 % 6 |
The reactions were carried out using 5b (0.1 mmol), Pd(OAc)2 (0.01 mmol), ligand (0.02 mmol), and base (0.2 mmol) in solvent (1.0 mL) under argon.
Isolated yield.
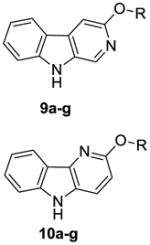
Since many 3-substituted β-carbolines exhibit subtype selectivity at α1β2/3γ2 BZR/GABAergic receptors it was decided to apply these conditions for the synthesis of various substituted β-carbolines previously reported.14,15,17, 34–36 In addition, it was important to evaluate the effect of different substitutions located ortho to the pyridyl nitrogen atom on the regioselectivity of the process (Scheme 3). The required bromopyridine ether derivatives 7a-g are commercially available and can be synthesized via known literature procedures.19 Buchwald-Hartwig aminations of 7a–g with 2-chloroaniline led to the chloro diarylamines 8a–g smoothly in 81–92 % yields. Heck-type cyclization of 8a–g afforded the regioisomers: β-carbolines 9a–g and δ-carbolines 10a–g in good yields (Table 2). The substituents ortho to the pyridyl nitrogen atom appeared to have some effect on the regioselectivity. The smaller substituents (Me, Et) tended to provide equimolar amounts of the regioisomers 9 and 10 (Table 2, entries 1 and 2); while the bulkier substituents (i-Pr, t-Bu) afforded the β-carbolines as the major product (entries 3 and 6). The best ratio of 9 to 10 for a β-carboline was obtained for the 5-bromo-2-t-butoxypyridine (1.9 : 1; entry 6).
Scheme 3.

Synthesis of substituted carboline analogs
Reagents and conditions: (a) 5 mol% Pd(OAc)2, 7.5 mol% X-Phos, 1.5 eq. Cs2CO3, toluene, 100°C, 15 h; (b) Pd(OAc)2, (t-Bu)3P.HBF4, K2CO3, DMA, 120°C, 16 h
Table 2.
Ratios of β (9) and δ (10) carbolines
| entry | R | Yielda (9 : 10) | |
|---|---|---|---|
|
| |||
| 1 | Me | 81 % (1.2 : 1) |
|
| 2 | Et | 85 % (1.25 : 1) | |
| 3 | i-Pr | 90 % (1.8 : 1) | |
| 4 | n-Bu | 87 % (1.35 : 1) | |
| 5 | i-Bu | 93 % (1.4 : 1) | |
| 6 | t-Bu | 85 % (1.9 : 1) | |
| 7 | Bn | 87 % (1.5 : 1) | |
Combined isolated yield
Large scale synthesis of 3-PBC (1) and βCCT (2)
Because 3-PBC·HCl (1·HCl) reduced alcohol self-administration in primates18 while both 3-PBC·HCl (1·HCl) and βCCt (2) had been shown to reduce a`lcohol self-administration in both alcohol preferring (P) and high alcohol drinking (HAD) rats when given orally and systemically, a large scale synthesis of 3-PBC (1) and βCCt (2) via this short route was required. This synthesis of 3-PBC (1) and βCCt (2) is illustrated in Scheme 4. In regard to the synthesis of 3-PBC (1), the Buchwald-Hartwig amination step was scaled up to the 50 gram level (87 % yield); while the Heck-type cyclization reaction was performed on a 20 gram scale. The overall conversion of N-(2-chlorophenyl)-6-propoxypyridin-3-amine (5b) into 3-PBC (1) via this two-step route was 50 %, as compared to 8 % previously obtained.14, 15 The large scale synthesis of βCCt (2) via this process is shown in Scheme 4. The Buchwald-Hartwig amination of tert-butyl 5-bromopicolinate 1137 with 2-chloroaniline afforded the coupled diarylamine 12 in 94 % yield on 50 gram scale. Heck-type cyclization of 12 was carried out on 10 gram scale and afforded a mixture of βCCT (2) and its regioisomer 13 in 83 % yield. (Ratio of 2:13 = 2:1). The overall yield of βCCT (2) from 11 on 10 gram scale was 52 %, as compared to the previous yield of 35 %.16
Scheme 4.

Large-scale synthesis of 3-PBC (1) and βCCt (2) using the new route
Reagents and conditions: (a) 5 mol% Pd(OAc)2, 7.5 mol% X-Phos, 1.5 eq. Cs2CO3, toluene, 100°C, 15 h; (b) Pd(OAc)2, (t-Bu)3P.HBF4, K2CO3, DMA, 120°C, 16 h
In summary, a two-step route to the two anti-alcohol agents of biological interest, 3-PBC (1) and βCCT (2), has been developed in much improved yields as compared to their earlier reported syntheses. This two step synthesis of 3-PBC (1) and βCCT (2) is capable of scale-up which cuts down the number of steps in the reported routes from 6 and 5, respectively, to 2. The yield of 3-PBC (1) via this two-step route was 50 %, as compared to the previously reported 8 %; while that for βCCT (2) was 52 %, as compared to the previous yield of 35 %. In addition, to extend the SAR in vivo, different β-carboline analogs 9a–g were synthesized in superior yields. It is easy to separate the β-carboline and δ-carboline regioisomers from each other by flash chromatography. Further studies are underway to render this transformation regiospecific.
Experimental Section
General Methods
All reactions were performed in oven-dried round bottomed flasks or in resealable screw-cap test tubes or heavy-wall pressure vessels. Stainless steel syringes or cannulae were used to transfer air-sensitive liquids. Chemical shifts are reported in δ (ppm) relative to TMS in CDCl3 as internal standard (1H NMR) or the residual CHCl3 signal (13C NMR).
General procedure for the synthesis of diarylamines: Representative procedure for the synthesis of N-(2-chlorophenyl)-6-propoxypyridin-3-amine (5b)
5-Bromo-2-propoxypyridine 319 (0.65 g, 3 mmol), Pd(OAc)2 (33.7 mg, 0.15 mmol), Cs2CO3 (1.17 g, 3.6 mmol) and X-Phos (107 mg, 0.225 mmol) were added to a screw-cap vial. The vial was fitted with a rubber septum; evacuated and back-filled with argon. 2-Chloroaniline 4b (0.4 g, 3.15 mmol) was injected into the vial with a syringe under a positive pressure of argon. Toluene (10 mL) was added via a syringe. The rubber septum was replaced with a screw-cap and the sealed vial was introduced into a pre-heated oil bath at 100 °C. After 15 h the reaction mixture was filtered through a short pad of celite, washed with water, brine, dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash chromatography (silica gel; 20:1; hexanes/ethyl acetate) to afford 5b (0.73 g, 93 %) as a pale yellow oil: TLC (20% EtOAc/hexanes), Rf: 0.73; 1H NMR (300 MHz, CDCl3): δ 8.06 (d, J = 2.7 Hz, 1H), 7.49 (dd, J1 = 8.7 Hz; J2 = 2.7 Hz, 1H), 7.34 (dd, J1 = 8.1 Hz; J2 = 1.5 Hz, 1H), 7.09 (dd, J1 = 8.4 Hz; J2 = 1.2 Hz, 1H), 6.86 (dd, J1 = 8.4 Hz; J2 = 1.5 Hz, 1H), 6.76 (m, 2H), 5.90 (br, 1H), 4.27 (t, J = 6.9 Hz, 2H), 1.84 (m, 2H), 1.06 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 161.2, 142.3, 142.0, 135.3, 131.0, 129.6, 127.6, 120.1, 119.5, 113.5, 111.4, 67.8, 22.4, 10.6; HRMS–ESI (m/z) calcd for C14H16ClN2O [M+H]+: 263.0951, found: 263.0961.
N-phenyl-6-propoxypyridin-3-amine (5a)
Following the general procedure, 319 (0.43 g, 2 mmol) with aniline 4a (0.2 g, 2.10 mmol), Pd(OAc)2 (22.5 mg, 0.10 mmol), X-Phos (71.4 mg, 0.15 mmol), and Cs2CO3 (0.78 g, 2.4 mmol) after flash chromatography (silica gel, 20:1 hexanes/ethyl acetate) afforded 5a (0.43 g, 93 %): off-white solid; TLC (20% EtOAc/hexanes), Rf: 0.33; mp: 60.6–61.9 °C; 1H NMR (300 MHz, CDCl3): δ 8.01 (d, J = 2.7 Hz, 1H), 7.47 (dd, J1 = 8.7 Hz, J2 = 3.0 Hz, 1H), 7.25 (m, 2H), 7.87 (m, 3H), 6.74 (d, J = 8.7 Hz, 1H), 5.46 (br, 1H), 4.25 (t, J = 6.6 Hz, 2H), 1.82 (m, 2H), 1.06 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 160.3, 145.0, 139.9, 133.3, 132.6, 129.4, 119.9, 115.3, 111.1, 67.7, 22.4, 10.5; Anal.: calcd for C14H16N2O: C, 73.66; H, 7.06; N, 12.27; found: C, 73.75; H, 7.08; N, 12.10.
N-(2-chlorophenyl)-6-methoxypyridin-3-amine (8a)
Following the general procedure, 5-bromo-2-methoxypyridine 7a19 (0.5 g, 2.66 mmol) with 4b (0.36, 2.8 mmol), Pd(OAc)2 (29.9 mg, 0.13 mmol), X-Phos (89.1 mg, 0.20 mmol), and Cs2CO3 (1.04 g, 3.2 mmol) after flash chromatography (silica gel, 20:1 hexanes/ethyl acetate) afforded 8a (0.56 g, 89 %): yellow oil; TLC (20% EtOAc/hexanes), Rf: 0.62; 1H NMR (300 MHz, CDCl3): δ 8.08 (d, J = 2.7 Hz, 1H), 7.50 (dd, J1 = 8.7 Hz; J2 = 2.7 Hz, 1H), 7.35 (dd, J1 = 7.9 Hz; J2 = 1.3 Hz, 1H), 7.09 (t, J = 7.6 Hz, 1H), 6.87 (dd, J1 = 8.2 Hz; J2 = 1.3 Hz, 1H), 6.75 (m, 2H), 5.92 (br, 1H), 3.97 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 161.1, 142.0, 141.8, 135.1, 131.2, 129.5, 127.5, 120.1, 119.5, 113.5, 111.2, 55.5; HRMS–ESI (m/z): calcd for C12H12ClN2O [M+H]+: 235.0638, found: 235.0628.
N-(2-chlorophenyl)-6-ethoxypyridin-3-amine (8b)
Following the general procedure, 5-bromo-2-ethoxypyridine 7b19 (0.303 g, 1.5 mmol) with 4b (0.2 g, 1.575 mmol), Pd(OAc)2 (16.8 mg, 0.075 mmol), X-Phos (26.7 mg, 0.112 mmol), and Cs2CO3 (0.85 g, 1.8 mmol) after flash chromatography (silica gel, 20:1 hexanes/ethyl acetate) afforded 8b (0.35 g, 94 %): light brown solid; TLC (20% EtOAc/hexanes), Rf: 0.68; mp: 49.5–51.0°C; 1H NMR (300 MHz, CDCl3): δ 8.06 (d, J = 2.7 Hz, 1H), 7.49 (dd, J1 = 8.7 Hz; J2 = 2.8 Hz, 1H), 7.35 (dd, J1 = 7.9 Hz; J2 = 1.4 Hz, 1H), 7.09 (t, J = 7.9 Hz, 1H), 6.86 (dd, J1 = 8.4 Hz; J2 = 1.5 Hz, 1H), 6.77 (m, 2H), 5.90 (br, 1H), 4.37 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 160.9, 142.2, 141.8, 135.1, 130.9, 129.5, 127.5, 120.0, 119.4, 113.4, 111.3, 61.8, 14.6; HRMS–ESI (m/z): calcd for C13H14ClN2O [M+H]+: 249.0795, found: 249.0799.
N-(2-chlorophenyl)-6-isopropoxypyridin-3-amine (8c)
Following the general procedure, 5-bromo-2-isoprpoxypyridine 7c19 (0.22 g, 1.0 mmol) with 4b (0.134 g, 1.05 mmol), Pd(OAc)2 (11.2 mg, 0.05 mmol), X-Phos (35.7 mg, 0.075 mmol), and Cs2CO3 (0.39 g, 1.2 mmol) after flash chromatography (silica gel, 20:1 hexanes/ethyl acetate) afforded 8c (0.247 g, 94 %): pale yellow oil; TLC (20% EtOAc/hexanes), Rf: 0.65; 1H NMR (300 MHz, CDCl3): δ 8.06 (d, J = 2.7 Hz, 1H), 7.47 (dd, J1 = 8.7 Hz; J2 = 2.8 Hz, 1H), 7.34 (dd, J1 = 7.9 Hz; J2 = 1.4 Hz, 1H), 7.09 (t, J = 7.6 Hz, 1H), 6.87 (dd, J1 = 8.2 Hz; J2 = 1.4 Hz, 1H), 6.61 (m, 2H), 5.89 (br, 1H), 5.20 (m, 1H), 1.38 (d, J = 6.0 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 160.5, 142.2, 141.9, 135.2, 130.6, 129.5, 127.5, 120.0, 119.3, 113.4, 111.8, 68.1, 22.0; HRMS–ESI (m/z): calcd for C14H16ClN2O [M+H]+: 263.0951, found: 263.0963.
6-butoxy-N-(2-chlorophenyl)pyridin-3-amine (8d)
Following the general procedure, 5-bromo-2-butoxypyridine 7d19 (0.23 g, 1.0 mmol) with 4b (0.134 g, 1.05 mmol), Pd(OAc)2 (11.2 mg, 0.05 mmol), X-Phos (35.7 mg, 0.075 mmol), and Cs2CO3 (0.39 g, 1.2 mmol) after flash chromatography (silica gel, 20:1 hexanes/ethyl acetate) afforded 8d (0.25 g, 91 %): colorless oil; TLC (20% EtOAc/hexanes), Rf: 0.71; 1H NMR (300 MHz, CDCl3): δ 8.06 (d, J = 2.8 Hz, 1H), 7.48 (dd, J1 = 8.8 Hz; J2 = 2.8 Hz, 1H), 7.34 (dd, J1 = 7.9 Hz; J2 = 1.4 Hz, 1H), 7.09 (t, J = 8.4 Hz, 1H), 6.86 (dd, J1 = 8.2 Hz; J2 = 1.4 Hz, 1H), 6.77 (m, 2H), 5.90 (br, 1H), 4.31 (t, J = 6.7 Hz, 2H), 1.79 (m, 2H), 1.52 (m, 2H), 1.01 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 161.1, 142.2, 141.9, 135.2, 130.9, 129.5, 127.5, 120.0, 119.4, 113.4, 111.3, 65.9, 31.1, 19.2, 13.8; HRMS–ESI (m/z): calcd for C15H18ClN2O [M+H]+: 277.1108, found: 277.1102.
N-(2-chlorophenyl)-6-isobutoxypyridin-3-amine (8e)
Following the general procedure, 5-bromo-2-isoprpoxypyridine 7e19 (0.23 g, 1.0 mmol) with 4b (0.134 g, 1.05 mmol), Pd(OAc)2 (11.2 mg, 0.05 mmol), X-Phos (35.7 mg, 0.075 mmol), and Cs2CO3 (0.39 g, 1.2 mmol), after flash chromatography (silica gel, 20:1 hexanes/ethyl acetate) afforded 8e (0.257 g, 93 %): light green oil; TLC (20% EtOAc/hexanes), Rf: 0.69; 1H NMR (300 MHz, CDCl3): δ 8.06 (d, J = 2.2 Hz, 1H), 7.49 (dd, J1 = 8.7 Hz; J2 = 2.6 Hz, 1H), 7.35 (d, J = 7.9 Hz, 1H), 7.09 (t, J = 7.8 Hz, 1H), 6.86 (d, J = 8.2 Hz, 1H), 6.77 (m, 2H), 5.90 (br, 1H), 4.08 (d, J = 6.7 Hz, 2H), 2.12 (m, 1H), 1.05 (d, J = 6.7 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 161.2, 142.2, 141.9, 135.2, 130.9, 129.5, 127.5, 120.0, 119.4, 113.4, 111.3, 72.5, 30.7, 19.2; HRMS–ESI (m/z): calcd for C15H18ClN2O [M+H]+: 277.1108, found: 277.1099.
6-(tert-butoxy)-N-(2-chlorophenyl)pyridin-3-amine (8f)
Following the general procedure, 2-(benzyloxy)-5-bromopyridine 7f19 (0.229 g, 1.0 mmol) with 4b (0.134 g, 1.05 mmol), Pd(OAc)2 (11.2 mg, 0.05 mmol), X-Phos (35.7 mg, 0.075 mmol), and Cs2CO3 (0.39 g, 1.2 mmol) after flash chromatography (silica gel, 20:1 hexanes/ethyl acetate) afforded 8f (0.252 g, 91 %): colorless oil; TLC (20% EtOAc/hexanes), Rf: 0.81; 1H NMR (300 MHz, CDCl3): δ 8.05 (d, J = 2.8 Hz, 1H), 7.44 (dd, J1 = 8.7 Hz; J2 = 2.7 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.10 (t, J = 8.7 Hz, 1H), 6.92 (d, J = 8.1 Hz, 1H), 6.77 (t, J = 7.2 Hz, 1H), 6.70 (d, J = 8.7 Hz, 1H), 5.89 (br, 1H), 1.60 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 160.6, 141.7, 141.6, 134.2, 130.9, 129.5, 127.5, 120.1, 119.4, 113.9, 113.6, 79.5, 28.6; HRMS–ESI (m/z): calcd for C15H18ClN2O [M+H]+: 277.1108, found: 277.1116.
6-(benzyloxy)-N-(2-chlorophenyl)pyridin-3-amine (8g)
Following the general procedure, 2-(benzyloxy)-5-bromopyridine 7g19 (0.263 g, 1.0 mmol) with 4b (0.134 g, 1.05 mmol), Pd(OAc)2 (11.2 mg, 0.05 mmol), X-Phos (35.7 mg, 0.075 mmol), and Cs2CO3 (0.39 g, 1.2 mmol) after flash chromatography (silica gel, 20:1 hexanes/ethyl acetate) afforded 8g (0.28 g, 90 %): off-white solid; TLC (10% EtOAc/hexanes), Rf: 0.63; mp: 101.2–102.3°C; 1H NMR (300 MHz, CDCl3): δ 8.09 (d, J = 2.7 Hz, 1H), 7.51 (m, 3H), 7.39 (m, 4H), 7.10 (t, J = 8.4 Hz, 1H), 6.90 (dd, J1 = 8.1 Hz, J2 = 1.2 Hz, 1H), 6.86 (d, J = 8.7 Hz, 1H), 6.78 (m, J1 = 7.5 Hz, J2 = 1.5 Hz, 1H), 5.92 (br, 1H), 5.40 (s, 2H); 13C NMR (75 MHz, CDCl3): δ 159.9, 141.2, 140.3, 136.8, 135.3, 131.9, 129.6, 128.8, 128.4, 127.9, 127.6, 120.6, 120.0, 114.1, 111.8, 68.4; Anal.: calcd for C18H15ClN2O 0.05 CH3(CH2)4CH3: C, 69.76; H, 5.02; N, 8.89; found: C, 69.98; H, 4.86; N, 9.01.
tert-butyl 5-[(2-chlorophenyl)amino]picolinate (12)
Following the general procedure, tert-butyl 5-bromopicolinate 1137 (50 g, 194 mmol) with 4b (26 g, 203 mmol), Pd(OAc)2 (2.18 g, 9.7 mmol), X-Phos (6.9 g, 14.55 mmol), and Cs2CO3 (75.9 g, 233 mmol) after flash chromatography (silica gel, 5:1 hexanes/ethyl acetate) afforded 12 (55.6 g, 94 %): lustrous off-white solid; TLC (50% EtOAc/hexanes), Rf: 0.46; mp: 147.8–149.4°C; 1H NMR (300 MHz, CDCl3): δ 8.51 (d, J = 2.7 Hz, 1H), 7.99 (d, J = 8.7 Hz, 1H), 7.44 (d, J = 8.1 Hz, 2H), 7.38 (d, J = 7.8 Hz, 1H), 7.24 (t, J = 7.2 Hz, 1H), 7.01 (t, J = 8.1 Hz, 1H), 6.34 (br, 1H), 1.63 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 164.1, 141.8, 141.3, 139.6, 137.4, 130.3, 127.7, 125.8, 124.4, 123.5, 122.5, 118.5, 81.7, 28.2; Anal.: calcd for C16H17ClN2O2: C, 63.05; H, 5.62; N, 9.19; found: C, 62.67; H, 5.65; N, 8.95.
General procedure for Heck cyclization: Representative procedure for the synthesis of 3-propoxy-9H-pyrido[3,4-b]indole (3-PBC; 1) and 2-propoxy-5H-pyrido[3,2-b]indole (6)
Pd(OAc)2 (22.4 mg, 0.1 mmol), (t-Bu)3P HBF4 (58 mg, 0.2 mmol), 5b (263 mg, 1.0 mmol) and K2CO3 (276 mg, 2.0 mmol) were added to a screw-cap vial. The vial was fitted with a rubber septum, evacuated and back-filled with argon. Degassed DMA (4.0 mL) was added via a syringe. The rubber septum was replaced with a screw cap and this sealed tube was introduced in a preheated oil bath at 120 °C. After stirring for 16 h, the reaction mixture was allowed to cool to rt. The reaction mixture was passed through a short pad of celite which was further washed with ethyl acetate until no product (TLC; silica gel) was detected in the eluent. The combined filtrates were washed with water, brine, dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash chromatography (silica gel; 5:1 hexanes/ethyl acetate) to afford 3-PBC (1) (127 mg): colorless crystals; mp 119.3–120.5°C.; Anal. calcd for C14H14N2O·0.33 H2O: C, 72.43; H, 6.36; N, 12.07; found: C, 72.48; H, 6.49; N, 11.78. A hydrochloride salt of 1 was prepared by the reported method to obtain 3-PBC·HCl (1·HCl): yellow solid; mp 194.7–195.6°C (lit.15 mp 194.0–195.0°C). The data for this compound matched in all aspects (1H NMR, mp) with that reported in the literature.15
And 6 (72 mg): white solid: TLC (50% EtOAc/hexanes) Rf: 0.63; mp 123.2–124.3 °C; 1H NMR (300 MHz, CDCl3): δ 8.27 (d, J = 7.8 Hz, 1H), 7.99 (br, 1H), 7.65 (d, J = 8.7 Hz, 1H), 7.46 (m, 2H), 7.27 (m, J1 = 7.1 Hz, J2 = 1.8 Hz, 1H), 6.84 (d, J = 8.7 Hz, 1H), 4.45 (t, J = 6.6 Hz, 2H), 1.91 (m, 2H), 1.11 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 159.7, 140.2, 138.4, 128.3, 126.8, 122.6, 121.5, 120.6, 119.8, 111.3, 108.8, 67.8, 22.7, 10.8; Anal. calcd for C14H14N2O: C, 74.31; H, 6.24; N, 12.38; found: C, 74.17; H, 6.30; N, 12.30.
[Combined yield = 199 mg, 88 %].
3-methoxy-9H-pyrido[3,4-b]indole (9a) and 2-methoxy-5H-pyrido[3,2-b]indole (10a)
Following the general procedure, 8a (130 mg, 0.55 mmol), Pd(OAc)2 (12.4 mg, 0.055 mmol), (t-Bu)3P HBF4 (31.9 mg, 0.11 mmol) and K2CO3 (152 mg, 1.11 mmol) after flash chromatography (silica gel, 5:1 hexanes/ethyl acetate) afforded 9a (48 mg): off-white solid; HRMS–ESI (m/z): calcd for C12H11N2O [M+H]+: 199.0871, found: 199.0877. Hydrochloride salt of 9a: light brown solid; mp: 214.8–216.0°C (lit.15 mp 215.0–217.0°C). The data for this compound matched in all aspects (1HNMR, mp) with that reported in the literature.15
And 10a (40 mg): light brown solid; TLC (50% EtOAc/hexanes), Rf: 0.72; mp: 94.5–97.0°C; 1H NMR (300 MHz, CDCl3): δ 8.28 (d, J = 7.9 Hz, 1H), 8.01 (br, 1H), 7.67 (d, J = 8.7 Hz, 1H), 7.46 (m, 2H), 7.29 (m, J1 = 6.7 Hz, J2 = 2.1 Hz, 1H), 6.85 (d, J = 8.7 Hz, 1H), 4.12 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 159.6, 140.0, 138.1, 129.3, 128.2, 126.7, 121.3, 120.4, 119.9, 111.1, 108.5, 53.5; HRMS–ESI (m/z): calcd for C12H11N2O [M+H]+: 199.0871, found: 199.0876.
[Combined yield = 88 mg, 81 %].
3-ethoxy-9H-pyrido[3,4-b]indole (9b) and 2-ethoxy-5H-pyrido[3,2-b]indole (10b)
Following the general procedure, 8b (75 mg, 0.3 mmol), Pd(OAc)2 (6.7 mg, 0.03 mmol), (t-Bu)3P HBF4 (17.4 mg, 0.06 mmol) and K2CO3 (83 mg, 0.6 mmol) after flash chromatography (silica gel, 5:1 hexanes/ethyl acetate) afforded 9b (30 mg) as yellow-brown solid; HRMS–ESI (m/z): calcd for C13H13N2O [M+H]+: 213.1028, found: 213.1018. Hydrochloride salt of 9b was a yellow solid; mp: 222.0–223.3°C (lit.15 mp 221.0–223.0°C). The data for this compound matched in all aspects (1HNMR, mp) with that reported in the literature.15
And 10b (24 mg) as off-white crystals; TLC (20% EtOAc/hexanes), Rf: 0.35; mp: 130.4–131.1°C; 1H NMR (300 MHz, CDCl3): δ 8.26 (d, J = 7.8 Hz, 1H), 7.91 (br, 1H), 7.68 (d, J = 8.7 Hz, 1H), 7.45 (m, 2H), 7.27 (m, 2H), 6.83 (d, J = 8.7 Hz, 1H), 4.56 (q, J = 6.9 Hz, 2H), 1.49 (t, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 159.3, 140.2, 128.3, 126.9, 122.4, 121.6, 120.7, 119.8, 111.2, 108.6, 61.9, 14.9; HRMS–ESI (m/z): calcd for C13H13N2O [M+H]+: 213.1028, found: 213.1019.
[Combined yield = 54 mg, 85 %].
3-isopropoxy-9H-pyrido[3,4-b]indole (9c) and 2-isopropoxy-5H-pyrido[3,2-b]indole (10c)
Following the general procedure, 8c (263 mg, 1.0 mmol), Pd(OAc)2 (22.5 mg, 0.1 mmol), (t-Bu)3P HBF4 (58 mg, 0.2 mmol) and K2CO3 (276 mg, 2.0 mmol) after flash chromatography (silica gel, 5:1 hexanes/ethyl acetate) afforded 9c (131 mg) as an off-white solid; HRMS–ESI (m/z): calcd for C14H15N2O [M+H]+: 227.1184, found: 227.1176. Hydrochloride salt of 9c: brown solid; mp: 169.5–171.3°C (lit.34 mp 168.0–172.0°C). The data for this compound matched in all aspects (1H NMR, mp) with that reported in the literature.34
And 10c (72 mg) as an off-white solid; TLC (20% EtOAc/hexanes), Rf: 0.38; mp: 105.6–107.7°C; 1H NMR (300 MHz, CDCl3): δ 8.28 (d, J = 7.8 Hz, 1H), 8.04 (br, 1H), 7.61 (d, J = 8.7 Hz, 1H), 7.44 (m, 2H), 7.27 (t, J = 7.4 Hz, 1H), 6.79 (d, J = 8.7 Hz, 1H), 5.56 (m, J = 6.3 Hz, 1H), 1.46 (d, J = 6.3 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 158.7, 140.1, 138.2, 128.1, 126.6, 122.4, 121.3, 120.4, 119.6, 111.1, 109.2, 68.0, 22.1; HRMS–ESI (m/z): calcd for C14H15N2O [M+H]+: 227.1184, found: 227.1173.
[Combined yield = 203 mg, 90 %].
3-butoxy-9H-pyrido[3,4-b]indole (9d) and 2-butoxy-5H-pyrido[3,2-b]indole (10d)
Following the general procedure, 8d (125 mg, 0.45 mmol), Pd(OAc)2 (10.1 mg, 0.045 mmol), (t-Bu)3P HBF4 (26.1 mg, 0.09 mmol) and K2CO3 (125 mg, 0.9 mmol) after flash chromatography (silica gel, 5:1 hexanes/ethyl acetate) afforded 9d (48.2 mg) as a buff colored solid; HRMS–ESI (m/z): calcd for C15H17N2O [M+H]+: 241.1341, found: 241.1346. Hydrochloride salt of 9d: light yellow solid; mp: 178.0–180.0°C (lit.34 mp 178.0–181.0°C). The data for this compound matched in all aspects (1HNMR, mp) with that reported in the literature.34
And 10d (35.8 mg): off-white solid; TLC (20% EtOAc/hexanes), Rf: 0.35; mp: 113.3–115.5°C; 1H NMR (300 MHz, CDCl3): δ 8.28 (d, J = 7.8 Hz, 1H), 7.05 (br, 1H), 7.63 (d, J = 8.7 Hz, 1H), 7.43 (m, 2H), 7.27 (m, J1 = 7.8 Hz, J2 = 1.5 Hz, 1H), 6.83 (d, J = 8.7 Hz, 1H), 4.50 (t, J = 6.6 Hz, 2H), 1.87 (m, 2H), 1.57 (m, 2H), 1.04 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 159.5, 140.0, 138.2, 128.2, 126.6, 122.3, 121.3, 120.4, 119.6, 111.1, 108.6, 65.8, 31.3, 19.3, 13.9; HRMS–ESI (m/z): calcd for C15H17N2O [M+H]+: 241.1341, found: 241.1347.
[Combined yield = 84 mg, 87 %].
3-Isobutoxy-9H-pyrido[3,4-b]indole (9e) and 2-isobutoxy-5H-pyrido[3,2-b]indole (10e)
Following the general procedure, 8e (139 mg, 0.50 mmol), Pd(OAc)2 (11.3 mg, 0.05 mmol), (t-Bu)3P HBF4 (29 mg, 0.10 mmol) and K2CO3 (138 mg, 1.0 mmol) after flash chromatography (silica gel, 5:1 hexanes/ethyl acetate) afforded 9e (63 mg) as an off-white crystalline solid; HRMS–ESI (m/z): calcd for C15H17N2O [M+H]+: 241.1341, found: 241.1337. Hydrochloride salt of 9e: light yellow solid; mp: 221.5–223.0°C (lit.35 mp 222.0–223.0°C). The data for this compound matched in all aspects (1HNMR, mp) with that reported in the literature.35
And 10e (45 mg): off-white solid; TLC (20% EtOAc/hexanes), Rf: 0.31; mp: 120.5–121.4°C; 1H NMR (300 MHz, CDCl3): δ 8.28 (d, J = 7.8 Hz, 1H), 7.04 (br, 1H), 7.64 (d, J = 8.7 Hz, 1H), 7.45 (m, 2H), 7.27 (m, J1 = 7.9 Hz, J2 = 1.5 Hz, 1H), 6.84 (d, J = 8.7 Hz, 1H), 4.27 (d, J = 6.7 Hz, 2H), 2.20 (m, 2H), 1.10 (d, J = 6.7 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 159.6, 140.0, 138.1, 128.2, 126.6, 122.3, 121.3, 120.4, 119.6, 111.1, 108.6, 72.4, 28.1, 19.4; HRMS–ESI (m/z): calcd for C15H17N2O [M+H]+: 241.1341, found: 241.1335.
[combined yield = 108 mg, 93 %].
3-(tert-butoxy)-9H-pyrido[3,4-b]indole (9f) and 2-(tert-butoxy)-5H-pyrido[3,2-b]indole (10f)
Following the general procedure, 8f (100 mg, 0.36 mmol), Pd(OAc)2 (8.0 mg, 0.036 mmol), (t-Bu)3P HBF4 (21 mg, 0.072 mmol) and K2CO3 (100 mg, 0.72 mmol) after flash chromatography (silica gel, 5:1 hexanes/ethyl acetate) afforded 9f (48 mg): colorless crystals; TLC (50% EtOAc/hexanes), Rf: 0.30; mp: 207.9–209.8°C (lit.36 208.0–212.0°C); HRMS–ESI (m/z): calcd for C15H17N2O [M+H]+: 241.1341, found: 241.1339. The data for this compound matched in all aspects (1HNMR, mp) with that reported in the literature.36
And 10f (25 mg): light brown solid; TLC (50% EtOAc/hexanes), Rf: 0.59; mp: 110.5–112.2°C; 1H NMR (300 MHz, CDCl3): δ 8.26 (d, J = 7.8 Hz, 1H), 7.94 (br, 1H), 7.63 (d, J = 8.7 Hz, 1H), 7.45 (m, 2H), 7.27 (t, J = 6.3 Hz, 1H), 6.80 (d, J = 8.7 Hz, 1H), 1.69 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 158.7, 140.1, 138.3, 128.2, 126.6, 122.7, 120.7, 120.4, 119.6, 111.8, 111.0, 79.3, 29.8; HRMS–ESI (m/z): calcd for C15H17N2O [M+H]+: 241.1341, found: 241.1349.
[Combined yield = 73.0 mg, 85 %].
3-(benzyloxy)-9H-pyrido[3,4-b]indole (9g) and 2-(benzyloxy)-5H-pyrido[3,2-b]indole (10g)
Following the general procedure, 8g (65 mg, 0.21 mmol), Pd(OAc)2 (4.7 mg, 0.021 mmol), (t-Bu)3P HBF4 (12.2 mg, 0.042 mmol) and K2CO3 (58 mg, 0.42 mmol) after flash chromatography (silica gel, 5:1 hexanes/ethyl acetate) afforded 9g (30 mg): off-white solid; HRMS–ESI (m/z): calcd for C18H15N2O [M+H]+: 275.1184, found: 275.1194. Hydrochloride salt of 9g: yellow solid; mp: 197.8–199.2°C (lit.35 mp 198.0–199.0°C). The data for this compound matched in all aspects (1HNMR, mp) with that reported in the literature.35
And 10g (20 mg): white solid; TLC (20% EtOAc/hexanes), Rf: 0.23; mp: 137.5–139.5°C; 1H NMR (300 MHz, CDCl3): δ 8.30 (d, J = 8.1 Hz, 1H), 7.96 (br, 1H), 7.67 (d, J = 8.7 Hz, 1H), 7.60 (d, J = 7.2 Hz, 2H), 7.40 (m, 6H), 6.91 (d, J = 8.7 Hz, 1H), 5.60 (s, 2H); 13C NMR (75 MHz, CDCl3): δ 159.0, 140.1, 138.1, 137.9, 128.4, 128.3, 127.7, 126.8, 122.4, 121.4, 120.5, 119.8, 111.2, 108.9, 67.7; HRMS–ESI (m/z): calcd for C18H15N2O [M+H]+: 275.1184, found: 275.1196.
[Combined yield = 50 mg, 87 %].
Large scale synthesis of 3-PBC (1) and βCCt (2)
Step 1: General procedure: Representative procedure for the synthesis of N-(2-chlorophenyl)-6-propoxypyridin-3-amine (5b)
Pd(OAc)2 (2.6 g, 11.6 mmol), 3 (50 g, 231.4 mmol), Cs2CO3 (90.5 g, 277.7 mmol) and X-Phos (8.3 g, 17.3 mmol) were added to a three-neck flask with a reflux condenser. The flask was evacuated and back-filled with argon. 4b (31 g, 25.6 mL, 243 mmol) was injected into the flask with a syringe. Toluene (500 mL) was added via a cannula and the flask was introduced into a pre-heated oil bath at 100°C. After 15 h at 100°C the reaction mixture was cooled down to rt, filtered through a short pad of celite, and the pad was washed with ethyl acetate. The combined organic eluents were washed with water, brine, dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica gel (20:1; hexanes/ethyl acetate) to afford 5b (52.9 g, 87 %).
Large scale synthesis of tert-butyl-5-[(2-chlorophenyl)amino]picolinate (12)
Following the general procedure, the tert-butyl 5-bromopicolinate 1137 (50 g, 194 mmol) was reacted with 4b (26 g, 25.6 mL, 203 mmol), Pd(OAc)2 (2.18 g, 9.7 mmol), Cs2CO3 (75.9 g, 233 mmol) and X-Phos (6.9 g, 14.55 mmol). Purification by flash chromatography (silica gel, 5:1 hexanes/ethyl acetate) afforded 12 (55.6 g, 94 %).
Step 2 general procedure: Representative procedure for large scale synthesis of 3-propoxy-9H-pyrido[3,4-b]indole (3-PBC; 1)
5b (20 g, 76.1 mmol), Pd(OAc)2 (1.71 g, 7.61 mmol), (t-Bu)3P HBF4 (4.41 g, 15.22 mmol) and K2CO3 (21 g, 152.2 mmol) were added to a heavy-wall pressure vessel (350 mL). The vessel was fitted with a rubber septum, evacuated and back-filled with argon. Degassed DMA (250 mL) was added to this vial via a cannula. The rubber septum was replaced with a Teflon screw cap and this sealed vessel was introduced in a pre-heated oil bath at 120°C. After stirring at this temperature for 16 h, the reaction mixture was allowed to cool to rt and was passed through a short pad of celite, which was further washed with ethyl acetate until no product (TLC; silica gel) was detected in the eluent. The combined filtrate was washed with water, brine, dried (Na2SO4) and concentrated under reduced pressure. The crude solid was purified by flash chromatography (5:1; hexanes/ethyl acetate) to afford 3-PBC (1) (10 g, 58 %).
Large scale synthesis of tert-butyl 9H-pyrido[3,4-b]indole-3-carboxylate (βCCt; 2)
Following the general procedure, 12 (10 g, 32.8 mmol), Pd(OAc)2 (735 mg, 3.28 mmol), (t-Bu)3P HBF4 (1.9 g, 6.56 mmol) and K2CO3 (9.06 g, 65.6 mmol) after flash chromatography (silica gel, 1:1 hexanes/ethyl acetate) afforded βCCt (2) (4.87 g, 55.6 %): white solid; mp 302.5–303.4°C (lit.17 mp 301–303°C). The spectral data for this compound matched in all aspects (1H NMR) with that reported in the literature.17
The regioisomer tert-butyl 5H-pyrido[3,2-b]indole-2-carboxylate (13): fluffy white solid (2.43 g, 27.4%); TLC (2:1 EtOAc/hexanes), Rf: 0.53; mp: 218–219.2°C; 1H NMR (300 MHz, CDCl3): δ 8.89 (br, 1H), 8.45 (d, J = 7.8 Hz, 1H), 8.19 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.52 (m, 2H), 7.31 (t, J = 7.2 Hz, 1H), 1.69 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 165.1, 142.8, 141.6, 141.2, 134.6, 128.6, 122.5, 122.4, 122.0, 120.9, 117.1, 111.3, 81.8, 28.3; Anal.: C16H16ClN2O2·0.1 CH2Cl2: C, 69.89; H, 5.90; N, 10.13; found: C, 69.93; H, 6.07; N, 9.89.
Supplementary Material
Acknowledgments
We thank the NIMH (MH 046851) (JMC), and NIAAA (AA016179) as well as the Lynde and Harry Bradley Foundation for generous financial support. The crystallographic analysis was carried out at the Naval Research Laboratory.
Footnotes
Supporting Information Available: Copies of spectra and crystallographic information files (CIFs). This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Sieghart W, Ernst M. Curr Med Chem– Central Nervous Syst Agents. 2005;5:217–242. [Google Scholar]
- 2.Clayton T, Chen JL, Ernst M, Richter L, Cromer BA, Morton HN, Kaczorowski CC, Helmstetter FJ, Furtmuller R, Ecker G, Parker MW, Sieghart W, Cook JM. Curr Top Med Chem. 2007;14:2755–2775. doi: 10.2174/092986707782360097. [DOI] [PubMed] [Google Scholar]
- 3.Kranzler HR. Alcohol. 2000;35:537–547. doi: 10.1093/alcalc/35.6.537. [DOI] [PubMed] [Google Scholar]
- 4.Johnson BA, Ait-Daoud N. Psychopharmacology. 2000;149:327–344. doi: 10.1007/s002130000371. [DOI] [PubMed] [Google Scholar]
- 5.Koob GF, Roberts AJ, Schulteis G. Alcohol Clin Exp Res. 1998;22:3–9. [PubMed] [Google Scholar]
- 6.June HL, Cason CR, Cheatham G, Liu RY, Gan T, Cook JM. Brain Res. 1998;794:103–118. doi: 10.1016/s0006-8993(98)00222-4. [DOI] [PubMed] [Google Scholar]
- 7.McBride WJ, Li T. Crit Rev Neurobiol. 1998;12:339–369. doi: 10.1615/critrevneurobiol.v12.i4.40. [DOI] [PubMed] [Google Scholar]
- 8.Allain H, Belliard S, Decertaines J, Bentueferrer D, Bureau M, Lacroix P. Dementia. 1993;4:347–352. doi: 10.1159/000107344. [DOI] [PubMed] [Google Scholar]
- 9.Heimer L, Alheid GF. In: The basal forebrain: anatomy and function. Napier TC, Kalivas PW, Hanin I, editors. Plenum Press; New York: 1991. pp. 1–42. [Google Scholar]
- 10.Harvey SC, Foster KL, McKay PF, Carroll MR, Seyoum R, Woods JE, Grey C, Jones CM, McCane S, Cummings R, Mason D, Ma CR, Cook JM, June HL. J Neurosci. 2002;22:3765–3775. doi: 10.1523/JNEUROSCI.22-09-03765.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carroll M, Woods JE, II , Seyoum RA, June HL. Alcohol Clin Exp Res. 2001;25:12A. [Google Scholar]
- 12.Cox ED, Hagen TJ, Mckernan RM, Cook JM. Med Chem Res. 1995;5:710–718. [Google Scholar]
- 13.Rowlett JK, Spealman RD, Lelas S, Cook JM, Yin WY. Psychopharmacol. 2003;165:209–215. doi: 10.1007/s00213-002-1275-z. [DOI] [PubMed] [Google Scholar]
- 14.Hagen TJ, Guzman F, Schultz C, Cook JM, Skolnick P, Shannon HE. Heterocycles. 1986;24:2845–2855. [Google Scholar]
- 15.Allen MS, Hagen TJ, Trudell ML, Codding PW, Skolnick P, Cook JM. J Med Chem. 1988;31:1854–1861. doi: 10.1021/jm00117a029. [DOI] [PubMed] [Google Scholar]
- 16.Yin W, Sarma PVVS, Ma J, Han D, Chen JL, Cook JM. Tetrahedron Lett. 2005;46:6363–6368. [Google Scholar]
- 17.Yin W, Majumder S, Clayton T, Petrou S, Van Linn ML, Namjoshi OA, Ma C, Cromer BA, Roth BL, Platt DM, Cook JM. Bioorg Med Chem. 2010;18:7548–7564. doi: 10.1016/j.bmc.2010.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weerts E, Kaminski B, Yin W, Sarma PVVS, Cook JM. Proceedings of the College on Problems of Drug Dependence; Orlando, FL. June 19–25, 2005; Philadelphia, PA: The College on Problems of Drug Dependence; [Google Scholar]
- 19.Jansen J-R, Fuesslein M, Hallenbach W, Ort O, Arnold C, Franken E-M, Malsam O, Reckmann U, Sanwald E, Goergens U. 2009068194. PCT Int Patent. 2009 June 4;
- 20.Charles MD, Schultz P, Buchwald SL. Org Lett. 2005;7:3965–3968. doi: 10.1021/ol0514754. [DOI] [PubMed] [Google Scholar]
- 21.Anderson KW, Tundel RE, Ikawa T, Altman RA, Buchwald SL. Angew Chem, Int Ed. 2006;45:6523–6527. doi: 10.1002/anie.200601612. [DOI] [PubMed] [Google Scholar]
- 22.Muci AR, Buchwald SL. Top Curr Chem. 2002;219:131–209. [Google Scholar]
- 23.Braese S, De Meijere A, EdNegishi E-I. From Handbook of Organopalladium Chemistry for Organic Synthesis. 2002;1:1223–1254. [Google Scholar]
- 24.Zeni G, Larock RC. Chem Rev. 2006;106:4644–4680. doi: 10.1021/cr0683966. [DOI] [PubMed] [Google Scholar]
- 25.Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem Rev. 2007;107:5318–5365. doi: 10.1021/cr068006f. [DOI] [PubMed] [Google Scholar]
- 26.Campeau L-C, Parisien M, Jean A, Fagnou K. J Am Chem Soc. 2006;128:581–590. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]
- 27.Liu Z, Larock RC. Tetrahedron. 2007;63:347–355. doi: 10.1016/j.tet.2006.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liégault B, Lee D, Huestis MP, Stuart DR, Fagnou K. J Org Chem. 2008;73:5022–5028. doi: 10.1021/jo800596m. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe T, Oishi S, Fujii N, Ohno H. J Org Chem. 2009;74:4720–4726. doi: 10.1021/jo9003376. [DOI] [PubMed] [Google Scholar]
- 30.Bedford RB, Betham M. J Org Chem. 2006;71:9403–9410. doi: 10.1021/jo061749g. [DOI] [PubMed] [Google Scholar]
- 31.Budén ME, Vaillard VA, Martin SE, Rossi RA. J Org Chem. 2009;74:4490–4498. doi: 10.1021/jo9006249. [DOI] [PubMed] [Google Scholar]
- 32.Hostyn S, Baelen GT, Lemiére GLF, Maes BUW. Adv Synth Catal. 2008;350:2653–2660. [Google Scholar]
- 33.Sridharan M, Martín MA, Menéndez JC. Eur J Org Chem. 2009;2009:4614–4621. [Google Scholar]
- 34.Allen MS, Tan Y-C, Trudell ML, Narayanan K, Schindler LR, Martin MJ, Schultz C, Hagen TJ, Koehler KF, Codding PW, Skolnick P, Cook JM. J Med Chem. 1990;33:2343–2357. doi: 10.1021/jm00171a007. [DOI] [PubMed] [Google Scholar]
- 35.Allen MS, LaLoggia AJ, Dorn LJ, Martin MJ, Costantino G, Hagen TJ, Koehler KF, Skolnick P, Cook JM. J Med Chem. 1992;35:4001–4010. doi: 10.1021/jm00100a004. [DOI] [PubMed] [Google Scholar]
- 36.Huth A, Rahtz D, Seidelmann D, Schmiechen R, Biere H, Braestrup CT. 3240514. German Patent DE. 1984 May 3;
- 37.Bailey JM, Bruton G, Huxley A, Milner PH, Orlek BS. 2005014571. PCT Int Patent. 2005 February 17;
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.