

Abstract
Background
Oxidative stress, resulting in a marked increase in the level of oxygen free radicals (OFR), has been implicated in the etiology of diabetic neuropathy (DN). Antioxidant enzymes may protect against the rapid onset and progression of DN, by reducing the excess of OFR and peroxide. Mutations and polymorphisms in the genes encoding such enzymes may therefore result in predisposition to DN. We investigated the role of genes encoding two antioxidant enzymes, mitochondrial (Mn-SOD) and extracellular (EC-SOD) superoxide dismutase, in DN pathogenesis in a Russian population. We studied Ala(-9)Val and Ile58Thr polymorphisms of the Mn-SOD gene and Arg213Gly dimorphism of the EC-SOD gene in type 1 diabetic patients with (n = 82) and without DN (n = 84).
Results
We developed and used a new polymerase chain reaction (PCR) assays for rapid detection of polymorphisms. These assays involved the use of mismatch PCR primers to create restriction sites in the amplified product only in presence of the polymorphic base. The PCR product was than digested with BshTI, Eco32I or Eco52I to detect Ala(-9)Val, Ile58Thr or Arg213Gly polymorphic site respectively. The frequencies of the Ala allele (50.6% vs. 68.5%, p < 0.002) and the Ala/Ala genotype (17.1% vs. 39.3%, p < 0.005) of the Mn-SOD gene were significantly lower in DN patients than in diabetic subjects without DN. In contrast, the Val allele (49.4% vs. 31.5%, p < 0.002) and the Val/Val genotype (15.9% vs. 2.4%, p < 0.01) were significantly more frequent in the DN patients than in the control group.
Conclusions
Ala(-9)Val substitution in the Mn-SOD gene was associated with DN in a Russian population
Background
Oxidative stress has been implicated in the etiology of long-term diabetic complications, including peripheral neuropathy [1]. Oxygen free radicals (OFR) may damage neurons by causing nerve lipid peroxidation, the breakdown of mitochondrial DNA and inhibition of the respiratory chain, and the cross-linking of the neurofilament protein [2-4]. Treatment with antioxidants (e.g. probucol, alpha-lipoic acid) decreases lipid peroxidation and oxidative stress in neural tissues and improves the condition of rats with streptozotocin-induced diabetic neuropathy [5,6].
Antioxidant enzyme activity is low in in peripheral nerves and even lower in diabetic nerves, possibly due non-enzymatic glycation and autooxidation of the glycated protein [7,8]. Antioxidant enzymes may protect against the rapid onset and progression of diabetic neuropathy (DN) by reducing the excess of both OFR and peroxide. Defects and mutations in the genes encoding these enzymes may therefore lead to suspectibility to DN.
Superoxide dismutase (SOD) is the key antioxidant enzyme involved in the detoxication of superoxide radicals. SOD is a metalloprotein and its manganese form (Mn-SOD) is present in mitochondria. Two other forms containing cooper and zinc (CuZn) have cytoplasmic or extracellular (EC-SOD) location.
An alanine (GCT) to valine (GTT) substitution at position -9 in the signal peptide of human Mn-SOD has been shown to change the structural conformation of the mitochondrial targeting sequence of the enzyme. Thissubstitution may lead to misdirected intracellular trafficking, followed by changes in Mn-SOD activity in the mitochondria [9,10]. Associations have been found between the Ala(-9)Val dimorphism in the SOD2 gene and such human neurodegenerative and ageing disorders as Parkinson's disease [9], tardive dyskinesia [11], sporadic motor neuron disease [12], and nonfamilial idiopathic dilated cardiomyopathy [13].
The Ile58Thr amino acid exchange destabilizes the tetrameric interface of Mn-SOD and reduces its enzymatic activity [14]. This finding suggests that the Ile58Thr polymorphism may be associated with neurodegenerative diseases involving a decrease in Mn-SOD levels [15]. However, no association between the Ile58Thr polymorphism and Parkinson's disease was found in German patients [16].
Extracellular superoxide dismutase (EC-SOD) has an amino acid substitution Arg213Gly in the heparin-binding domain [17]. The glycine variant of the enzyme is responsible for high EC-SOD levels in serum [18,19] that are correlated with a decrese in nitric oxide production in epithelial cells [20] and various other metabolic cardiovascular risk factors [21]. A relationship was found between the Arg213Gly dimorphism and familial amyloidotic (non-diabetic) in a Japanese population, involving greater dissociation of EC-SOD from the vascular wall followed by extensive oxidative stress and cardiac, renal, and autonomic nervous system failure due to massive amyloid deposition [22].
In this study, we investigated whether allelic variants of the Mn-SOD and EC-SOD genes were involvedin the etiology of DN in Russian type 1 diabetic patients. To do this, we developed new methods for the simple and rapid detection of polymorphisms in the Mn-SOD and EC-SOD genes.
Materials and Methods
Subjects
In this study, 88 healthy Russian donors selected at random (59 males and 29 females, mean age 28.3 ± 8.4 years, mean ± SEM) were examined. Healthy subjects had no autoimmune, cardiovascular, or other diseases. All study participants lived in Moscow or the Moscow region. Blood samples were collected in the Department of Endocrinology and Diabetology of the Russian Academy for Advanced Medical Studies. This study was approved by the Academy Review Board and was carried out in accordance with the principles of the second Helsinki Declaration. Informed consent was obtained from all subjects before participation in this study.
We studued a total of 166 genetically unrelated Russian patients affected with type 1 diabetes mellitus (97 males and 69 females, mean age 25.0 ± 13.4 years, mean duration of diabetes 9.6 ± 9.2 years). None of the patients studied were treated with antioxidants. Patients with causes of neuropathy other than diabetes (e. g. chronic alcohol abuse, drug-induced neuropathy), truncal neuropathy, and significant neurological diseases (e.g. Parkinson's disease, epilepsy, multiple sclerosis) were excluded from the association study. DN was diagnosed on the basis of symptomatic symmetrical distal neuropathy (reduced/absent ankle reflexes, reduced vibration, thermal, tactile, pin-prick, and/or position sensation) with one or more typical symptoms (burning pains or cramps, paresthesiae, numbness) of at least moderate severity in the feet. DN patients were also identified by measuring the motor conduction velocity of the peroneal nerve, with a velocity of <42 m/s (mean 31.6 ± 1.0 m/s) used to identify patients with DN. Accompanying retinopathy was diagnosed based on the presense of microaneurysms together with hard exudates and retinal hemorrhages, new vessel formation and/or fibrous retiniis proliferans Clinical nephropathy was diagnosed on basis of persistent proteinuria (urinary protein excretion rate > 0.3 g/24 h). The 166 patients tested were assigned to two groups according to the duration of diabetes. The groups were formed using principal of extreme phenotype and nonoverlapping selection criteria to minimize the masking effects of non-genetic factors. The first group consisted of 82 patients with diabetes of short duration (<3 years) and DN. The other group contsisted of the ramaining 84 diabetic patients, all of whom had diabetes of long duration (>10 years) and no neuropathy. The characteristics of the two groups are shown in Table 1.
Table 1.
Clinical characteristics of the patients
| Characteristic | Neuropathy (n = 82) | No neuropathy (n = 84) |
| Sex (male/female) | 48/34 | 49/35 |
| Age (years) | 23.5 ± 14.3 | 32.1 ± 14.2 |
| Duration of diabetes (years) | 1.1 ± 0.9 | 20.1 ± 8.4 |
| Glycated haemoglobin, HbA1 (%) | 10.6 ± 3.0 | 10.8 ± 3.8 |
| Retinopathya | 31.7 | 92.8 |
| Nephropathy a | 2.4 | 59.5 |
Values are mean ± SEM or a percentage of patients
Genomic DNA isolation and genotyping of Mn-SOD
DNA was extracted from whole human blood with phenol and chloroform [23]. Polymorphic regions were amplified by polymerase chain reaction (PCR) in 50 μl reaction mixture consisting of 67 mM Tris-HCl (pH 8.8), 16.7 mM ammonium chloride, 1.0 mM magnesium chloride, 0.1% Tween-20, 10% dimethyl sulfoxide, 0.2 mM of each dNTP, 5 pmol of each primer, 100 ng of genomic DNA, and 2.5 units of Taq polymerase (Biotekh, Russia). To amplify the polymorphic Ala(-9)Val region, we used primers SOD2-16F 5'-CCAGCAGGCAGCTGGCACCG-3' and SOD2-16R 5'-TCCAGGGCGCCGTAGTCGTAGG-3'. For the polymorphic Ile58Thr region of the Mn-SOD gene, PCR primers SOD2-58F 5'-AAGCTCCTCCCATTATCTAATAGC-3' and SOD2-58R 5'-TCAGTGCAGGCTGAAGAGAT-3' were used. PCR was carried out in a PHC-2 thermal cycler (Techne, UK) with 35 cycles of denaturation for 1 min at 94°C, annealing for 1 min at 55°C (Ile58Thr) or 60°C (Ala(-9)Val), and extension for 1 min at 72°C.
To detect Ala(-9)Val dimorphism, the PCR product was digested with BshTI. For the Ile58Thr polymorphism, the amplified fragment was digested with Eco32I (both enzymes from Fermentas, Lithuania). For digestion, 17 μl of PCR product, 2 μl of buffer O+/Tango™ (BshTI) or R+/Tango™ (Eco32I) (all buffers from Fermentas, Lithuania), and 1 μl of restriction endonuclease (1 unit/μl) were mixed and incubated for 12 h at 37°C. Digested DNA products were separated by electrophoresis in a 3% agarose gel with ethidium bromide or in an 8% polyacrylamide gel stained with silver [24].
Genotyping of Arg213Gly EC-SOD
The polymorphic region was amplified in a 50 μl reaction mixture consisting of 10 mM Tris-HCl (pH 8.8), 50 mM KCl, 1.5 mM magnesium chloride, 0.1% Tween-20, 10% dimethyl sulfoxide, 0.2 mM of each dNTP, 5 pmol of each primer SOD3-213F 5'-GGCTGGCCTGCTGCGTGGTGG-3' and SOD3-213R 5'-CCTTGCACTCGCTCTCGCGCG-3', 100 ng of genomic DNA, and 2.5 units of Taq polymerase. The PCR cycling profile was as described above except that annealing was carried out for 1 min at 65°C the amplicon was cleaved with restriction endonuclease Eco52I (Fermentas, Lithuania) in Eco52I+ specific buffer according to the manufacturer's recommendations. Digested fragments were analyzed as described by electrophoresis for the genotyping of Mn-SOD.
Statistical analysis
Genotype frequencies were checked for deviation from Hardy-Weinberg equilibrium by the χ2 and G-statistic tests, using the Rows and Columns program based on the Roff and Bentzen algorithm [25]. Genotype and allele frequencies in the groups studied were compared by Fisher's exact test The p value was corrected by multiplying it by the number of alleles (2) for each locus to obtain the pc value. Odds ratios and 95% confidence interval (95% CI) were calculated to assess the strength of the relationship between the Mn-SOD or EC-SOD polymorphisms and DN.
Results
The Ala(-9)Val polymorphism of the Mn-SOD gene
The first adenosine residue from 3' end of the forward primer, SOD2-16F, is mismatched with genomic DNA. The original sequence, near the polymorphic site of the Mn-SOD gene, is 5'-TCCGGT-3' with the most variable 3' thymidine (Val) to cytidine (Ala) at position -9 [26]. Use of the mismatched primer, SOD2-16F, results in a 91-bp PCR product. This product may or may not contain a BshTI restriction site (5'-ACCGGT-3'), depending on the sequence. If the -9 codon is GTT (Val), than digestion with BshTI produces two DNA fragments, 17 and 74 bp in length. If codon -9 is GCT (Ala), the amplified product not digested with this enzyme (Fig. 1).
Figure 1.
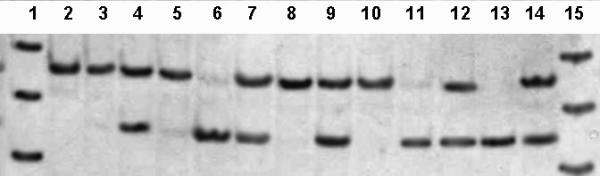
Restriction analysis of Ala(-9)Val polymorphism in the SOD2 gene. Lanes 2–14: 13 samples after digestion with BshT1; lanes 2, 3, 5, 8, and 10, Ala/Ala genotype; lanes 4, 7, 9, 12, and 14, Ala/Val genotype; lanes 6, 11, and 13, Val/Val genotype. Lanes 1 and 15: molecular weight DNA marker φX174/HinfI; 100-, 82- and 60-bp DNA fragments are shown.
The Ala allele and the Ala/Val genotype were the most common in healthy subjects (Table 2). The observed level of heterozygosity was 53.4%. The observed genotype frequency was consistent with Hardy-Weinberg equilibrium (χ2 = 2.0976 and G-statistic = 2.1149, p = 0.3930 ± 0.0154).
Table 2.
Allele and genotype distribution of the Ala(-9)Val SOD2 and Arg213Gly SOD3 polymorphic markers in healthy subjects
| Ala(-9)Val SOD2 | Frequency n (%) | Arg213Gly SOD3 | Frequency n (%) |
| Allele distribution | |||
| Ala | 119 (67.6) | Arg | 116 (65.9) |
| Val | 57 (32.4) | Gly | 60 (34.1) |
| Genotype distribution | |||
| Ala/Ala | 36 (40.9) | Arg/Arg | 34 (38.6) |
| Ala/Val | 47 (53.4) | Arg/Gly | 48 (54.5) |
| Val./Val | 5 (5.7) | Gly/Gly | 6 (6.8) |
The frequencies of the Ala allele and the Ala/Ala genotype were significantly lower in patients with DN than in patients without DN whrereas the frequency of the Val allele and the homozygous Val/Val genotype was significantly higher in patients with DN (Table 3). This suggests that the Ala(-9)Val dimorphism in the Mn-SOD gene is associated with neuropathy in type 1 diabetes mellitus. The Ala allele (odds ratio = 0.48, 95% CI; 0.31–0.74) and the Ala/Ala genotype (odds ratio = 0.34, 95% CI; 0.17–0.66) are associated with lower risk of DN development in than the Val allele (odds ratio = 2.09, 95% CI; 1.35–3.24) and the Val/Val genotype (odds ratio = 5.10, 95% CI; 1.77–14.69).
Table 3.
Allele and genotype distribution of the Ala(-9)Val SOD2 polymorphic marker in type 1 diabetic patients with and without neuropathy
| Group | Allele frequency n (%) | Genotype frequency n (%) | |||
| Ala | Val | Ala/Ala | Ala/Val | Val/Val | |
| Neuropathy (n = 82) | 83a (50.6) | 81b (49.4) | 14c (17.1) | 55 (67.0) | 13d (15.9) |
| No neuropathy (n = 84) | 115 (68.5) | 53 (31.5) | 33 (39.3) | 49 (58.3) | 2 (2.4) |
a Ala allele frequency was significantly lower in patients with diabetic neuropathy than in subjects without this complication (odds ratio = 0.48, p = 0.00067, pc = 0.00134). b Val allele frequency was significantly higher in patients with diabetic neuropathy than in subjects without this complication (odds ratio = 2.10, p = 0.00067, pc = 0.00134). c Ala/Ala genotype frequency was significantly lower in patients with diabetic neuropathy than in subjects without this complication (odds ratio = 0.34, p = 0.00122, pc = 0.00366). d Val/Val genotype frequency was significantly higher in patients with diabetic neuropathy tha in subjects without this complication (odds ratio = 5.10, p = 0.00219, pc = 0.00657).
The Ile58Thr polymorphism of the Mn-SOD gene
The penultimate adenosine residue at 3' end of the reverse primer SOD2-58R used is not complemetary to the genomic sequence (5'-GATAGC-3'), creating an Eco32I restriction site (5'-GATATC-3') during amplification if codon 58 is ATA (Ile) [26]. In this case, a 140-bp PCR product is produced, which yields two fragments, 20-bp and 120-bp in size, on digestion with Eco32I. If codon 58 is ACA (Thr allele) the amplified product not cleaved by this enzyme.
Eighty-seven of the 88 healthy subjects displaied the Ile/Ilegenotype, the remaining subject was heterozygous, Ile/Thr. All of the diabetic patients without complications were homozygous (Ile/Ile), as were all but one of the patients with DN, the remaining patient being heterozygous. So, no association between the Ile58Thr polymorphism of the Mn-SOD gene and DN was observed in type 1 diabetic patients.
The Arg213Gly polymorphism of the EC-SOD gene
The last guanosine residue at the 3' end of the reverse primer, SOD3-231R, displays a mismatch with the original sequence, 5'-CGGCGG-3', close to codon 213 of the EC-SOD gene. This resulted (depending on the sequence) in creation of an Eco52I restriction site (5'-CGGCCG-3') in the PCR product [19]. If codon 213 was CGG (Arg), a 104-bp PCR product was obtained that yielded two DNA fragments of 23 and 81 bp in size on digestion with Eco52I. In contrast, if codon 213 was GGG (the Gly allele), the 104-bp product was not digested with the enzyme (Fig. 2).
Figure 2.
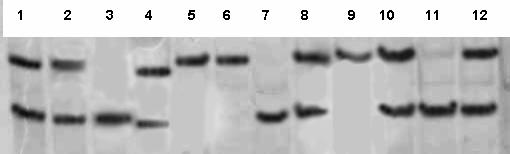
Restriction analysis of Arg213Gly polymorphism at the SOD3 gene. Lanes 1–3, 5–12: 11 samples after digestion with Eco521; lanes 3, 7, and 11, Arg/Arg genotype; lanes 1, 2, 8, 10, and 12, Arg/Gly genotype; lanes 5, 6, and 9, Gly/Gly genotype. Lane 4: molecular weight DNA marker pBR322/MspI; 90- and 76-bp DNA fragments are shown.
The Arg allele was 1.9 times more frequent than the Gly allele in healthy subjects. Arg/Arg homozygotes were 5.7 times more frequent than Gly/Gly homozygotes (Table 2). The observed level of heterozygosity was 54.6%. The observed genotype distribution was consisted with Hardy-Weinberg equilibrium (χ2 = 1.9495 and G-statistic = 1.9614, p = 0.3980 ± 0.0155). No significant difference in allele and genotype distribution was found between DN patients and patients without DN (Table 4). Thus, Arg213Gly substitution in the EC-SOD gene is not associated with nerve lesions in type 1 diabetes mellitus.
Table 4.
Allele and genotype distribution of the Arg213Gly SOD3 polymorphic marker in type 1 diabetic patients with and without neuropathy
| Group | Allele frequency n (%) | Genotype frequency n (%) | |||
| Arg | Gly | Arg/Arg | Arg/Gly | Gly/Gly | |
| Neuropathy (n = 82) | 82 (50.0) | 82 (50.0) | 16 (19.5) | 50 (61.0) | 16 (19.5) |
| No neuropathy (n = 84) | 95 (56.5) | 73 (43.5) | 21 (25.0) | 53 (63.1) | 10 (11.9) |
Discussion and Conclusions
A number of techniques have been described for detection of the Ala (-9)Val Mn-SOD gene dimorphism. One such method is PCR amplification of the polymorphic region, followed by BsaWI treatment (i. e., PCR/restriction fragment length polymorphism, PCR/RFLP) [16]. Another method is single-strand conformational polymorphism (SSCP) analysis of the product amplified by PCR [9]. A third method is two-step PCR using allele-specific primers for a second round of PCR [10]. Finally, the PCR product can be hybridized with allele-specific oligonucleotide (ASO) probes [13]. We have decided to use restriction endonuclease BshT1 for detection of the Ala(-9)Val polymorphism because this enzyme is commercially available in Russia whereas BsaWI is not. Our detection method has advantages over other techniques because is no need to carry out using the second round of PCR or laborious procedure of SSCP analysis after PCR amplification. Our molecular assay involves use of the mismatch-PCR/RFLP approach, which can be applied to the detection of any polymorphic single base substitution that fails to create or eliminates restriction sites. This rapid assay requires no special equipment or expertise. The use of a mismatch PCR primer makes it possible to create a restriction site in the amplified product only in the presence of the polymorphic base. This approach has been used to genotype the M235T variant of angiotensinogen [27] and the C1166T nucleotide variation in the angiotensinogen II (type 1) receptor gene [28].
The Ala allele of the Mn-SOD gene was more widespread than the Gly allele in healthy Russian subjects. This feature is common to all other Caucasian population samples tested (Germans, Swedes, Lithuanians, Finns, and Saamis) [16,29]. In contrast, the frequency of the Ala variant is significantly lower in Asian populations (Chinese and Japanese) than in most European populations [9,11,13,29].
The Ala/Val variation in the Mn-SOD leader signal affects the processing efficiency of the enzyme. The conformations of the Ala-type and Val-type leader signals have been predicted: the Ala form has an alpha-helical structure, a common conformation for mitochondrial leader signals, whereas the Val form may change its conformation from alpha-helix to beta-sheet starting from the position 16 due to amino acid substitution [9]. The Val form is less efficiently transported into mitochondria than the Ala form of the enzyme [9]. Poor signal sequence recognition by a receptor in the inner mitochondrial membrane may result in mistargeting. In addition, inefficient cleavage of a particular signal may reduce the level of enzymatic activity of an imported protein, such as Mn-SOD, within the mitochondrial compartment [30]. Studies of processing have suggested basal level of the Mn-SOD activity may be highest for Ala/Ala, followed by Ala/Val, and then Val/Val [13].
The Val variant of the Mn-SOD may be present in a lower concentration in mitochondria. If this is the case, then homozygous Val/Val should have lower resistance to oxidative stress than patients with other Mn-SOD variants. Such lower resistance is a common feuature of diabetes mellitus and various ageing and neurological disorders. This study showed that the ValMn-SOD allele predisposed patients to the development of DN in type 1 diabetes mellitus. Inefficient targeting of Mn-SOD may leave mitochondria inabequately defended against superoxide radicals. This may lead to protein oxidation, mitochondrial DNA mutations and damage, common in the pathogenesis of diabetic neuropathy [2,3] and neurodegenerative disorders such as Alzheimer and Parkinson's disease [31].
The Ala/Val dimorphic site is located within exon 2 of the Mn-SOD gene. Knock-out mice lacking exons 1 and 2 of Mn-SOD have been found to display progressive motor disturbances due to neuronal degeneration [32]. This observation provides future evidence for the involvement of the SOD2 gene in neuropathology.
Another functional polymorphism of the Mn-SOD gene, Ile58Thr in exon 3, affects the stability of the Mn-SOD tetramer and reduces the activity of the enzyme [14]. However, the Thr allele has been shown to be extremely rare. Our data consistent with those of Grasbon-Frodl et al. [16] who detected the Ile/Ile variant in only 63 randomly selected German subjects. The Ile58Thr variation of Mn-SOD probably not involved in DN pathogenesis. However this mutation is more likely to be involve in hereditary aging-related neurodegenerative disease, as shown by the finding that leukocytes lose the ability to induce Mn-SOD expression in response to increases in reactive oxygen species levels in tissue, in subjects over the age of 55 years [14]. Thus, older people with genetic defects in the four-helix bundle tetrameric interface of Mn-SOD may be especially prone to degenerative diseases.
An Arg213Gly substitution in the extracellular isoform (EC-SOD) does not affect enzymatic activity but does affect the amount of the enzyme on the external endothelial cell surface [33]. The Arg variant of EC-SOD is the most common in varios populations [17,18] including the Russian population (Table 2). Arginine-to-glycine substitution occurs in the center of the carboxyl-terminal cluster of positively charged amino acid residues defining the heparin-binding domain [17]. This amino acid substitution results in a higher level of dissociation of the enzyme from the cell surface into serum. Thus, the lower levels of EC-SOD on vascular walls must be positively correlated with a higher risk of progression of oxidative stress in affected patients with the Gly/Gly genotype. However, this finding was not supported by our data which showed a lack of association of the polymorphic marker with diabetic neuropathy. Such a relationship has been found only in Japanese patients with non-diabetic form of neuropathy resulting from massive amyloid deposition in nervous tissues [22].
Finally, we have no comparative data concerning the role of genetic factors directly related to oxidative stress in DN formation and progression. It would be of value to compare our findings with those of similar association studies in other populations.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
We thank Dr. I.A. Strokov for the collection of blood samples.
Contributor Information
Dimitry A Chistyakov, Email: dimitry.chistiakov@eudoramail.com.
Kirill V Savost'anov, Email: kir_savostianov@yahoo.com.
Elena V Zotova, Email: lena-zotova@mail.ru.
Valery V Nosikov, Email: nosikov@genetika.ru.
References
- Hunt JV, Wolff SP. Oxidative glycation and free radical production: a causal mechanism of diabetic complications. Free Radic Res Commun. 1991;12-13:115–123. doi: 10.3109/10715769109145775. [DOI] [PubMed] [Google Scholar]
- Low PA, Nickander KK, Tritschler HJ. The roles of oxidative stress and antioxidant treatment in experimental diabetic neuropathy. Diabetes. 1997;46:S38–S42. doi: 10.2337/diab.46.2.s38. [DOI] [PubMed] [Google Scholar]
- Zhu M, Spink DC, Yan B, Bank S, DeCaprio AP. Formation and structure of cross-linking and monomeric pyrrole autoxidation products in 2,5-hexanedione-treated amino acids, peptides, and protein. Chem Res Toxicol. 1994;7:551–558. doi: 10.1021/tx00040a011. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA. Metabolic and vascular factors in the pathogenesis of diabetic neuropathy. Diabetes. 1997;46:S31–37. doi: 10.2337/diab.46.2.s31. [DOI] [PubMed] [Google Scholar]
- Nagamatsu M., Nickander KK, Schmelzer JD, Raya A, Wittrock DA, Tritschler H, Low PA. Lipoic acid improves nerve blood flow, reduces oxidative stress, and improves distal nerve conduction in experimental diabetic neuropathy. Diabetes Care. 1995;18:1160–1167. doi: 10.2337/diacare.18.8.1160. [DOI] [PubMed] [Google Scholar]
- Van Dam PS, Van Asbeck BS, Bravenboer B, Van Oirschot JF, Marx JJ, Gispen WH. Nerve conduction and antioxidant levels in experimentally diabetic rats: effects of streptozotocin dose and diabetes duration. Metabolism. 1999;48:442–447. doi: 10.1016/s0026-0495(99)90101-4. [DOI] [PubMed] [Google Scholar]
- Martinez-Blasco A, Bosch-Morell F, Trenor C, Romero FJ. Experimental diabetic neuropathy: role of oxidative stress and mechanisms involved. Biofactors. 1998;8:41–43. doi: 10.1002/biof.5520080108. [DOI] [PubMed] [Google Scholar]
- Yan H, Harding JJ. Glycation-induced inactivation and loss of antigenicity of catalase and superoxide dismutase. Biochem J. 1997;328:599–605. doi: 10.1042/bj3280599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda-Matsubayashi S, Matsumine H, Kobayashi T, Nakagawa-Hattori Y, Shimizu Y, Mizino Y. Structural dimorphism in the mitochondrial targeting sequence in the human manganese superoxide dismutase gene. Biochem Biophys Res Commun. 1996;226:561–565. doi: 10.1006/bbrc.1996.139410.1006/bbrc.1996.1394. [DOI] [PubMed] [Google Scholar]
- Rosenblum JS, Gilula NB, Lerner RA. On signal sequence polymorphisms and diseases of distribution. Proc Natl Acad Sci USA. 1996;93:4471–4473. doi: 10.1073/pnas.93.9.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori H, Ohmori O, Shinkai T, Kojima H, Okano C, Suzuki T, Nakamura J. Manganese superoxide dismutase gene polymorphism and schizophrenia. Relation to tardive dyskinesia. Neuropsychopharmacol. 2000;23:170–177. doi: 10.1016/S0893-133X(99)00156-6. [DOI] [PubMed] [Google Scholar]
- Van Landeghem GF, Tabatabaie P, Beckman G, Beckman L, Andersen PM. Manganese-containing superoxide dismutase signal sequence polymorphism associated with sporadic motor neuron disease. Eur J Neurol. 1999;6:639–644. doi: 10.1046/j.1468-1331.1999.660639.x. [DOI] [PubMed] [Google Scholar]
- Hiroi S, Harada H, Nishi H, Satoh M, Nagai R, Kimura A. Polymorphisms in the SOD2 and HLA-DRB1 genes are associated with nonfamilial idiopathic dilated cardiomyopathy in Japanese. Biochem Biophys Res Commun. 1999;261:332–339. doi: 10.1006/bbrc.1999.1036. [DOI] [PubMed] [Google Scholar]
- Borgstahl GE, Parge HE, Hickey MJ, Johnson MJ, Boissinot M, Hallewell RA, Lepock JR, Cabelli DE, Tainer JA. Human mitochondrial manganese superoxide dismutase polymorphic variant Ile58Thr reduces activity by destabilizing the tetrameric interface. Biochemistry. 1996;35:4287–4297. doi: 10.1021/bi951892w. [DOI] [PubMed] [Google Scholar]
- Checkoway H, Farin FM, Costa-Mallen P, Kirchner SC, Costa LG. Genetic polymorphisms in Parkinson's disease. Neurotoxicology. 1998;19:635–643. [PubMed] [Google Scholar]
- Grasbon-Frodl EM, Kosel S, Riess O, Muller U, Mehraein P, Graeber MB. Analysis of mitochondrial targeting sequence and coding region polymorphisms of the manganese superoxide dismutase gene in German Parkinson disease patients. Biochem Biophys Res Commun. 1999;255:749–752. doi: 10.1006/bbrc.1998.9998. [DOI] [PubMed] [Google Scholar]
- Sandstrom J, Nilsson P, Karlsson K, Marklund SL. 10-fold increase in human plasma extracellular superoxide dismutase content caused by a mutation in heparin-binding domain. J Biol Chem. 1994;269:19163–19166. [PubMed] [Google Scholar]
- Yamada H, Yamada Y, Adachi T, Goto H, Ogasawara N, Futenma A, Kitano M, Hirano K, Kato K. Molecular analysis of extracellular-superoxide dismutase gene associated with high level in serum. Jpn J Hum Genet. 1995;40:177–184. doi: 10.1007/BF01883574. [DOI] [PubMed] [Google Scholar]
- Yamada H, Yamada Y, Adachi T, Goto H, Ogasawara N, Futenma A, Kitano M, Miyai H, Fukatsu A, Hirano K, Kakumu S. Polymorphism of extracellular superoxide dismutase (EC-SOD) gene: relation to the mutation responsible for high EC-SOD level in serum. Jpn J Hum Genet. 1997;42:353–356. doi: 10.1007/BF02766958. [DOI] [PubMed] [Google Scholar]
- Adachi T, Wang XL. Association of extracellular-superoxide dismutase phenotype with the endothelial constitutive nitric oxide synthase polymorphism. FEBS Lett. 1998;433:166–168. doi: 10.1016/S0014-5793(98)00903-X. [DOI] [PubMed] [Google Scholar]
- Marklund SL, Nilsson P, Israelsson K, Schampi I, Peltonen M, Asplund K. Two variants of extracellular-superoxide dismutase: relationship to cardiovascular risk factors in an unselected middle-aged population. J Intern Med. 1997;242:5–14. doi: 10.1046/j.1365-2796.1997.00160.x. [DOI] [PubMed] [Google Scholar]
- Sakashita N, Ando Y, Marklund SL, Nilsson P, Tashima K, Yamashita T, Takahashi K. Familial amyloidotic polyneuropathy type I with extracellular superoxide dismutase mutation: a case report. Hum Pathol. 1998;29:1169–1172. doi: 10.1016/s0046-8177(98)90433-6. [DOI] [PubMed] [Google Scholar]
- Mathew CGP. The isolation of high molecular weight eukaryotic DNA. In: Walker J, editor. In Methods of Molecular Biology. Vol. 2. Humana Press; 1984. pp. 31–34. [DOI] [PubMed] [Google Scholar]
- Budowle B, Chakraborty R, Giusti AM, Eisenberg AJ, Allen RC. Analysis of the VNTR locus D1S80 by PCR followed by high-resolution PAGE. Am J Hum Genet. 1991;48:137–144. [PMC free article] [PubMed] [Google Scholar]
- Roff DA, Bentzen P. The statistical analysis of mitochondrial DNA polymorphisms: χ2 and the problem of small samples. Mol Biol Evol. 1989;6:539–545. doi: 10.1093/oxfordjournals.molbev.a040568. [DOI] [PubMed] [Google Scholar]
- Church SL, Grant JW, Meese EU, Trent JM. Sublocalization of the gene encoding manganese superoxide dismutase (MnSOD/SOD2) to 6q25 by fluorescence in situ hybridization and somatic cell hybrid mapping. Genomics. 1992;14:823–825. doi: 10.1016/s0888-7543(05)80202-2. [DOI] [PubMed] [Google Scholar]
- Caulfield M, Lavender P, Farral M, Nunroe P, Lawson M, Turner P, Clark AJI. Linkage of the angiotensinogen gene to essential hypertension. N Engl J Med. 1994;330:1629–1633. doi: 10.1056/NEJM199406093302301. [DOI] [PubMed] [Google Scholar]
- Hingorani A, Brown MJ. A simple molecular assay for the C1166 variant of the angiotensin II type 1 receptor gene. Biochem Biophys Res Commun. 1995;213:725–729. doi: 10.1006/bbrc.1995.2190. [DOI] [PubMed] [Google Scholar]
- Van Landeghem GF, Tabatabaie P, Kucinskas V, Saha N, Beckman G. Ethnic variation in the mitochondrial targeting sequence polymorphism of MnSOD. Hum Hered. 1999;49:190–193. doi: 10.1159/000022873. [DOI] [PubMed] [Google Scholar]
- Shimoda-Matsubayashi S, Hattori Y, Matsumine H, Shinohara A, Yoritaka A, Mori H, Kondo T, Chiba M, Mizuno Y. MnSOD activity and protein in a patient with chromosome 6-linked autosomal recessive parkinsonism in comparison with Parkinson's disease and control. Neurology. 1997;49:1257–1262. doi: 10.1212/wnl.49.5.1257. [DOI] [PubMed] [Google Scholar]
- Jenner P. Oxidative stress in Parkinson's disease and other neurodegenerative disorders. Pathol Biol (Paris) 1996;44:57–64. [PubMed] [Google Scholar]
- Melov S, Schneider J, Day B, Hinerfield D, Coscun P, Mirra SS, Crapo ID, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- Adachi T, Yamada H, Yamada Y, Morihara N, Yamazaki N, Murakami T, Futenma A, Kato K, Hirano K. Substitution of glycine for arginine-213 in extracellular-superoxide dismutase impairs affinity for heparin and endothelial cell surface. Biochem J. 1996;313:235–239. doi: 10.1042/bj3130235. [DOI] [PMC free article] [PubMed] [Google Scholar]