

Abstract
Dendritic cell (DC) vaccines have shown great promise in generating anti-tumor immune responses but have generally fallen short of producing durable cures. Determining mechanisms by which these vaccines fail will provide one strategy towards improving their success. Several manipulations of DCs have improved their migration and longevity, but the immune inhibitory environment surrounding tumors provides a powerful suppressive influence. To determine the mechanisms by which DCs at the site of the tumor convert to a suppressive phenotype, we evaluated pathways in DCs that become expressed at the tumor site. Our results revealed that tumors lead to induction of the glucocorticoid induced leucine zipper (GILZ) gene in DCs, and that this gene is critical for the development of tumor induced tolerance of both DCs and T cells. Previous data suggested that GILZ is a pivotal gene in the balance between activation and tolerance of DCs. Our new data show that GILZ is highly upregulated in DCs in the tumor microenvironment in vivo and that blockade of this gene in DC vaccines significantly improves long term survival. These results suggest that GILZ may be an ideal candidate gene to target for novel immune-based tumor therapies.
Keywords: Dendritic cell vaccine, GILZ, Dendritic cell, immunotherapy, tumor vaccine
Introduction
The immune system has long been recognized for its powerful potential to recognize and eliminate tumor cells with a high level of specificity. Because of their potent antigen presentation capacity, dendritic cells (DCs) have been utilized as a platform for a number of different immunotherapies. DC vaccines have now been tested in numerous animal models and clinical trials. Positive results from animal models led to a large number of clinical trials, which have been planned and/or tested for many different types of tumors, including glioblastoma 1–3, astrocytoma 4 melanoma 5, 6, pancreatic 5, 7, renal cell 8colorectal 9 prostate, (reviewed in 10), hepatocellular 11chronic lymphocytic leukemia 12, 13 medullary thyroid 14, breast 15; and acute myelogenous leukemia 16. Despite this potential and widespread use, many immunotherapies have not met with the degree of success that was hoped. While the results have been varied, there has been a recurrent theme in that there has been success in inducing immune responses to tumor antigens and in some cases, tumor regression, but ultimately, durable cures have not been commonly achieved. In part, tumor evasion of immunotherapies, which include suppression of DCs, induction of Tregs, and secretion of toxic factors (reviewed in 17), have limited the long term success of these strategies. Many summaries of these trials highlight the necessity of determining the physiologic characteristics of DCs in order to improve the outcomes. A number of investigations have been undertaken in which specific limitations of the DCs have been manipulated. Some examples of approaches that have arisen from these investigations include: extending the lifespan of DCs 18–21, enhancing antigen targeting and delivery 22 and improving trafficking of DCs 23. Thus, while the potential for tumor vaccines is great, the need to improve them is also apparent.
One mechanism by which the tumor environment induces suppression in DCs is through altering their pattern of gene expression such that the phenotype and function of the DCs are changed. In the present study, we have identified one critical change in gene expression of the glucocorticoid induced leucine zipper (GILZ), which has been shown to mediate the immunosuppressive effects of glucocorticoids in T cells, at the site of the tumor that may help to account for the conversion of DCs to a tolerogenic nature.
GILZ has been previously shown to mediate immunosuppressive effects in both T cells and DCs. The initial study that identified a role for GILZ in T cells was based on the hypothesis that glucocorticoids are critical regulators of T cell survival. In this study, the genes that became upregulated in T cells after treatment with dexamethasone (DEX) were analyzed, which led to the identification of GILZ as a new gene expressed by T cells 24. A later study ultimately identified the gene as the mediator of glucocorticoid (GC) action in T cells 25. GILZ is a member of the TSC family (TGFβ-stimulated clone-22, 26 (Tsc22d3) and contains a leucine zipper region and a putative Foxo binding site/response element. Interestingly the same gene was described in 197227and studied in the nervous system as “delta sleep inducing peptide.” GILZ appears to be a pivotal gene in the regulation of activation and survival of T cells and its expression is mediated antagonistically by IL-2 and GCs. 28. Evidence for its therapeutic use was shown by experiments in which overexpressing GILZ in T cells led to an improvement in the course of Th1-mediated autoimmune colitis 29.
While much less is known about the role of GILZ in DCs, it has been reported to mediate a similar immunosuppressive effect. Overexpression of GILZ led to a decrease in antigen specific immunity but an increase in regulatory T cell function 30, 31. Importantly in the context of tumors, other immunosuppressive factors such as IL-10 can also lead to GILZ upregulation 32, 33, and our preliminary data show that tumors upregulate GILZ expression in DCs that are found at the site of the tumor. These findings together indicate that GILZ expression may be pivotal in determining whether DCs ultimately generate an activating effector or tolerogenic response in T cells, particularly in an immunosuppressive setting such as a tumor.
Materials and Methods
Mice
BALB/c and C57BL/6 mice were obtained from NIH, Frederick MD or Jackson Laboratories, Bar Harbor, ME and maintained in the JHU Animal Care Facilities. All protocols were approved by the institutional review committee.
Preparation of DCs
Bone marrow–derived DCs (BMDCs) were generated by standard methods as follows: bones were flushed with RPMI/10% FCS (both from Invitrogen, Carlsbad, CA), and a single cell suspension was prepared. Following centrifugation, cells were resuspended in DC medium (RPMI1640 containing: 10% FBS, Sodium Pyruvate (Sigma Aldrich, St. Louis, MO), Penicillin/Streptomycin (Quality Biological Inc, Gaithersburg, MD) and 1% HEPES buffer (Invitrogen, Carlsbad, CA), with 20 ng/ml GM-CSF (PeproTech, Rocky Hill, NJ). Cells were plated in non tissue culture Petri dishes (100mm) at 2-5e6/ plate. On day 8, DCs were collected for fluorescence-activated cell sorting (FACS) based on CD86 and major histocompatibility complex class II (MHC II) expression. Immature and mature DCswere classified based on expression of markers as follows: CD11c+CD86lowMHCIIlow and CD11c+CD86highMHCIIhigh cells, respectively. For transduction with lentiviral vectors, DCs were infected for 12 to 24 hours with self-inactivating LV-GILZ-small interfering RNA (siRNA) or LV-control-siRNA ata MOI of infection of 2–5 on days 7 and 8 with polybrene(8 μg/mL) (Sigma Chemical Co, St. Louis, MO). As each vector contains a GFP reporter, the efficiency of transduction was analyzedby flow cytometry.
A20HA and B16 are a mouse B cell lymphoma modified to express hemagglutinin and a murine melanoma line, respectively (derived from BALB/c for the A20 and B6 mice for the B16). All antibodies were obtained from BD-Pharmingen, San Jose, CA.
Antibodies and peptide sequences
Anti-mouse CD11c APC (clone HL3), anti-mouse I-A[b] PE (clone AF6-120.1), anti-mouse CD86 PE-Cy5 (clone GL1) and Annexin V APC were purchased from BD Biosciences PharMingen (San Diego, CA). Flow cytometry analyses were performed on a FACSCalibur instrument (BD Biosciences PharMingen) and analyzed using CellQuest (BD Biosciences PharMingen) and FlowJo (TreeStar, Ashland, OR). The Class II (110–120) peptide sequence was SFEREIFPKE.
siRNA design and generation of lentivirus-based siRNA
siRNAs were designed corresponding to the mouse GILZ gene (GenBank accession no. NP_056558). Sequences were chosen using the Oligoengine software (Oligoengine, Seattle, WA). siRNAs with no sequence homology to any known mouse gene were used as negative controls. All siRNA sequences were BLAST searched in the National Center for Biotechnology Information’s (NCBI) “search for short nearly exact matches” mode against all mouse sequences deposited in the GenBank and were not found to have significant homology to genes other than GILZ. To generate a vector-based suppression of GILZ expression, the construct pSUPER-retro (Oligoengine) was used as a template. The siRNA oligonucleotides designed contained a sense strand of 19-nucleotide sequence followed by a short spacer (TTCAAGAGA), the reverse complement of the sense strand, and 5 thymidines as a RNA polymerase III transcriptional stop signal. Briefly, the pSUPER-retro vector was digested with BglII and HindIII and the annealed oligos (5′-GAT CCC CTG CCC TTG TCC GAG CTT TAT TCA AGA GAT AAA GCT CGG ACA AGG GCA TTT TTG GAA A-3′; forward and 5′-AGC TTT TCC AAA AAT GCC CTT GTC CGA GCT TTA TCT CTT GAA TAA AGC TCG GAC AAG GGC AGG G-3′; reverse were ligated into the vector. For generating lentivectors encoding siRNA construct (LV-siRNA), the complete human H1-RNA promoter and the siRNA cassette as well as the PGK promoter were subcloned at XhoI and NheI sites before the reporter eGFP gene of the third generation self-inactivating lentiviral vector, Sin-18 provided by D. Trono (University of Geneva, Geneva, Switzerland). All inserts were confirmed by sequencing. Efficiency of knockdown was confirmed by qPCR (details in next section), and a clone with 90% knockdown was selected for further experimentation. Methods for viral production are detailed in our previous work. 34
Quantification of transcripts
Quantitative polymerase chain reaction (PCR) analysis was conducted using the Bio-Rad iCycler system (Bio-Rad, Hercules, CA). The GILZ primer sequences are: forward: 5′-TGTATCAGACCCCCATGGAG-3′ and reverse: 5′-TCCATGGCCTGCTCAATCTTG -3′. Values have been normalized to β-actin. Oligos were obtained from IDT Technologies, Coralville, IA 52241; LPS and dexamethasone were obtained from Sigma Chemical Co, St. Louis, MO.
Phagocytosis
DCs were collected and resuspended at a concentration of 2 × 106 in 200μl PBS per sample. PE-labeled Ova 555 (Molecular Probes) was diluted to 20 μg/ml and 20μl was added to each sample. The cells were incubated at 37° for 1 hour. At the end of incubation, cells were washed twice with FACS buffer and stained for CD11c and Annexin V; samples were acquired on a FACSCalibur, and AxV negative, CD11c+/PE+ populations were identified.
DC Vaccine
Mice with a CD4+ T cell receptor transgenic for HA (6.5 mice) on a congenic thy1.1 background were used for T cell effector studies. One day prior to DC vaccine, 25 million 6.5 spleen T cells2illion transgenic T cells). For tumor therapy studies, 4 × 105 A20-HA cells (from a mouse B cell lymphoma modified to express hemagglutinin) were injected 7 days before first DC vaccine. The control HA expressing vaccine (C5A) and the GILZ-silenced HA expressing vaccine were created by transducing DCs on days 7 and 8 as above. For an additional control, BALB/c DCs were generated and pulsed with HA Class II peptide by adding 1 μg/ml and incubating cells at 37°C for 1 hour. All DC vaccine cells were collected on day 9, washed twice with PBS and resuspended at 5 × 106 in 200μl / mouse and s.c. injected into two flanks. Cell viability and DC phenotype were assessed before each vaccine administration using flow cytometry analysis based on CD11c, CD86 and Annexin V. On day 7, 14, 21, 28 DC vaccines were given. Mouse survival was then monitored (10 mice/group).
Statistics
All statistical analyses were conducted using GraphPad Prism (GraphPad Software, San Diego, CA). Either ANOVA or t tests were conducted, depending on the experiments. Survival analysis was conducted using both t tests to determine significance at a given time point and Kaplan-Meier to determine significance at completion of experiments.
Results
Tumors and immunosuppressive stimuli upregulate GILZ expression in DCs while activation decreases it
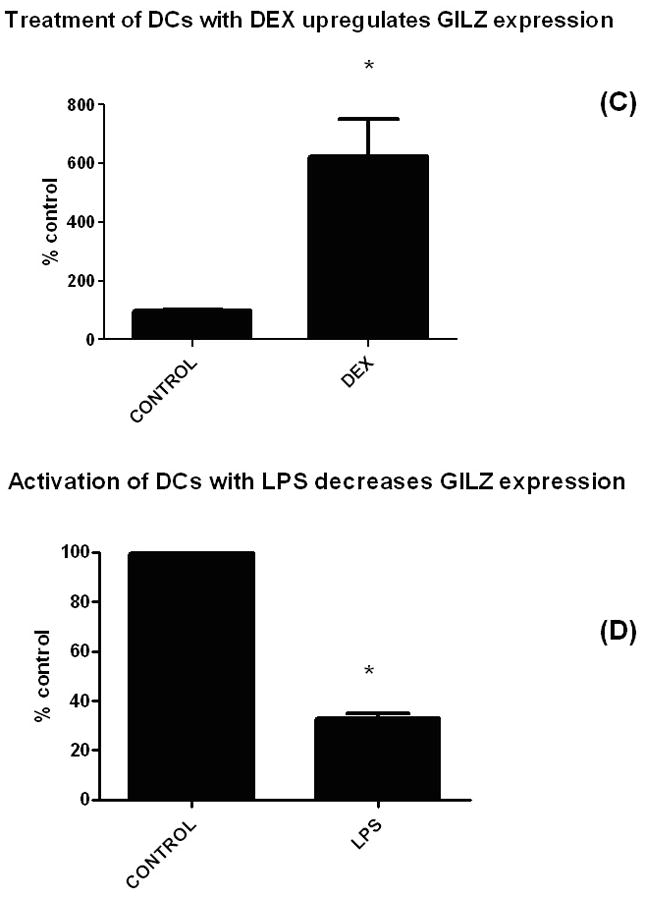
To test the hypothesis that tumors lead to an increase in GILZ expression by DCs, we compared expression from tumor-infiltrating DCs to that of naïve cells. Following injection of A20-HA B cell lymphoma tumor cells into Balb/C mice, splenic DCs were isolated and their level of expression of GILZ quantified via qPCR and compared to the expression of splenic DCs from naïve animals (with the assay normalized to actin). Results of those experiments showed that GILZ was highly upregulated in DCs found at the site of the tumor (Figure 1A). We next determined whether other types of tumors would also lead to upregulation of GILZ in DCs and whether the factor leading to this upregulation was soluble by assessing whether cell-cell contact was required. Tumor cells were cultured from both the A20 and also a B16 melanoma cell line and filtered supernatant was added to cultures of DCs. qPCR analysis (Figure 1B) shows that supernatant obtained from both types of tumors led to upregulation of the gene and thus that a soluble factor rather than cellular contact was necessary. To assess the pivotal nature of this gene, we sought to determine whether GILZ was responsive to signals from an activating stimulus. Thus, we next compared the relative expression levels of GILZ in BMDCs treated with either dexamethasone (DEX) or LPS. As figures 1C and 1D show, while DEX dramatically increased GILZ expression, LPS led to a significant decrease, indicating that this gene responded inversely in these opposing settings.
Figure 1. Tumors and immunosuppressive stimuli upregulate GILZ expression in DCs while activation decreases it.

(A) To determine whether the immunosuppressive environment of tumors would upregulate GILZ expression in DCs, mice were injected with 1 × 106 A20-HA B cell lymphoma cells. Ten days later, tumor-containing spleens were harvested, single cell suspensions prepared, stained with CD11c, and FACS sorted into a CD11c+ population. RNA was prepared from the DCs from the tumor site, reverse transcribed into cDNA and subjected to qPCR. Shown are the relative values of GILZ expression, normalized to actin, with naïve DCs set to control values of 100%. Minimum of three experiments were combined for each subset of figure 1; statistics shown are paired t tests. For each panel, asterisks denote differences that are significantly different (p<.05) from control.
(B) Tumors upregulated GILZ in DCs via a soluble factor. To assess whether cell-cell contact was required between tumor cells and DCs, and to assess whether other tumor types would increase expression of GILZ, the supernatants from either A20 or B16 melanoma tumor cells were harvested 48 hours after medium change, filtered, and added to cultures of DCs. Twenty four hours later, DCs were harvested and RNA and cDNA prepared for the qPCR assay as in 1A. A paired t test shows significant differences for both tumor types compared to controls.
(C) Dexamethasone upregulated GILZ in BMDCs and (D) LPS decreased GILZ expression. BMDCs were exposed to a suppressive (DEX) and an activating (LPS) agent and expression levels of GILZ were measured by qPCR (normalized to expression of actin). For the cultures, the following concentrations were used: DEX treatment (100 nM, overnight); LPS (100 ng/ml, overnight).
Blockade of GILZ inhibits DEX-mediated downregulation of CD86
To evaluate the role of GILZ in producing a suppressive phenotype, we assessed whether blockade of GILZ would prevent a known consequence of DEX treatment, i.e., decrease in co-stimulatory molecule expression. For these studies, we first gene modified BMDCs with a lentiviral vector that expressed either a control GFP or an siRNA against GILZ-GFP (via a similar strategy to our previous studies testing the role of the MINOR gene in DCs 21. Following overnight treatment with DEX, DC expression of CD11c, CD86 and MHCII was analyzed via FACS (gating on Annexin V negative cells). While treatment of DCs with DEX downregulated CD86 expression, the GILZ blocked DCs maintained their high expression of CD86 (Figure 2).
Figure 2.
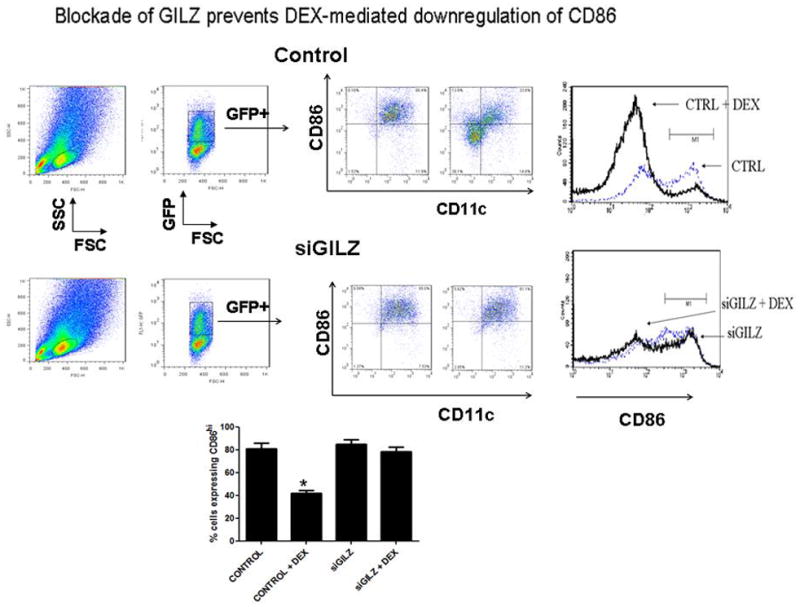
Blockade of GILZ inhibits DEX-mediated downregulation of CD86. BMDCs were transduced with a lentiviral vector expressing an siRNA for GILZ or a control vector, both of which expressed GFP. DCs were then activated with LPS (100 ng/ml) and then treated with DEX (100nM) or vehicle control for 16 hours. Cells were stained with an antibody for CD86 and Annexin V, and FACS analysis was then conducted, with gene-modified CD11c+ cells identified by gating on GFP+ AxV− cells. As shown, while DEX downregulated CD86 expression in the control group of DCs, the GILZ-blocked (GILZD) retained their high expression of CD86. Shown are FACS plots from one of three different experiments and a graph of the different experiments combined, analyzed by a paired t test.
GILZ inhibition increases phagocytic capacity of DCs
As DCs often decrease their phagocytic capability with increased maturation, we next sought to determine whether an unexpected adverse consequence of GILZ blockade might be a decrease in antigen uptake. To test this possibility, BMDCs were cultured, gene modified with either control or with the siRNA against GILZ, and incubated with a fluorescently labeled peptide so that quantitative comparisons of antigen uptake in gene modified cells could be made. Figure 3 summarizes the percentages of CD11c+ cells that phagocytosed labeled peptide. While blocking GILZ showed no effect on phagocytosis at day 6, by days 8 and 10, there was a significant increase in phagocytosis in the GILZ blocked cells compared to control.
Figure 3. GILZ inhibition increases phagocytic capacity of DCs.

BMDCs were cultured, gene modified with either control (labeled CTRL) or with the siGILZ. Cultures were maintained in DC medium and analyzed at days 6, 8, and 10 of development, as shown. PE-labeled OVA (20 μg/ml) was added for 1 hour, and cells were stained for CD11c and Annexin V. Shown are summaries of the percentages of live CD11c+ cells that phagocytosed labeled peptide (identified by PE-positive cells). The bars are gated on GFP+ fractions of control vs siRNA-GILZ. Significant differences (p<.05, paired t tests with control) are marked with an asterisk: GILZ inhibition at days 8 and 10 led to an increase in phagocytosis compared to control. Three different experiments are combined in the graph and the two conditions for each timepoint are shown.
GILZ blockade enhanced T cell effector function in vivo
To assess the relative ability of GILZ-silenced DCs to activate an effector T cell response, BMDCs were transduced with the control-HA or the siGILZ-HA for use as a vaccine. Five days following the DC vaccination, spleens and lymph nodes were collected and analyzed by FACS. Antigen specific effector T cells were identified by their expression of thy1.1 and CD4, and intracellular cytokine staining was conducted for IFNγ. The total percentage of IFNγ-secreting Ag specific T cells for both groups is shown (Figure 4). Mice that received the DC vaccine with blocked GILZ had an increase in the total number of antigen-reactive T cells that were IFNγ-secreting.
Figure 4. GILZ blockade enhanced T cell effector function in vivo.

BMDCs were generated and gene modified with a dual promoter lentivirus that expresses the influenza hemagluttinin (HA) gene along with either a control siRNA or the siRNA for GILZ. These vectors were validated for both knockdown of GILZ and equivalent expression of HA (testing both by staining for HA and qPCR for levels of HA). DCs were transduced with the control-HA or the siGILZ-HA to generate the DC vaccine. 2.5 × 107 spleen cells from 6.5 mice were injected into mice on day 0, 1 million transduced DCs were administered on day 1, and spleens and lymph nodes were harvested on day 5. Numbers of antigen specific T cells were quantified, and their secretion of IFNγ determined by intracellular cytokine staining. Mice that received the DC vaccine with the siRNA blocking GILZ expression had significantly more effector cells in the lymph nodes. Shown is a representative graph of three different experiments. A paired t test shows a significant difference for this comparison.
Antigen-expressing/GILZ blocked DCs significantly prolong survival of mice with a pre-existing tolerogenic tumor
To assess whether the increase in effector T cell function would enhance the therapeutic efficacy of a GILZ-blocked DC vaccine, we first established tumor in BALB/c mice by inoculating A20-HA tumor cells prior to the induction of therapy. After tumor was established, mice were injected weekly, for five weeks, with DCs prepared under the following conditions: DCs pulsed with HA peptide (BALB/c), DCs transduced with a lentiviral vector expressing the control siRNA and the HA gene (C5A) or DCs transduced with the siRNA for GILZ and HA gene (GH8). Figure 5 reports the curves of the survival time from the beginning of treatment versus the percentage of living GH8, C5A or animals treated with only BALBc DCs. Shown is a representative survival curve, indicating that blocking GILZ mice in the DC vaccine (GH8) provided a significant advantage over traditional DC vaccine strategies.
Figure 5. Antigen-expressing/GILZ blocked DCs significantly prolong survival of mice with a pre-existing tolerogenic tumor.

4 × 105 A20-HA cells were injected into mice (10/group) and allowed to grow for 7 days. On day 7, the first of 5 weekly vaccines was administered. Mice were treated with 1 million DCs of the following types: BALB/c DCs that were (1) pulsed with HA peptide (2) transduced with the control siRNA-HA (C5A) or (3) transduced with the siRNA-GILZ-HA (GH8). Mice were then followed for survival (euthanized per institutional guidelines, as appropriate). Shown is one representative of three experiments. Statistical analysis was conducted using a Kaplan-Meier survival analysis.
Discussion
It has long been appreciated that tumor cells have a number of mechanisms by which they can evade treatment strategies, whether through drug resistance or immune suppression. There is now an extensive literature on DC and T cell vaccines, with many vaccines now in clinical trials (described above and recently reviewed in 35). We hypothesized that identifying potentially suppressive genes that tumors upregulated in DCs could lead to improvements in DC vaccines. Our previously published work showed that upon activation, DCs upregulated a gene termed MINOR that induced apoptosis. While our results showed that blockade of this gene led to a delay in tumor progression, ultimately all the mice succumbed to tumor 21. In order to assess whether additional genes were upregulated in vivo that might account for the ultimate lack of cure, we first isolated DCs from tumor-bearing mice and conducted a targeted micro array to identify apoptosis-related genes and thus identified expression of GILZ (not shown), which we hypothesized might contribute to the suppressive phenotype that DCs acquire at the tumor site. Our results indicating that different tumor types do lead to an increase in GILZ expression, which is analogous to the results previously obtained in T cells supports the contention that in situ modification of DCs might contribute to their loss of efficacy. While previous studies have shown that cytokines secreted by tumors can suppress DC function, changes in genetic expression of DCs as a result of tumor interaction have not been fully elucidated. The combination of the results indicating that GILZ is upregulated in DCs both by the immunosuppressive DEX and by tumor supernatant and, conversely, downregulated by LPS, suggest a role for GILZ in the response of DCs to environmental cues within the tumor setting.
While the pattern of GILZ expression indicated a correlation with immunosuppressive phenotypes, we sought to demonstrate that to have utility as a therapeutic target by which we might decrease the immunosuppressive effects of tumors. A number of studies have shown that gene-modified DCs could enhance anti-tumor immunity either by increasing their recruitment of T cells 23 or their expression of stimulatory cytokines such as TNFα 36. The latter study provides further evidence of the importance of maintained expression of stimulatory molecules at the site of the tumor as forced expression of TNFα- in DCs was more effective than the addition of cytokine to the culture. Along these lines, we sought to determine a functional assessment of GILZ, namely that its blockade would prevent DCs from converting to a more suppressive phenotype in an immunosuppressive setting. We evaluated the role of GILZ in mediating the decrease in DEX-mediated CD86 immunosuppression and assessed the ability of blocking this gene to maintain high levels. Our result that blocking GILZ expression in DCs prevented DEX-mediated downregulation of CD86 is consistent with previously published findings showing that overexpression of GILZ led to a more tolerogenic phenotype in human DCs and that blockade of GILZ increased the activation state 30. These outcomes might be expected, since as mentioned, GILZ had been previously identified as the mediator of glucocorticoid-induced immunosuppression in T cells and further suggest that inhibiting expression of this single gene may be sufficient to maintain a high level of stimulatory function of DCs, which could potentially improve one major shortcoming of current DC vaccine strategies.
For DCs to initiate a successful immune response antigen must be taken up as well as presented; we thus next evaluated whether blockade of GILZ would impact on uptake of antigen. While activation of DCs has generally been associated with a decrease in phagocytic capacity, the direct relationship has not clearly been defined. To test whether GILZ silencing would affect antigen uptake, we compared the relative abilities of control and siRNA- GILZ transduced cells to phagocytose fluorescently labeled peptide. We assessed uptake on successive days of maturation of DCs to better define a role of GILZ in this process. Interestingly, antigen uptake was increased in GILZ-blocked DCs, but only at later timepoints, which suggests that there may be multiple roles for this gene in DC effector function and further, that an increase in activational state does not necessitate a downregulation of phagocytic function, if additional genes are manipulated. The mechanisms behind this increase are less clear, although it is likely that activation state and phagocytic ability are not directly linked. Further support of this contention was shown in a previous report in which results demonstrated that while GILZ impacted on activation state, it had no impact on phagocytosis 30 (which we also observed at one early timepoint). Thus, depending on the state of maturation of the cells, processes of activation and antigen uptake may be differentially affected.
Ultimately, for therapeutic efficacy, modifications must translate into an improvement in immune stimulatory function in vivo. We therefore conducted a series of experiments in which we analyzed the ability of these DCs to first activate antigen-specific T cells in vivo and second to eliminate pre-existing tumors. Our initial lentiviral vector for gene modification contained only the siRNA for GILZ, which required that in order to introduce antigen, we needed to pulse cells with peptide. In order to bypass the limitation of not achieving 100% transduction efficiency and to generate DCs that only expressed siGILZ in HA-expressing cells, we engineered two vectors, both of which expressed HA, one with the control siRNA and one with the siRNA for GILZ. These vectors were validated for both knockdown of GILZ and equivalent expression of HA (testing both by staining for HA and qPCR for levels of HA). Inhibition of GILZ expression led to a significant increase in expansion of effector T cells and a significant prolongation of survival in the group administered siGILZ-vaccine when compared to control.
In summary, when taken together, our results indicate that one mechanism by which tumors can dampen the efficacy of DC vaccines is through altering their patterns of gene expression, leading to their conversion away from an immunostimulatory capacity. By manipulating vaccines such that this compensatory change in gene expression by DCs is inhibited, it may be possible to maintain the initial level of effector function that the DCs provided. Thus, both the identification of such genes and therapeutic measures to manipulate them will likely provide new avenues by which immunotherapies may be improved in the future.
Acknowledgments
This work was funded by R01CA111989.
Footnotes
Disclosure of Conflicts of Interest: The authors have no conflict of interest to disclose.
References
- 1.Wheeler CJ, Black KL, Liu G, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008 Jul 15;68(14):5955–5964. doi: 10.1158/0008-5472.CAN-07-5973. [DOI] [PubMed] [Google Scholar]
- 2.Yu JS, Wheeler CJ, Zeltzer PM, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001 Feb 1;61(3):842–847. [PubMed] [Google Scholar]
- 3.Yamanaka R, Homma J, Yajima N, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005 Jun 1;11(11):4160–4167. doi: 10.1158/1078-0432.CCR-05-0120. [DOI] [PubMed] [Google Scholar]
- 4.Walker DG, Laherty R, Tomlinson FH, Chuah T, Schmidt C. Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: potential interaction with adjuvant chemotherapy. J Clin Neurosci. 2008 Feb;15(2):114–121. doi: 10.1016/j.jocn.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Mule JJ. Dendritic cell-based vaccines for pancreatic cancer and melanoma. Ann N Y Acad Sci. 2009 Sep;1174:33–40. doi: 10.1111/j.1749-6632.2009.04936.x. [DOI] [PubMed] [Google Scholar]
- 6.Nestle FO, Alijagic S, Gilliet M, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998 Mar;4(3):328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 7.Pecher G, Haring A, Kaiser L, Thiel E. Mucin gene (MUC1) transfected dendritic cells as vaccine: results of a phase I/II clinical trial. Cancer Immunol Immunother. 2002 Dec;51(11–12):669–673. doi: 10.1007/s00262-002-0317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwaab T, Schwarzer A, Wolf B, et al. Clinical and immunologic effects of intranodal autologous tumor lysate-dendritic cell vaccine with Aldesleukin (Interleukin 2) and IFN-{alpha}2a therapy in metastatic renal cell carcinoma patients. Clin Cancer Res. 2009 Aug 1;15(15):4986–4992. doi: 10.1158/1078-0432.CCR-08-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgdorf SK, Fischer A, Myschetzky PS, et al. Clinical responses in patients with advanced colorectal cancer to a dendritic cell based vaccine. Oncol Rep. 2008 Dec;20(6):1305–1311. [PubMed] [Google Scholar]
- 10.Lehrfeld TJ, Lee DI. Dendritic cell vaccines for the treatment of prostate cancer. Urol Oncol. 2008 Nov-Dec;26(6):576–580. doi: 10.1016/j.urolonc.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Palmer DH, Midgley RS, Mirza N, et al. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology. 2009 Jan;49(1):124–132. doi: 10.1002/hep.22626. [DOI] [PubMed] [Google Scholar]
- 12.Hus I, Schmitt M, Tabarkiewicz J, et al. Vaccination of B-CLL patients with autologous dendritic cells can change the frequency of leukemia antigen-specific CD8+ T cells as well as CD4+CD25+FoxP3+ regulatory T cells toward an antileukemia response. Leukemia. 2008 May;22(5):1007–1017. doi: 10.1038/leu.2008.29. [DOI] [PubMed] [Google Scholar]
- 13.Palma M, Adamson L, Hansson L, et al. Development of a dendritic cell-based vaccine for chronic lymphocytic leukemia. Cancer Immunol Immunother. 2008 Nov;57(11):1705–1710. doi: 10.1007/s00262-008-0561-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papewalis C, Wuttke M, Jacobs B, et al. Dendritic cell vaccination induces tumor epitope-specific Th1 immune response in medullary thyroid carcinoma. Horm Metab Res. 2008 Feb;40(2):108–116. doi: 10.1055/s-2007-1022565. [DOI] [PubMed] [Google Scholar]
- 15.Dees EC, McKinnon KP, Kuhns JJ, et al. Dendritic cells can be rapidly expanded ex vivo and safely administered in patients with metastatic breast cancer. Cancer Immunol Immunother. 2004 Sep;53(9):777–785. doi: 10.1007/s00262-004-0520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Driessche A, Van de Velde AL, Nijs G, et al. Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy. 2009;11(5):653–668. doi: 10.1080/14653240902960411. [DOI] [PubMed] [Google Scholar]
- 17.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008 Sep 19;29(3):372–383. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Kim SO, Ono K, Tobias PS, Han J. Orphan nuclear receptor Nur77 is involved in caspase-independent macrophage cell death. J Exp Med. 2003 Jun 2;197(11):1441–1452. doi: 10.1084/jem.20021842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim TW, Hung CF, Boyd D, et al. Enhancing DNA vaccine potency by combining a strategy to prolong dendritic cell life with intracellular targeting strategies. J Immunol. 2003 Sep 15;171(6):2970–2976. doi: 10.4049/jimmunol.171.6.2970. [DOI] [PubMed] [Google Scholar]
- 20.Kim TW, Lee JH, He L, et al. Modification of professional antigen-presenting cells with small interfering RNA in vivo to enhance cancer vaccine potency. Cancer Res. 2005 Jan 1;65(1):309–316. [PubMed] [Google Scholar]
- 21.Wang T, Jiang Q, Chan C, et al. Inhibition of activation-induced death of dendritic cells and enhancement of vaccine efficacy via blockade of MINOR. Blood. 2009 Mar 26;113(13):2906–2913. doi: 10.1182/blood-2008-08-176354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shortman K, Lahoud MH, Caminschi I. Improving vaccines by targeting antigens to dendritic cells. Exp Mol Med. 2009 Feb 28;41(2):61–66. doi: 10.3858/emm.2009.41.2.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terando A, Roessler B, Mule JJ. Chemokine gene modification of human dendritic cell-based tumor vaccines using a recombinant adenoviral vector. Cancer Gene Ther. 2004 Mar;11(3):165–173. doi: 10.1038/sj.cgt.7700671. [DOI] [PubMed] [Google Scholar]
- 24.D’Adamio F, Zollo O, Moraca R, et al. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity. 1997 Dec;7(6):803–812. doi: 10.1016/s1074-7613(00)80398-2. [DOI] [PubMed] [Google Scholar]
- 25.Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. Faseb J. 2009 Jun 30; doi: 10.1096/fj.09-134684. [DOI] [PubMed] [Google Scholar]
- 26.Shibanuma M, Kuroki T, Nose K. Isolation of a gene encoding a putative leucine zipper structure that is induced by transforming growth factor beta 1 and other growth factors. J Biol Chem. 1992 May 25;267(15):10219–10224. [PubMed] [Google Scholar]
- 27.Schoenenberger GA, Cueni LB, Monnier M, Hatt AM. Humoral transmission of sleep. VII. Isolation and physical-chemical characterization of the “sleep inducing factor delta”. Pflugers Arch. 1972;338(1):1–17. doi: 10.1007/BF00586851. [DOI] [PubMed] [Google Scholar]
- 28.Asselin-Labat ML, Biola-Vidamment A, Kerbrat S, Lombes M, Bertoglio J, Pallardy M. FoxO3 mediates antagonistic effects of glucocorticoids and interleukin-2 on glucocorticoid-induced leucine zipper expression. Mol Endocrinol. 2005 Jul;19(7):1752–1764. doi: 10.1210/me.2004-0206. [DOI] [PubMed] [Google Scholar]
- 29.Cannarile L, Cuzzocrea S, Santucci L, et al. Glucocorticoid-induced leucine zipper is protective in Th1-mediated models of colitis. Gastroenterology. 2009 Feb;136(2):530–541. doi: 10.1053/j.gastro.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 30.Cohen N, Mouly E, Hamdi H, et al. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood. 2006 Mar 1;107(5):2037–2044. doi: 10.1182/blood-2005-07-2760. [DOI] [PubMed] [Google Scholar]
- 31.Hamdi H, Bigorgne A, Naveau S, et al. Glucocorticoid-induced leucine zipper: A key protein in the sensitization of monocytes to lipopolysaccharide in alcoholic hepatitis. Hepatology. 2007 Dec;46(6):1986–1992. doi: 10.1002/hep.21880. [DOI] [PubMed] [Google Scholar]
- 32.Berrebi D, Bruscoli S, Cohen N, et al. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood. 2003 Jan 15;101(2):729–738. doi: 10.1182/blood-2002-02-0538. [DOI] [PubMed] [Google Scholar]
- 33.Godot V, Garcia G, Capel F, et al. Dexamethasone and IL-10 stimulate glucocorticoid-induced leucine zipper synthesis by human mast cells. Allergy. 2006 Jul;61(7):886–890. doi: 10.1111/j.1398-9995.2006.01065.x. [DOI] [PubMed] [Google Scholar]
- 34.Whartenby KA, Straley EE, Kim H, et al. Transduction of donor hematopoietic stem-progenitor cells with Fas ligand enhanced short-term engraftment in a murine model of allogeneic bone marrow transplantation. Blood. 2002 Nov 1;100(9):3147–3154. doi: 10.1182/blood-2002-01-0118. [DOI] [PubMed] [Google Scholar]
- 35.Smits EL, Anguille S, Cools N, Berneman ZN, Van Tendeloo VF. Dendritic Cell-Based Cancer Gene Therapy. Hum Gene Ther. 2009 Sep 9; doi: 10.1089/hum.2009.145. [DOI] [PubMed] [Google Scholar]
- 36.Zhang W, Chen Z, Li F, et al. Tumour necrosis factor-alpha (TNF-alpha) transgene-expressing dendritic cells (DCs) undergo augmented cellular maturation and induce more robust T-cell activation and anti-tumour immunity than DCs generated in recombinant TNF-alpha. Immunology. 2003 Feb;108(2):177–188. doi: 10.1046/j.1365-2567.2003.01489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]