

Abstract
Mycophenolic acid (MPA) is commonly used to treat patients with solid organ transplants during maintenance immunosuppressive therapy. Response to MPA varies widely, both for efficacy and drug-induced toxicity. A portion of this variation can be explained by pharmacokinetic and pharmacodynamic factors, including genetic variation in MPA-metabolizing UDP-glucuronyltransferase isoforms and the MPA targets, inosine monophosphate dehydrogenase 1 and 2. However, much of the variation in MPA response presently remains unexplained. We set out to determine whether there might be additional genes that modify response to MPA by performing a genome-wide association study between basal gene mRNA expression profiles and an MPA cytotoxicity phenotype using a 271 human lymphoblastoid cell line model system to identify and functionally validate genes that might contribute to variation in MPA response. Our association study identified 41 gene expression probe sets, corresponding to 35 genes, that were associated with MPA cytotoxicity as a drug response phenotype (p < 1 × 10−6). Follow-up siRNA-mediated knockdown-based functional validation identified four of these candidate genes, C17orf108, CYBRD1, NASP, and RRM2, whose knockdown shifted the MPA cytotoxicity curves in the direction predicted by the association analysis. These studies have identified novel candidate genes that may contribute to variation in response to MPA therapy and, as a result, may help make it possible to move toward more highly individualized MPA-based immunosuppressive therapy.
Keywords: Mycophenolic acid, mycophenolate, immunosuppression, pharmacogenomics, cell line model system, C17orf108, CYPRD1, NASP, RRM2
1. Introduction
Mycophenolic acid (MPA) is an anti-metabolite that is used as a component of immunosuppressive therapy for patients receiving solid organ transplants [1]. Because of its favorable response rates and side-effect profile [2, 3], MPA has supplanted azathioprine for this purpose [1]. In addition to its use for rejection prophylaxis in solid organ transplant patients, MPA is also being tested in the treatment of autoimmune disease [4, 5] and hematological malignancies such as multiple myeloma [6].
MPA is an uncompetitive inhibitor of the inosine monophosphate dehydrogenases, IMPDH1 and IMPDH2, enzymes that catalyze the rate-limiting step in de novo purine nucleotide synthesis [7]. Inhibition of IMPDH causes an imbalance between adenosine and guanosine nucleotides, resulting in feedback inhibition of the synthesis of the purine nucleotide precursor, 5-phosphoribosyl 1-pyrophosphate. This sequence of events can result in the inhibition of DNA synthesis and cell proliferation [8, 9]. Whereas most cells can utilize recycled purine nucleotides generated by the purine salvage pathway, lymphocytes require the de novo purine nucleotide synthesis pathway to provide adequate purine nucleotides for proliferation [8]. As a result, inhibition of the de novo pathway by MPA inhibits lymphocyte proliferation.
Despite the success of MPA therapy in rejection prophylaxis, treatment response can still be quite variable, with the occurrence of drug-induced toxicity, chronic rejection, and excessive immunosuppression [10]. A portion of this variation results from pharmacokinetic factors. MPA is metabolized by several uridine 5′-diphospho-glucuronosyltransferase (UGT) isoforms and undergoes enterohepatic recirculation, which results in variable plasma drug concentrations [11–13]. In addition, the genes encoding many of the UGT isoforms that metabolize MPA have functional polymorphisms, resulting in altered MPA metabolism and, thus, plasma levels [14]. Furthermore, although some studies have reported correlations between MPA plasma levels and treatment response [15, 16], the issue of whether therapeutic drug monitoring for MPA might be useful remains controversial [17, 18].
Pharmacodynamic factors also contribute to variable MPA response. IMPDH activity varies widely among patients [19, 20], and there is evidence that MPA therapy can induce IMPDH1 and IMPDH2 mRNA expression [21]. Genetic polymorphisms in IMPDH1 and IMPDH2 have also been associated with variable MPA response by altering expression or levels of enzyme activity [22–26]. Therefore, it has been suggested that pharmacodynamic monitoring of MPA treatment by the determination of IMPDH activity might help reduce the incidence of the neutropenia/leukopenia in MPA-treated patients [20, 27]. Since many additional, presently unknown factors may be responsible for variable MPA response, we set out to identify additional genes that contribute to this variation. To do that, we used a well-established approach to pharmacogenomic studies by performing a genome-wide association study with basal gene mRNA expression profiles in a cell-line model system [28, 29]. Specifically, we obtained basal gene expression data for 271 human lymphoblastoid cell lines (LCLs) obtained from healthy subjects using Affymetrix U133 Plus 2.0 GeneChips, and performed MPA cytotoxicity assays with these LCLs to obtain an in vitro MPA response phenotype. We then performed an association study to identify biomarkers for MPA response, followed by functional validation of candidates identified during the association study. These studies represent a step toward the identification of novel mechanisms that might contribute to variation in MPA response.
2. Materials and methods
2.1. Reagents, cell lines and cell culture
Unless otherwise noted, all chemicals were obtained from Sigma-Aldrich (St. Louis, MO). LCLs from 91 African-American (AA), 88 European-American (EA), and 92 Han Chinese-American (HCA) unrelated healthy individuals from Coriell sample sets HD100AA, HD100CAU, and HD100CHI were purchased from the Coriell Cell Repositories (Camden, NJ). These samples had been collected and anonymized by the National Institute of General Medical Sciences, and all subjects provided written consent for the use of their LCLs for research purposes. This study was reviewed and approved by the Mayo Clinic Institutional Review Board. The human non-small cell lung cancer A549 cell line, human embryonic kidney 293T (293T) cell line, the human cervical cancer HeLa cell line, and the human breast carcinoma Hs578T cell line were obtained from ATCC (Manassas, VA). The human ovarian adenocarcinoma IGROV1 and human ovarian carcinoma OVCAR10 cell lines were obtained from Dr. Liewei Wang. LCLs were cultured in RPMI-1640 media (Thermo Scientific, Logan, UT) supplemented with 15% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA). The A549 and OVCAR10 cell lines were grown in RPMI-1640 supplemented with 10% FBS, and 293T, HeLa, Hs578T, and IGROV1 cell lines were grown in DMEM media (Thermo Scientific) supplemented with 10% FBS.
2.2. MPA cytotoxicity assay
MPA (Sigma M3536, BioReagent, ≥ 98%, suitable for cell culture) was dissolved in dimethyl sulfoxide (Sigma D2650, ≥ 99.7%, Hybri-Max™, sterile-filtered, hybridoma tested) to make a 100 mM stock solution. The MPA involved two separate lots, 019k4045 and 119k4014. These stock solutions were stored at −20°C in single-use aliquots. Subsequent dilutions of MPA were made in the appropriate cell culture media supplemented with 1% dimethyl sulfoxide.
Cytotoxicity assays at each MPA dose studied were performed in triplicate. Specifically, 90 μl of cells (5 × 105 cells/ml) were plated into 96-well plates (Corning, Lowell, MA), and were treated with 8 concentrations of MPA ranging from 3.0 nM to 30 μM. After incubation for 3 days, 20 μl of the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) solution (Promega, Madison, WI) was added to each well. Plates were read in a Safire2 microplate reader (Tecan, Durham, NC). MPA cytotoxicity after siRNA knockdown for the A549, 293T, HeLa, and IGROV1 cell lines was determined in the same manner except the cells were incubated with siRNA for 24 h after transfection prior to treatment with 8 concentrations of MPA ranging from 43 nM to 100 μM.
2.3. Expression array assays
Total RNA was extracted from each of the cell lines using RNeasy Mini kits (Qiagen, Valencia, CA). RNA quality was ascertained with an Agilent 2100 Bioanalyzer, and mRNA expression was determined using Human Genome U133 Plus 2.0 Arrays (Affymetrix, Santa Clara, CA). A total of 54,613 probe sets were used in these analyses. The expression array data have been described in detail previously [28–30].
2.4. Candidate gene knockdown by transient siRNA transfection
siRNA pools from the siGENOME series that targeted the candidate genes and the siGENOME Non-Targeting siRNA Pool #1 were obtained from Dharmacon (Lafayette, CO). Transfection using Lipofectamine™ RNAiMAX (Invitrogen, Carlsbad, CA) was performed in 96-well plates. Specifically, 75 μl of cells, 6 × 104 cells/ml, were plated into wells that contained 0.9 pmol of the siRNA pool and 0.1 μl of Lipofectamine™ RNAiMAX dissolved in 15 μl of Opti-MEM® I Reduced-Serum Medium (Invitrogen). The final concentration of the siRNA pool was 10 nM.
2.5. Quantitative real-time reverse transcription-PCR
Total RNA was isolated from cultured cells transfected with control or experimental siRNAs with the Quick-RNA™ MiniPrep kit (Zymo Research, Orange, CA), and knockdown efficiency was assessed using the ΔΔCT method with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the reference gene. Specifically, the Power SYBR® Green RNA-to-CT™ 1-Step kit (Applied Biosystems, Foster City, CA) was used with validated primers obtained from Qiagen, and reactions were performed with the StepOnePlus™ Real-Time PCR System (Applied Biosystems). No-template reactions were included as negative controls.
2.6. Statistical methods and data analysis
The MPA cytotoxicity phenotype, area under the curve (AUC), was calculated per cell line based on a logistic model. Three different logistic functions (four parameter, three free parameters with an asymptote fixed at 0%, and three free parameters with an asymptote fixed at 100%) were fitted to the data using the R package ‘drc‘ (http://cran.r-project.org/doc/packages/drc.pdf). The logistic model with the lowest mean square error was used to determine the AUC cytotoxicity phenotype. AUC was determined by numerically computing the area under the estimated dose-response curve from 3.0 nM to 30 μM. Logarithmic transformed AUC values were then compared between genders, pairs of races and batches of drugs using an independent samples t-test. An overall comparison of transformed AUC values among ethnic groups was performed using an F-test based on ANOVA. AUC values were log-transformed and regressed on gender, ethnic group, and drug batch. Expression array data were normalized on a log2 scale using GCRMA [31]. The normalized expression data were regressed on gender, ethnic group and drug batch effect. The association analyses of adjusted expression and adjusted AUC were then performed using Pearson correlations. False discovery q-values [32] were also computed for each test. To assess repeatability of AUC values, an intraclass correlation coefficient was estimated. Intraclass correlation measures the agreement between repeated AUC measures (i.e. how closely do they follow the line of equality AUCoriginal=AUCreassayed). Strict agreement is not necessary to assure that the relationship between genotypes and AUC is preserved, only correlation between AUCoriginal and AUCreassayed would be required. Therefore, a Pearson correlation coefficient was also calculated to assess repeatability of the relationship between genotype and AUC. Genes were annotated using NCBI Build 36.3.
3. Results
3.1. MPA cytotoxicity
MPA cytotoxicity assays were performed to determine the range of variation in MPA AUC values in the cell lines studied. Six representative MPA cytotoxicity curves are shown in Fig. 1. MPA AUC data from the 271 cell lines studied displayed a skewed distribution and approximately a 7-fold difference between the most and least sensitive cell lines (Fig. 2A). Logarithmic transformation normalized the distribution (Fig. 2B). Logarithmic transformed AUC was not significantly associated with gender data (p = 0.116). However, AUC values differed among ethnic groups (ANOVA p = 0.0163; Fig. 2D). This difference was driven by enhanced sensitivity of the HCA samples when with the EA population (p = 0.00434). There was also a significant “batch effect” for the two different batches of MPA used to perform the cytotoxicity assays (p = 0.0494; Fig. 1E). Because of these differences, the AUC data were adjusted for gender, ethnic group, and batch effects. To test replicability of the cytotoxicity assays, 50 randomly selected cell lines were assayed a second time at a later date. The AUC values for the second assay were significantly correlated with those obtained initially (rp = 0.304, p = 0.0321).
Fig. 1.
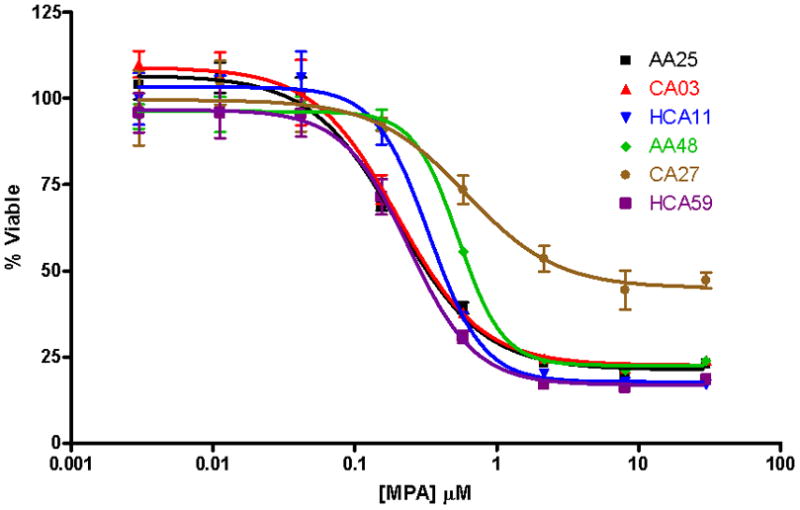
MPA cytotoxicity curves for six lymphoblastoid cell lines. The cells were treated in triplicate with concentrations of MPA ranging from 3.0 nM to 30 μM, and cell viability was determined 3 days later with the MTS assay. Cell viability was always normalized to a no-drug control. Values represent mean ± SD. AA: African-American; EA: European-American; HCA: Han Chinese-American.
Fig. 2.
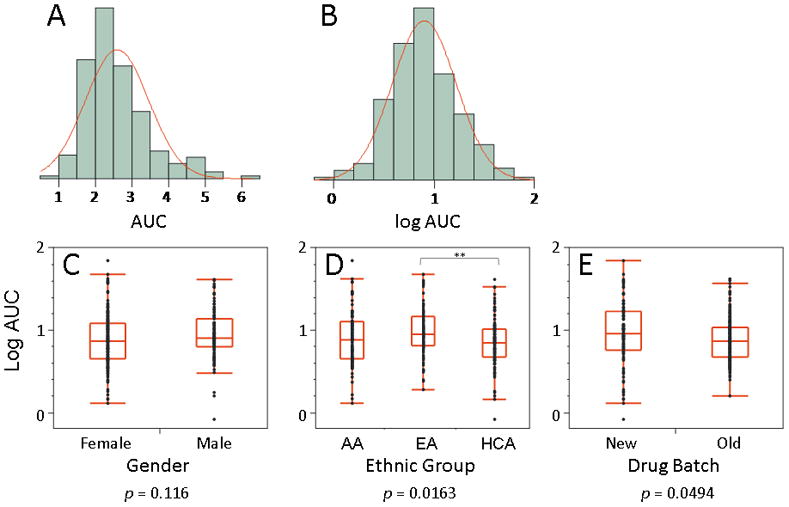
MPA cytotoxicity. (A) The distribution of AUC values was skewed and ranged from 0.91 to 6.37 (approximately a 7-fold difference), with a mean ± SD of 2.59 ± 0.87, and a median value of 2.40. (B) Logarithmic transform of the AUC data. (C) The log-transformed AUC data did not differ by gender (p = 0.116). (D) The log-transformed AUC data differed across ethnic group as assessed by one way ANOVA (p = 0.0163). This difference was mainly due to a pairwise difference between the EA and the HCA cell lines (p = 0.00434). (E) Two batches of MPA were used to perform these studies, and there was a significant difference in mean AUC between batches (p = 0.0494).
3.2. Association between basal gene expression and MPA cytotoxicity
An association analysis between the MPA AUC and genome-wide basal gene expression data was performed to identify genes with expression levels that might be associated with MPA sensitivity. A Manhattan plot of the results showing the significance of the association of each probe set with MPA AUC plotted by chromosomal location of the gene is shown in Fig. 3. The association analysis identified 53 probe sets, corresponding to 44 gene loci, that were associated with MPA AUC at a Bonferroni-corrected significance level of α < 0.05 (p < 1 × 10−6). Probe sets known to cross-hybridize with sequences in non-targeted genes on the basis of Affymetrix annotations were excluded from further analysis, resulting in 41 probe sets corresponding to 35 genes (Table 1). All probe sets with p < 1 × 10−4 are listed in Supplementary Table 1.
Fig. 3.
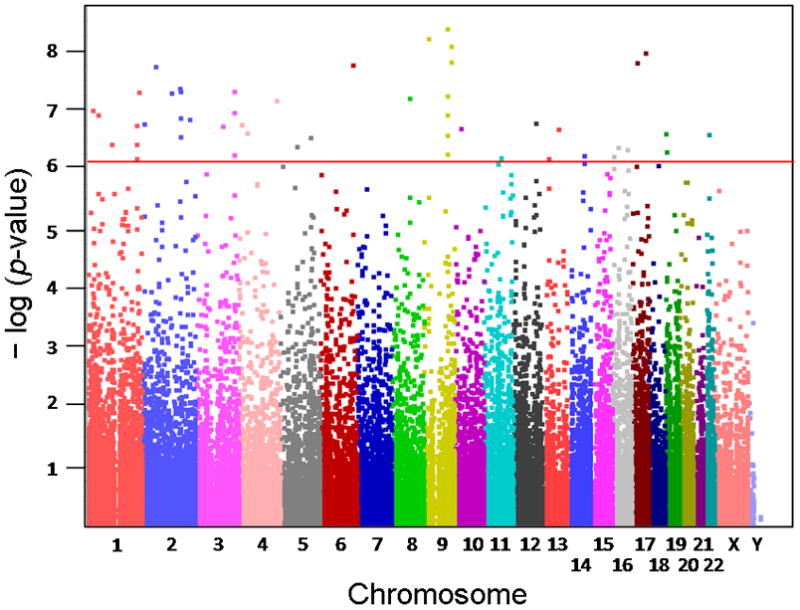
Manhattan plot of the results of the association study between basal mRNA expression levels and MPA AUC cytotoxicity. Each of the probe sets from the Affymetrix Human Genome U133 Plus 2.0 Array is represented as a square. The x-axis represents the chromosomal location of the gene represented by each of the probe sets, and the y-axis shows the negative logarithm of the p-value for the association between basal mRNA expression level and MPA cytotoxicity AUC data. Probe sets that were significant after a Bonferroni correction of α < 0.05 are located above the red horizontal line.
Table 1.
The top 41 probe sets from the basal mRNA expression and MPA AUC cytotoxicity phenotype association analysis with α <0.05. The gene symbols used are the official symbols approved by the HUGO Gene Nomenclature Committee, and the gene IDs correspond to those used by the NCBI Entrez Gene database. The q-values correspond to Storey and Tibshirani’s false discovery q-values [32]. The q-value of a particular association is the expected fraction of false positives in all the other associations with a more significant p-value than this particular association.
| Probe ID | Gene Symbol | Gene ID | Chromosome Location | p-value | r | Bonferroni p-value | q-value |
|---|---|---|---|---|---|---|---|
| 227620_at | SLC44A1 | 23446 | 9q31.2 | 4.02 × 10−9 | −0.348 | 2.20 × 10−4 | 8.75 × 10−5 |
| 231984_at | MTAP | 4507 | 9p21 | 5.98 × 10−9 | −0.344 | 3.26 × 10−4 | 8.75 × 10−5 |
| 223158_s_at | NEK6 | 10783 | 9q33.3-q34.11 | 7.97 × 10−9 | 0.342 | 4.35 × 10−4 | 8.75 × 10−5 |
| 213353_at | ABCA5 | 23461 | 17q24.3 | 1.04 × 10−8 | 0.339 | 5.70 × 10−4 | 8.75 × 10−5 |
| 223159_s_at | NEK6 | 10783 | 9q33.3-q34.11 | 1.47 × 10−8 | 0.336 | 8.03 × 10−4 | 8.75 × 10−5 |
| 242009_at | SLC6A4 | 6532 | 17q11.2 | 1.50 × 10−8 | 0.336 | 8.21 × 10−4 | 8.75 × 10−5 |
| 226024_at | COMMD1 | 150684 | 2p15 | 1.75 × 10−8 | 0.334 | 9.56 × 10−4 | 8.75 × 10−5 |
| 222453_at | CYBRD1 | 79901 | 2q31.1 | 4.09 × 10−8 | 0.326 | 2.24 × 10−3 | 1.53 × 10−4 |
| 219628_at | ZMAT3 | 64393 | 3q26.32 | 4.59 × 10−8 | 0.325 | 2.51 × 10−3 | 1.53 × 10−4 |
| 211932_at | HNRNPA3 | 220988 | 2q31.2 | 4.60 × 10−8 | −0.325 | 2.51 × 10−3 | 1.53 × 10−4 |
| 204137_at | GPR137B | 7107 | 1q42-q43 | 4.82 × 10−8 | 0.324 | 2.63 × 10−3 | 1.53 × 10−4 |
| 201930_at | MCM6 | 4175 | 2q21 | 4.96 × 10−8 | −0.324 | 2.71 × 10−3 | 1.53 × 10−4 |
| 224596_at | SLC44A1 | 23446 | 9q31.2 | 5.53 × 10−8 | −0.323 | 3.02 × 10−3 | 1.58 × 10−4 |
| 201689_s_at | TPD52 | 7163 | 8q21 | 5.96 × 10−8 | −0.322 | 3.25 × 10−3 | 1.59 × 10−4 |
| 208808_s_at | HMGB2 | 3148 | 4q31 | 6.56 × 10−8 | −0.321 | 3.58 × 10−3 | 1.64 × 10−4 |
| 228361_at | E2F2 | 1870 | 1p36 | 9.64 × 10−8 | −0.317 | 5.26 × 10−3 | 2.27 × 10−4 |
| 201970_s_at | NASP | 4678 | 1p34.1 | 1.15 × 10−7 | −0.315 | 6.27 × 10−3 | 2.30 × 10−4 |
| 228485_s_at | SLC44A1 | 23446 | 9q31.2 | 1.15 × 10−7 | −0.315 | 6.29 × 10−3 | 2.30 × 10−4 |
| 201266_at | TXNRD1 | 7296 | 12q23-q24.1 | 1.59 × 10−7 | −0.312 | 8.66 × 10−3 | 2.64 × 10−4 |
| 201890_at | RRM2 | 6241 | 2p25-p24 | 1.61 × 10−7 | −0.312 | 8.81 × 10−3 | 2.64 × 10−4 |
| 205692_s_at | CD38 | 952 | 4p15 | 1.69 × 10−7 | −0.311 | 9.25 × 10−3 | 2.64 × 10−4 |
| 201795_at | LBR | 3930 | 1q42.1 | 1.71 × 10−7 | −0.311 | 9.36 × 10−3 | 2.64 × 10−4 |
| 219874_at | SLC12A8 | 84561 | 3q21.2 | 1.79 × 10−7 | 0.311 | 9.79 × 10−3 | 2.65 × 10−4 |
| 235421_at | MAP3K8 | 1326 | 10p11.23 | 1.94 × 10−7 | 0.310 | 0.0106 | 2.78 × 10−4 |
| 225312_at | COMMD6 | 170622 | 13q22 | 2.02 × 10−7 | 0.309 | 0.0110 | 2.79 × 10−4 |
| 212518_at | PIP5K1C | 23396 | 19p13.3 | 2.37 × 10−7 | 0.308 | 0.0129 | 3.03 × 10−4 |
| 228486_at | SLC44A1 | 23446 | 9q31.2 | 2.50 × 10−7 | −0.307 | 0.0137 | 3.03 × 10−4 |
| 219293_s_at | OLA1 | 29789 | 2q31.1 | 2.69 × 10−7 | 0.306 | 0.0147 | 3.16 × 10−4 |
| 206879_s_at | NRG2 | 9542 | 5q23-q33 | 2.79 × 10−7 | 0.306 | 0.0152 | 3.18 × 10−4 |
| 205240_at | GPSM2 | 29899 | 1p13.3 | 3.61 × 10−7 | −0.303 | 0.0197 | 3.90 × 10−4 |
| 206668_s_at | SCAMP1 | 9522 | 5q14.1 | 3.96 × 10−7 | −0.302 | 0.0216 | 4.07 × 10−4 |
| 224984_at | NFAT5 | 10725 | 16q22.1 | 4.34 × 10−7 | 0.301 | 0.0237 | 4.33 × 10−4 |
| 219424_at | EBI3 | 10148 | 19p13.3 | 4.77 × 10−7 | 0.300 | 0.0260 | 4.65 × 10−4 |
| 222364_at | SLC44A1 | 23446 | 9q31.2 | 5.22 × 10−7 | −0.299 | 0.0285 | 4.96 × 10−4 |
| 244033_at | C14orf145 | 145508 | 14q31.1 | 5.54 × 10−7 | −0.299 | 0.0303 | 5.04 × 10−4 |
| 209460_at | ABAT | 18 | 16p13.2 | 5.75 × 10−7 | 0.298 | 0.0314 | 5.11 × 10−4 |
| 229873_at | KCTD21 | 283219 | 11q14.1 | 6.00 × 10−7 | 0.298 | 0.0327 | 5.20 × 10−4 |
| 232635_at | C14orf145 | 145508 | 14q31.1 | 7.35 × 10−7 | −0.296 | 0.0401 | 6.00 × 10−4 |
| 214369_s_at | RASGRP2 | 10235 | 11q13 | 7.73 × 10−7 | −0.295 | 0.0422 | 6.18 × 10−4 |
| 223071_at | IER3IP1 | 51124 | 18q12 | 8.06 × 10−7 | −0.295 | 0.0440 | 6.32 × 10−4 |
| 213195_at | C17orf108 | 201229 | 17q11.2 | 8.43 × 10−7 | 0.294 | 0.0460 | 6.36 × 10−4 |
3.3. Functional screening of candidate genes
We next set out to functionally validate the 35 candidate genes listed in Table 1 by performing siRNA knockdown, followed by cytotoxicity studies. These studies were performed with cancer cell lines to test the generalizability of the LCL data. We began by using mRNA expression array data generated for a series of commonly-used immortalized cell lines to choose those to include in our functional screen; i.e. we selected cell lines that expressed the gene of interest. We used only adherent cell lines for the functional screen, in part because of their relative ease of transfection, which made it possible to use identical experimental conditions during the screening of each cell line.
Of the 35 genes screened, four, C17orf108, CYBRD1, NASP, and RRM2 displayed significant and replicable shifts in their MPA cytotoxicity curves after siRNA knockdown (Fig. 4 and Fig. 5). For all genes studied, we also determined the extent of “knockdown” by performing qRT-PCR of the target gene. Those values are listed for each of the genes shown in Fig. 4 and Fig. 5. Knockdown of C17orf108 in A549 cells (Fig. 4A) and OVCAR10 cells (Fig. 4B) resulted in a significant (p < 0.05) leftward shift of the cytotoxicity curve as compared to the non-targeting siRNA control. The dotted lines in both Fig. 4 and Fig. 5 represent the 95% confidence intervals for the cytotoxicity curves. In both cases, they did not overlap at MPA concentrations at the IC50. Knockdown efficiency as assessed by qRT-PCR showed that only 5% of C17orf108 mRNA remained in A549 cells transfected with the pooled C17orf108 siRNA when compared to that in cells transfected with the non-targeting control siRNA—a knockdown efficiency of 95%. In the OVCAR10 cells, a knockdown efficiency of 83% was achieved. The left shift of the two dose response curves agreed with the direction of the effect size predicted by the association analysis, r = 0.294. In the association analysis, a positive r-value indicated that, as the basal mRNA expression level of a gene increased, the cells became more resistant to MPA. Conversely, a negative r-value indicated that as the basal mRNA expression of a gene decreased, the cells became more sensitive to MPA. In this case, the positive r-value from the association study meant that a decrease of mRNA expression levels as a result of siRNA transfection would be expected to cause the siRNA-transfected cells to become more sensitive to MPA, exactly what we observed. A 98% knockdown of CYBRD1 in A549 cells also showed a similar left shift, confirming the association study r of 0.326 (Fig. 4C). A similar shift was also observed in OVCAR10 cells after a 68% knockdown of CYBRD1 (Fig. 4D). However, candidate gene knockdowns did not just result in MPA sensitization. A 96% knockdown of NASP in IGROV1 cells resulted in a right shift in the cytotoxicity curve (Fig. 5A), agreeing with the directionality predicted by the association analysis (r = −0.315). Knockdown of NASP in Hs578T cells confirmed this observation (Fig. 5B). A 97% knockdown of RRM2 in IGROV1 cells also resulted in a right shift (Fig. 5C) that agreed with the association analysis results (r = −0.312). Similar observations were made with Hs578T cells (Fig. 5D). Unfortunately, we isolated too little RNA from the Hs578T cells to measure knockdown efficiency, but previous experience has shown the RRM2 siRNA pool to be very efficient.
Fig. 4.
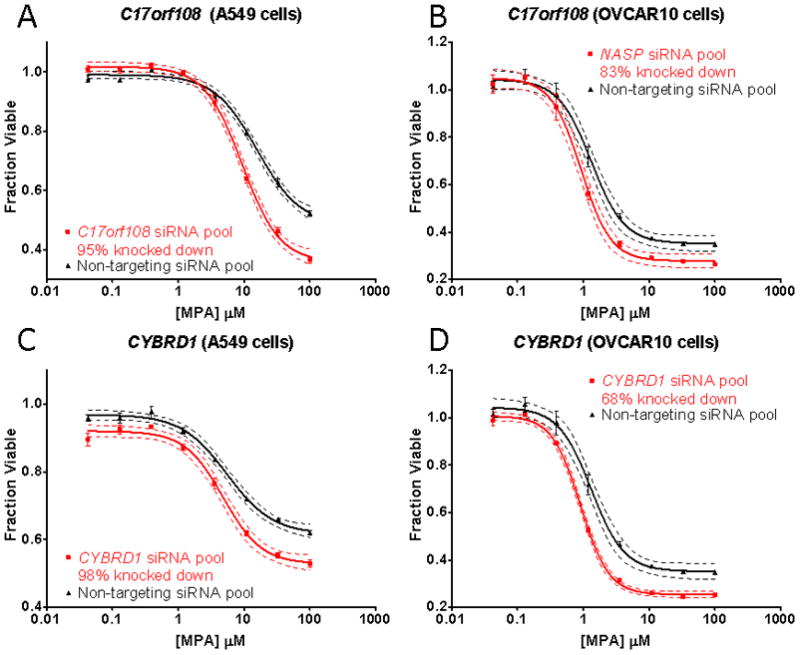
Candidate gene functional validation using A549 and OVCAR10 cells. A pooled siRNA knockdown of C17orf108 in (A) A549 and (B) OVCAR10 cells sensitized the cells to MPA as compared to control cells transfected with non-targeting pooled siRNA. qRT-PCR showed that only 5% of the C17orf108 mRNA remained in the A549 cells as compared to the control; the OVCAR10 cells had 17% of C17orf108 mRNA remaining CYBRD1 functional validation in (C) A549 cells and (D) OVCAR10 cells. Each of the cytotoxicity curves was performed in triplicate. Bars represent the SEM, and dashed curves represent 95% confidence intervals for each cytotoxicity curve.
Fig. 5.
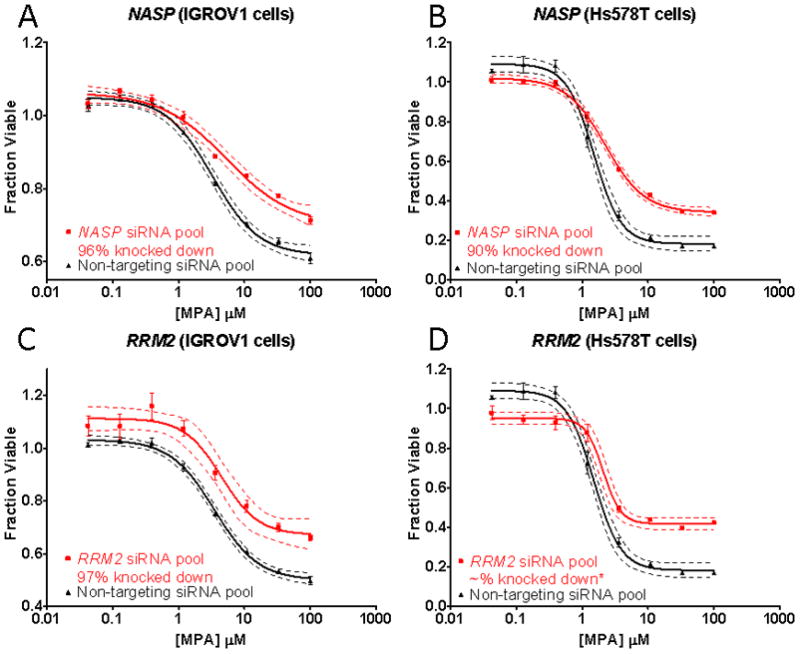
Candidate gene functional validation using IGROV1 and Hs578T cells. (A) NASP functional validation in (A) IGROV1 cells and (B) Hs578T cells. (C) RRM2 functional validation in IGROV1 cells and (D) Hs578T cells. Each of the cytotoxicity curves was performed in triplicate. Bars represent the SEM, and dashed curves represent 95% confidence intervals for each cytotoxicity curve. *We were unable to perform qRT-PCR assays to determine RRM2 mRNA levels because of insufficient template.
4. Discussion
MPA is a major component of immunosuppressive therapy for solid organ transplant patients and is administered to a majority of these patients. There is mounting evidence that “standard doses” of MPA might not be the best way to use this drug. Proponents of individualized MPA dosing have proposed therapeutic drug monitoring and/or pharmacodynamic monitoring of IMPDH activity as possible ways to individualize MPA therapy [17, 27]. Because it remains unclear whether either of these approaches might be clinically useful in reducing rejection rates or adverse drug reactions in transplant patients, there is increasing interest in genotyping for IMPDH1 and IMPDH2 variants that might be associated with differences in rejection rates in patients treated with MPA [10]. However, studies of the effect of IMPDH1 and IMPDH2 genetic variants on rejection rates have identified only a small number of variant alleles with modest functional impact. Therefore, we set out to use genome-wide association in an attempt to identify novel genes that might influence MPA response which might, in turn, influence rejection rates in patients treated with this drug. Specifically, we hypothesized that basal levels of mRNA expression for these genes might influence MPA sensitivity, and that it might be possible to use individual variation in basal mRNA expression to identify these genes during the association study.
Specifically, we performed a genome-wide association analysis using 271 LCLs for which we had obtained basal genome-wide basal mRNA expression data and generated MPA cytotoxicity phenotypes for each of these cell lines. This approach has been used successfully in many previous studies designed to identify genetic biomarkers associated with variation in drug response [28–30, 33, 34]. We should emphasize that LCLs differ biologically from lymphocytes, but LCLs are much more similar to the cells inhibited by MPA in vivo than was the case for many of the previous successful studies of antineoplastic drugs performed using this approach. Our association analysis identified 41 probe sets, corresponding to 35 genes, all of which passed a stringent Bonferroni correction at α < 0.05.
The LCL model system, like all model systems, has limitations. For instance, cellular changes and chromosomal instability have been shown to occur during the EBV transformation used to generate LCLs [35], and non-genetic factors such as cell growth rates and baseline ATP levels might contribute to variation in drug response in these cells [36]. Therefore, to eliminate false positives, we also functionally validated the 35 candidate genes identified during the association study by performing siRNA gene knockdown studies. We performed these screening studies using adherent cells that expressed the gene being tested. That was done because we could then perform an identical functional screen across the different cell lines chosen for all of the candidate genes. Even though only 4 of the 35 genes were unequivocally validated during our siRNA-mediated knockdown screen, the other 31 genes should not be discounted as potentially viable candidates.
Among the 4 validated candidates, RRM2 is a subunit for the ribonucleotide reductase holoenzyme that is responsible for the conversion of nucleotide triphosphates to deoxynucleotide triphosphates [37]. Ribonucleotide reductase is also linked to the effects of many cancer chemotherapeutic and antiviral agents because of its crucial role in DNA synthesis [38], so it was not surprising that variation in the expression of one of its subunits, RRM2, might be associated with variable MPA response. Our identification and functional validation of RRM2 should also enhance confidence that our genome-wide screen for potential biomarkers for MPA response was capable of identifying plausible candidates.
Although some functional information is available for CYBRD1 and NASP, exactly how these genes might influence MPA response is less clear than is the case for RRM2. CYBRD1 is known to have ferric and cupric reductase activity [39], is highly expressed in the duodenal brush border membrane, and might be involved in dietary iron absorption [40]. Because of its ability to catalyze redox reactions, CYBRD1 might alter the redox state of cells, and, hence affect IMPDH activity through NADH/NAD+ balance. Because of the expression pattern for CYBRD1, it is tempting to speculate whether this gene might be associated with the adverse gastrointestinal reactions experienced by a significant portion of patients treated with MPA. NASP is a histone chaperone that preferentially binds to histones H3, H4 and the linker histone H1, thus promoting nucleosome assembly [41]. Because NASP is required for cell cycle progression and cell proliferation [42], it is possible that alterations in NASP expression might influence MPA response, especially since MPA’s main function is to inhibit lymphocyte proliferation. Even less is known about C17orf108. This gene encodes a validated cDNA for a hypothetical 77 amino acid protein, LOC201229, of unknown function [43]. However, C17orf108 is highly conserved through evolution, and human LOC201229 is 91.0% identical in protein sequence with the sequence of the homologous M. musculus 1810012P15Rik protein and 64.9% identical with the homologous D. rerio LOC563525 protein sequence.
In summary, we have performed a “discovery” association study using basal mRNA expression array and MPA cytotoxicity data for 271 human lymphoblastoid cell lines in an attempt to identify biomarkers that might help to predict response to MPA, followed by functional validation for selected candidate genes. Our studies have identified and functionally validated genes and mechanisms that might contribute to variation in response to MPA therapy. These observations represent a step toward individualized therapy with this important immunosuppressant agent.
Supplementary Material
Acknowledgments
Role of the Funding Sources
This work was supported in part by a Predoctoral Fellowship Award from the American Heart Association to TYW (09PRE2080377); by NIH grants R01 GM28157, U19 GM61388 (The Pharmacogenomics Research Network), R01 CA132780, K22 CA130828 and R01 CA138461; and by a PhRMA Foundation “Center of Excellence Award in Clinical Pharmacology”. The study sponsors had no role in the study design; in the collection, analysis, and interpretation of data; in the writing of this report; and no role in the decision to submit this manuscript for publication.
Footnotes
Conflicts of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Tse-Yu Wu, Email: Wu.TseYu@mayo.edu.
Brooke L. Fridley, Email: Fridley.Brooke@mayo.edu.
Gregory D. Jenkins, Email: Jenkins.Gregory@mayo.edu.
Anthony Batzler, Email: Batzler.Anthony@mayo.edu.
Liewei Wang, Email: Wang.Liewei@mayo.edu.
Richard M. Weinshilboum, Email: Weinshilboum.Richard@mayo.edu.
References
- 1.HRSA. 2008 Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients: Transplant Data 1998–2007. U.S. Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation; Rockville, MD: 2008. [Google Scholar]
- 2.A blinded, randomized clinical trial of mycophenolate mofetil for the prevention of acute rejection in cadaveric renal transplantation. The Tricontinental Mycophenolate Mofetil Renal Transplantation Study Group. Transplantation. 1996;61:1029–37. [PubMed] [Google Scholar]
- 3.Sollinger HW. Mycophenolate mofetil for the prevention of acute rejection in primary cadaveric renal allograft recipients. U.S. Renal Transplant Mycophenolate Mofetil Study Group. Transplantation. 1995;60:225–32. doi: 10.1097/00007890-199508000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Hiemstra TF, Jones RB, Jayne DR. Treatment of primary systemic vasculitis with the inosine monophosphate dehydrogenase inhibitor mycophenolic acid. Nephron Clin Pract. 2010;116:c1–10. doi: 10.1159/000314543. [DOI] [PubMed] [Google Scholar]
- 5.Mak A, Cheak AA, Tan JY, Su HC, Ho RC, Lau CS. Mycophenolate mofetil is as efficacious as, but safer than, cyclophosphamide in the treatment of proliferative lupus nephritis: a meta-analysis and meta-regression. Rheumatology (Oxford) 2009;48:944–52. doi: 10.1093/rheumatology/kep120. [DOI] [PubMed] [Google Scholar]
- 6.Takebe N, Cheng X, Wu S, Bauer K, Goloubeva OG, Fenton RG, et al. Phase I clinical trial of the inosine monophosphate dehydrogenase inhibitor mycophenolate mofetil (cellcept) in advanced multiple myeloma patients. Clin Cancer Res. 2004;10:8301–8. doi: 10.1158/1078-0432.CCR-04-0747. [DOI] [PubMed] [Google Scholar]
- 7.Wu JC. Mycophenolate mofetil: Molecular mechanisms of action. Perspectives in Drug Discovery and Design. 1994;2:185–204. [Google Scholar]
- 8.Allison AC, Eugui EM. Mechanisms of action of mycophenolate mofetil in preventing acute and chronic allograft rejection. Transplantation. 2005;80:S181–90. doi: 10.1097/01.tp.0000186390.10150.66. [DOI] [PubMed] [Google Scholar]
- 9.Villarroel MC, Hidalgo M, Jimeno A. Mycophenolate mofetil: An update. Drugs Today (Barc) 2009;45:521–32. doi: 10.1358/dot.2009.45.7.1384878. [DOI] [PubMed] [Google Scholar]
- 10.Barraclough KA, Lee KJ, Staatz CE. Pharmacogenetic influences on mycophenolate therapy. Pharmacogenomics. 2010;11:369–90. doi: 10.2217/pgs.10.9. [DOI] [PubMed] [Google Scholar]
- 11.Shipkova M, Armstrong VW, Wieland E, Niedmann PD, Schutz E, Brenner-Weiss G, et al. Identification of glucoside and carboxyl-linked glucuronide conjugates of mycophenolic acid in plasma of transplant recipients treated with mycophenolate mofetil. Br J Pharmacol. 1999;126:1075–82. doi: 10.1038/sj.bjp.0702399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez A, Martins J, Villlafruela JJ, Marcen R, Pascual J, Cano T, et al. Variability of mycophenolate mofetil trough levels in stable kidney transplant patients. Transplant Proc. 2007;39:2185–6. doi: 10.1016/j.transproceed.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 13.Pisupati J, Jain A, Burckart G, Hamad I, Zuckerman S, Fung J, et al. Intraindividual and interindividual variations in the pharmacokinetics of mycophenolic acid in liver transplant patients. J Clin Pharmacol. 2005;45:34–41. doi: 10.1177/0091270004270145. [DOI] [PubMed] [Google Scholar]
- 14.Levesque E, Delage R, Benoit-Biancamano MO, Caron P, Bernard O, Couture F, et al. The impact of UGT1A8, UGT1A9, and UGT2B7 genetic polymorphisms on the pharmacokinetic profile of mycophenolic acid after a single oral dose in healthy volunteers. Clin Pharmacol Ther. 2007;81:392–400. doi: 10.1038/sj.clpt.6100073. [DOI] [PubMed] [Google Scholar]
- 15.Le Meur Y, Buchler M, Thierry A, Caillard S, Villemain F, Lavaud S, et al. Individualized mycophenolate mofetil dosing based on drug exposure significantly improves patient outcomes after renal transplantation. Am J Transplant. 2007;7:2496–503. doi: 10.1111/j.1600-6143.2007.01983.x. [DOI] [PubMed] [Google Scholar]
- 16.Weber LT, Shipkova M, Armstrong VW, Wagner N, Schutz E, Mehls O, et al. The pharmacokinetic-pharmacodynamic relationship for total and free mycophenolic Acid in pediatric renal transplant recipients: a report of the german study group on mycophenolate mofetil therapy. J Am Soc Nephrol. 2002;13:759–68. doi: 10.1681/ASN.V133759. [DOI] [PubMed] [Google Scholar]
- 17.Barraclough KA, Staatz CE, Isbel NM, Johnson DW. Therapeutic monitoring of mycophenolate in transplantation: is it justified? Curr Drug Metab. 2009;10:179–87. doi: 10.2174/138920009787522205. [DOI] [PubMed] [Google Scholar]
- 18.Knight SR, Morris PJ. Does the evidence support the use of mycophenolate mofetil therapeutic drug monitoring in clinical practice? A systematic review. Transplantation. 2008;85:1675–85. doi: 10.1097/TP.0b013e3181744199. [DOI] [PubMed] [Google Scholar]
- 19.Glander P, Braun KP, Hambach P, Bauer S, Mai I, Roots I, et al. Non-radioactive determination of inosine 5′-monophosphate dehydro-genase (IMPDH) in peripheral mononuclear cells. Clin Biochem. 2001;34:543–9. doi: 10.1016/s0009-9120(01)00267-3. [DOI] [PubMed] [Google Scholar]
- 20.Glander P, Hambach P, Braun KP, Fritsche L, Giessing M, Mai I, et al. Pre-transplant inosine monophosphate dehydrogenase activity is associated with clinical outcome after renal transplantation. Am J Transplant. 2004;4:2045–51. doi: 10.1111/j.1600-6143.2004.00617.x. [DOI] [PubMed] [Google Scholar]
- 21.Sanquer S, Breil M, Baron C, Dhamane D, Astier A, Lang P. Induction of inosine monophosphate dehydrogenase activity after long-term treatment with mycophenolate mofetil. Clin Pharmacol Ther. 1999;65:640–8. doi: 10.1016/S0009-9236(99)90085-1. [DOI] [PubMed] [Google Scholar]
- 22.Sombogaard F, van Schaik RH, Mathot RA, Budde K, van der Werf M, Vulto AG, et al. Interpatient variability in IMPDH activity in MMF-treated renal transplant patients is correlated with IMPDH type II 3757T > C polymorphism. Pharmacogenet Genomics. 2009;19:626–34. doi: 10.1097/FPC.0b013e32832f5f1b. [DOI] [PubMed] [Google Scholar]
- 23.Wang J, Yang JW, Zeevi A, Webber SA, Girnita DM, Selby R, et al. IMPDH1 gene polymorphisms and association with acute rejection in renal transplant patients. Clin Pharmacol Ther. 2008;83:711–7. doi: 10.1038/sj.clpt.6100347. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Zeevi A, Webber S, Girnita DM, Addonizio L, Selby R, et al. A novel variant L263F in human inosine 5′-monophosphate dehydrogenase 2 is associated with diminished enzyme activity. Pharmacogenet Genomics. 2007;17:283–90. doi: 10.1097/FPC.0b013e328012b8cf. [DOI] [PubMed] [Google Scholar]
- 25.Winnicki W, Weigel G, Sunder-Plassmann G, Bajari T, Winter B, Herkner H, et al. An inosine 5′-monophosphate dehydrogenase 2 single-nucleotide polymorphism impairs the effect of mycophenolic acid. Pharmacogenomics J. 2010;10:70–6. doi: 10.1038/tpj.2009.43. [DOI] [PubMed] [Google Scholar]
- 26.Gensburger O, Van Schaik RH, Picard N, Le Meur Y, Rousseau A, Woillard JB, et al. Polymorphisms in type I and II inosine monophosphate dehydrogenase genes and association with clinical outcome in patients on mycophenolate mofetil. Pharmacogenet Genomics. 20:537–43. doi: 10.1097/FPC.0b013e32833d8cf5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glander P, Budde K. Target enzyme activity as a biomarker for immunosuppression. Ther Drug Monit. 2010;32:257–60. doi: 10.1097/FTD.0b013e3181ddd7b3. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Fridley B, Kalari K, Jenkins G, Batzler A, Safgren S, et al. Gemcitabine and cytosine arabinoside cytotoxicity: association with lymphoblastoid cell expression. Cancer Res. 2008;68:7050–8. doi: 10.1158/0008-5472.CAN-08-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niu N, Qin Y, Fridley BL, Hou J, Kalari KR, Zhu M, et al. Radiation pharmacogenomics: a genome-wide association approach to identify radiation response biomarkers using human lymphoblastoid cell lines. Genome Res. 2010;20:1482–92. doi: 10.1101/gr.107672.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, Fridley BL, Kalari K, Jenkins G, Batzler A, Weinshilboum RM, et al. Gemcitabine and arabinosylcytosin pharmacogenomics: genome-wide association and drug response biomarkers. PLoS One. 2009;4:e7765. doi: 10.1371/journal.pone.0007765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu ZJ, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J Am Stat Assoc. 2004;99:909–17. [Google Scholar]
- 32.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bleibel WK, Duan S, Huang RS, Kistner EO, Shukla SJ, Wu X, et al. Identification of genomic regions contributing to etoposide-induced cytotoxicity. Hum Genet. 2009;125:173–80. doi: 10.1007/s00439-008-0607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang RS, Duan S, Kistner EO, Hartford CM, Dolan ME. Genetic variants associated with carboplatin-induced cytotoxicity in cell lines derived from Africans. Mol Cancer Ther. 2008;7:3038–46. doi: 10.1158/1535-7163.MCT-08-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sie L, Loong S, Tan EK. Utility of lymphoblastoid cell lines. J Neurosci Res. 2009;87:1953–9. doi: 10.1002/jnr.22000. [DOI] [PubMed] [Google Scholar]
- 36.Choy E, Yelensky R, Bonakdar S, Plenge RM, Saxena R, De Jager PL, et al. Genetic analysis of human traits in vitro: drug response and gene expression in lymphoblastoid cell lines. PLoS Genet. 2008;4:e1000287. doi: 10.1371/journal.pgen.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jordan A, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- 38.Cerqueira NM, Fernandes PA, Ramos MJ. Ribonucleotide reductase: a critical enzyme for cancer chemotherapy and antiviral agents. Recent Pat Anticancer Drug Discov. 2007;2:11–29. doi: 10.2174/157489207779561408. [DOI] [PubMed] [Google Scholar]
- 39.Wyman S, Simpson RJ, McKie AT, Sharp PA. Dcytb (Cybrd1) functions as both a ferric and a cupric reductase in vitro. FEBS Lett. 2008;582:1901–6. doi: 10.1016/j.febslet.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Latunde-Dada GO, Simpson RJ, McKie AT. Duodenal cytochrome B expression stimulates iron uptake by human intestinal epithelial cells. J Nutr. 2008;138:991–5. doi: 10.1093/jn/138.6.991. [DOI] [PubMed] [Google Scholar]
- 41.Osakabe A, Tachiwana H, Matsunaga T, Shiga T, Nozawa RS, Obuse C, et al. Nucleosome formation activity of human somatic nuclear autoantigenic sperm protein (sNASP) J Biol Chem. 2010;285:11913–21. doi: 10.1074/jbc.M109.083238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richardson RT, Alekseev OM, Grossman G, Widgren EE, Thresher R, Wagner EJ, et al. Nuclear autoantigenic sperm protein (NASP), a linker histone chaperone that is required for cell proliferation. J Biol Chem. 2006;281:21526–34. doi: 10.1074/jbc.M603816200. [DOI] [PubMed] [Google Scholar]
- 43.Ota T, Suzuki Y, Nishikawa T, Otsuki T, Sugiyama T, Irie R, et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat Genet. 2004;36:40–5. doi: 10.1038/ng1285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.