

Abstract
Lack of target specificity by existing matrix metalloproteinase (MMP) inhibitors has hindered anti-metastatic cancer drug discovery. Inhibitors that bind to non-catalytic sites of MMPs and disrupt protease signaling function have the potential to be more specific and selective. In this work, compounds that target the hemopexin (PEX) domain of MMP-9 were identified using an in silico docking approach and evaluated using biochemical and biological approaches. Two of the selected compounds interfere with MMP-9-mediated cancer cell migration and proliferation in cells expressing exogenous or endogenous MMP-9. Furthermore, these inhibitors do not modulate MMP-9 catalytic activity. The lead compound, N-[4-(difluoromethoxy)phenyl]-2-[(4-oxo-6-propyl-1H-pyrimidin-2-yl)sulfanyl]-acetamide, specifically binds to the PEX domain of MMP-9, but not other MMPs. This interaction between the compound and the PEX domain results in the abrogation of MMP-9 homodimerization and leads to blockage of a downstream signaling pathway required for MMP-9-mediated cell migration. In a tumor xenografic model, this pyrimidinone retarded MDA-MB-435 tumor growth and inhibited lung metastasis. Thus, we have demonstrated for the first time that a novel small molecule interacts specifically with the PEX domain of MMP-9 and inhibits tumor growth and metastasis by reducing cell migration and proliferation.
Keywords: MMP-9, small molecule compound, hemopexin, migration, metastasis
Introduction
Mortality in cancer is primarily due to failure to prevent metastasis. Much attention has been focused on targeting tumor growth; drug discovery targeting metastasis has lagged far behind. Thus, there is a pressing need for novel treatment strategies to prevent metastasis. Emerging evidence has emphasized the role of matrix metalloproteinases (MMPs) in early aspects of cancer dissemination (1–3). The demonstration that several MMPs display pro-tumor, as well as anti-tumor effects (4), highlights that more specific inhibitory drugs are required for clinical development.
MMPs have also been implicated in other disease entities, leading to the development of numerous drugs, which interfere with MMP enzymatic activity (5). Several classes of compounds, including peptidomimetics, tetracyclines and bisphosphonates, have been designed to bind and inhibit the catalytic activity of MMPs (6, 7). However, the catalytic domains of all MMPs share a highly conserved binding site and lack of specificity of these MMP inhibitors (MMPIs) has hindered their development as drugs. After the failure of broad-spectrum MMPIs in the treatment of cancer in phase III clinical trials, a re-evaluation of the biological roles of the MMPs has been undertaken (8).
A major conceptual advance in the development of novel MMPIs is to target less conserved, non-catalytic domains of the proteases to increase target specificity and selectivity. Exosites are crucial for the catalytic functions of most MMPs; enzyme lacking their PEX domain or the addition of an exogenous PEX domain greatly inhibits the proteolytic efficiency of the enzyme (9–11). Because the PEX domains of MMPs are not as highly conserved as the catalytic sites (Supplemental Table 1), the PEX domain is an alternative site that can inhibit the biological roles of MMPs with greater selectivity (12, 13). Novel therapeutics targeting MMP exosites are currently being evaluated with a focus to develop drugs with fewer side effects than previously developed broad-spectrum catalytic-site inhibitors (8, 14).
MMP-9 is linked to many pathological processes including cancer invasion, metastasis, and angiogenesis, as well as cardiovascular, neurologic and inflammatory diseases (2, 3, 15). Elevated levels of MMP-9 in tissue and blood are observed in these conditions. Active MMP-9 is an attractive target for cancer therapy development (16). The ability of MMP-9 to degrade collagen and laminin correlates with its ability to regulate cell migration, increase angiogenesis and affect tumor growth (15, 17). In addition to the effects of activated MMP-9 in degrading substrates and cleaving biologically relevant proteins, proMMP-9 induces cell migration independent of any proteolytic activity (12, 13, 17). Enhanced epithelial cell migration is linked to the formation of homodimers through the MMP-9 PEX domain, as well as heterodimers with other cell surface molecules (12, 13).
In the present study, we utilized an in silico docking approach to screen for novel compounds that bind to the MMP-9 PEX domain. Experimental assay of the best fitting compounds identified a small molecule with micromolar affinity for MMP-9. This small molecule selectively inhibits cell migration, proliferation, invasion, tumor growth, and metastasis induced by MMP-9.
Materials and Methods
Cell Culture, Reagents and Transfection
COS-1 monkey epithelial, human HT-1080, and MDA-MB-435 cancer cell lines, and murine macrophage-like RAW246.7 cell line were purchased from ATCC (Manassas, VA) and were maintained in DMEM (Invitrogen) containing 10% fetal calf serum. Transfection of plasmid DNA (human) into cells was achieved using polyethylenimine (Polysciences) and the transfected cells were incubated for 48 h at 37 °C followed by assay. MMP-9 and MMP-9/MMP-2PEX (13) proteins were purified from transfected cell-conditioned media by gelatin-Sepharose chromatography. Compounds 1–5 (Supplemental Table 2) were purchased from Enamine Ltd. (Kiev, Ukraine) and their purity was verified by LC/MS to be greater than 98%. Anti-tubulin, anti-AKT, anti-pAKT, anti-ERK1/2, and anti-pERK1/2 antibodies were purchased from Cell Signaling Technology (Davers, MA). Mac-P-L-G-L-Dpa-A-R-NH2 fluorogenic peptide was obtained from R & D Systems (Minneapolis, MN, USA).
Fluorogenic Assay of Enzyme Activity
Fluorogenic peptide substrate (50 μM) (18) was incubated with the compounds either in the presence or absence of latent MMP-9 and APMA-activated MMP-9 for 30 min at 25 °C before detection. Fluorescence emission at 393 nm with excitation at 328 nm was measured in a fluorescent plate reader (Gemini EM, Molecular Devices).
Fluorescence Spectroscopy
Binding of compound 2 to MMP-9 was assayed by observing the change of tryptophan emission upon binding. Purified recombinant MMP-9 (50 nM) or MMP-9/MMP-2PEX (50 nM) was diluted in buffer (50 mM Tris-HCl, 60 mM KCl and 0.05% Tween 20, pH 7.4) in the presence or absence of compound 2. As a control for protein stability and loss, an analogous buffer solution was added to the protein. The protein sample was excited at 280 nm and emission scans were collected from 290 to 400 nm, using slit widths of 0.3 nm on a QM-4/200SE spectrofluorimeter with double excitation and emission monochromators. Three emission scans were collected and averaged at each concentration. The Kd was determined using the Prism software package (GraphPad V5) to fit the data to equation (1).
| (1) |
in which λmax is the wavelength at which maximal fluorescence of the protein was observed.
Cell Viability
Compound cytotoxicity was determined using the CellTiter-Glo™ Luminescent Cell Viability Assay (Promega Corporation, Madison, WI). 2.5 × 104 COS-1 cells were plated to a 96-well plate and incubated for 18 h with compounds 1–5. Luminescence was recorded using a SpectraMax Microplate Reader (Molecular Devices). LD50’s of the compounds were measured over a 100 pM to 10 mM concentration range. The LD50 was determined using the Prism software package (GraphPad V5) and fitting to equation (2).
| (2) |
in which L = the measured luminescence.
Cell Proliferation
Cell proliferation was determined using the CellTiter-Glo™ Luminescent Assay. Cells (5 × 103) were added to a 96-well plate in the presence or absence of the compounds and monitored for 9 days by luminescence assay.
In vivo Study
Human MDA-MB-435 cancer cells (2 × 106) expressing green fluorescent protein (GFP) cDNA were inoculated subcutaneously into 4–5 week-old female NCR-Nu mice with 5 mice per group (Taconic). Once palpable, tumors were measured twice/week and volume was calculated using the following formula: length × width × height × 0.5236. Mice were treated with a vehicle control (DMSO/PBS), compound 2, or 4 (20 mg/kg) via intraperitoneal and intratumoral injection alternately (6 days/week). At 14 weeks, the mice were sacrificed and the tumors and lungs were dissected. Fresh lung sections were cut (~3 mm thick) and examined for the presence of GFP-expressing tumor foci. The area of metastatic foci per field of examination was quantified from 10 random sites of three different slides for each mouse using NIH ImageJ software.
Statistical Analysis
Data are expressed as the mean ± standard error of triplicates. Each experiment was repeated as least 3 times. Student’s t-test and analysis of variants (ANOVA) were used to assess differences with * P < 0.05, **: P < 0.01, and ***: P < 0.001.
Transwell Chamber Migration Assay, Construction of Plasmids of MMP-9/MMP-2PEX, Gelatin Zymography, Co-immunoprecipitation, and Three Dimensional (3D) Invasion Assay
These techniques have been described previously (13, 19). DOCK 6.0 Calculations can be found in supplemental information.
Results
MMP-9 expression correlates with the survival probability of patients
MMP-9 is one of the Rosetta 70 genes, serving as a poor prognosis signature for patients with breast cancer (20). To gain insight into the clinical significance of MMP-9 in patients with breast cancer, we analyzed two additional publicly available DNA microarray datasets, which contain a large number of breast cancer patient samples, in order to establish a correlation between MMP-9 expression and the probability of disease-free survival.
Patient samples were grouped based on MMP-9 RNA expression dichotomized at the mean value in the Van de Vijver cohort, which contains 295 breast cancer patients (21). High expression of MMP-9 was found to be significantly associated with shortened overall survival rate by Kaplan-Meier analysis (P= 0.0143) (Fig. 1A). In lymph node negative group patient samples (120 cases) in the same cohort, high expression of MMP-9 correlated with lower patient survival probability (P=0.0203, data not shown). In analysis of the Stockholm cohort (GSE 1456) (22), similar survival probability results were obtained when MMP-9 RNA expression was dichotomized at the mean from 159 breast cancer patient samples (P=0.0126) (Fig. 1B). In addition, patients in the high MMP-9 expression group had a higher cumulative incidence of relapse (P=0.0059) (Fig. 1C). Hence, elevated expression levels of MMP-9 in breast cancer correlate with a poor prognosis and suppressing MMP-9 may improve patient outcome.
Figure 1. Clinical relevance of MMP-9 in patients with breast cancer.
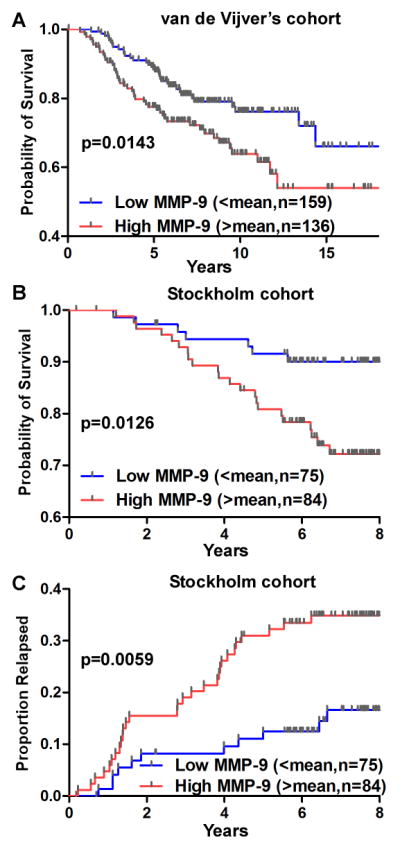
DNA microarray data mining of van de Vijver (21) and Stockholm (22) cohorts was performed using Kaplan-Meier survival analysis for correlation of MMP-9 expression with breast cancer survival rate (A & B) and recurrence (C). Levels of MMP-9 RNA were dichotomized at mean. n=cases.
Identification of small molecules targeting the PEX domain of MMP-9 using DOCK
We utilized DOCK 6.0 (23) to map potential ligand binding sites in the PEX domain of human MMP-9, which contains the dimerization interface. Homodimerization is observed under X-ray crystallographic conditions (PDB: 1ITV) (24) and in cell culture (12). The structures of the two subunits are similar, but not perfectly symmetric, therefore we used subunit A in its entirety for docking. A large cavity in the center of the top face of the barrel (24) formed by the innermost strands of all four blades and the loops which connect them to the second β-strand of each blade was identified (Fig. 2A & B).
Figure 2. Identification of small molecular weight compounds bound to the MMP-9 PEX domain.

A) Ribbon model of the PEX domain of MMP-9 with the compounds 1, 2, 4 and 5 docked. The sulfate ions are shown for references, but were not included as part of the docking receptor. B) The same docked structure as shown in (A) rotated 90° about the X-axis with a solvent-accessible surface on the protein. C) Compounds 1, 2, 4, and 5 overlaid in their docked conformations. Atom colors: Carbon (grey), oxygen (red), nitrogen (blue), sulfur (yellow), and fluorine (green). The images and the solvent-accessible surface of MMP-9 PEX monomer were generated in UCSF Chimera (42). D) Structures of the best five docked molecules.
As proof-of-principle that docking to the PEX domain was feasible, 100 commercially available compounds were selected from the ZINC 2007 database (25) and docked. Docked molecules were ranked based on their cluster size, grid score (energies), van der Waals energies and electrostatic energies. The top five hits docked to the cavity (Fig. 2A & B) and are shown in Fig. 2D and in Supplemental Table 2. Four of five hits contain a 4-pyrimidone core with variable substitutions at the 2- and 6-carbons and docked in the cavity (Fig. 2C).
Inhibition of MMP-9-induced cell migration by the identified compounds
COS-1 cells expressing MMP-9 cDNA, or GFP cDNA as a control, were pre-incubated with or without the compounds (doses ranging from 100 nM to 100 μM) for 30 minutes, and examined by a Transwell chamber migration assay. Compounds 1, 2, 3 and 5 inhibited the migration of MMP-9-expressing COS-1 cells, whereas compound 4 showed no activity (Fig. 3A–E). Compounds 3 and 5, but not 1 and 2, inhibited the migration of control cells (GFP-transfected) as well as MMP-9 transfected cells (Fig. 3C & E).
Figure 3. Inhibition of migration of cells expressing MMP-9 by the selected compounds.

A–E) Cell migration assay: Transfected COS-1 cells were pre-incubated with the compounds at different concentration for 30 minutes followed by a Transwell chamber migration assay. Each concentration was assayed in triplicate and the experiments were repeated three times. F) Cell cytotoxic assay: COS-1 cells were incubated with the five compounds (100 μM) for 24 hours followed by a cell viability assay. DMEM media alone and media containing thapsigargin (1 μM) were included as negative and positive controls, respectively.
To rule out the possibility that the reduction of cell migration by these compounds was due to cytotoxicity, a cell viability assay was performed. COS-1 cells were treated with the compounds for 24 hours followed by a cytotoxicity assay. Thapsigargin, an ER stress inducer that inhibits intracellular Ca2+-ATPases (26), was used as a positive control to trigger cell death. Treatment with compounds 1, 2 and 4 did not cause notable cytotoxicity at the maximum concentration used, whereas treatment with compounds 3 and 5 induced cell death (Fig. 3F). Compounds 3 and 5 were therefore excluded from further evaluation.
The lethal dose (LD50) of compounds 1 and 2 was determined in COS-1 cells. Cells were treated with increasing doses of the compounds for 24 hours followed by cell viability assay (Suppl. Fig. 1). The LD50 of compounds 1 and 2 were 360 ± 2 μM and 3.5 ± 0.3 mM, respectively, suggesting that their inhibition of MMP-9-induced cell migration was not due to cytotoxicity.
To further determine the specificity and selectivity of compound 1 and 2 for MMP-9-induced cell migration, we examined the effect of compounds 1 and 2 on cell migration induced by other MMPs in which the PEX domain has been reported to play a critical role in enhanced cell migration (27), e.g., MMP-2 and MT1-MMP (MMP-14). In contrast to MMP-9 expressing cells, neither compound inhibited the migration of MMP-2 or MT1-MMP ectopically expressing COS-1 cells (Fig. 4A). In addition, compound 2 did not interfere with MT1-MMP-mediated cancer cell invasion examined by a 3D invasion assay (Suppl. Fig. 2). Thus, small synthetic compounds that potentially bind to the PEX domain of MMP-9, inhibit MMP-9-induced cell migration with enhanced-specificity and -selectivity.
Figure 4. Specific inhibition of MMP-9-induced cell migration and invasion by the selected compounds.

A) Specific inhibition of MMP-9-induced cell migration: COS-1 cells transfected with cDNAs as indicated were incubated with compound 1 or 2 (100 μM) for 30 minutes followed by a Transwell chamber migration assay. B–C) Dose-dependent inhibition of cancer cell migration by the selected compounds. HT-1080 cells and MDA-MB-435 cells were incubated with 1% DMSO, or different concentrations of compound 1 or 2 for 30 minutes followed by a Transwell chamber migration assay. D–E) Reduction of HT-1080 cell invasion by the selected compounds. HT-1080 cells (1 × 104) were pre-treated with DMSO (1%), compound 1 or 2 (100 μM) for 30 minutes followed by 3D type I collagen invasion assay in the presence or absence of the compounds for additional 18 hours. Invading cells at the cell-collagen interface were microscopically counted.
Inhibition of migration in cancer cells that produce endogenous MMP-9 by compounds 1 and 2
We next investigated whether compounds 1 and 2 inhibit migration of cells producing a pathologically relevant level of endogenous MMP-9. Two human invasive cancer cell lines, HT-1080 and MDA-MB-435, expressing high endogenous levels of MMP-9 were employed. Treatment of the cells with compounds 1 and 2 significantly reduced cell migratory abilities. Furthermore, both compounds inhibited the migration of HT-1080 and MDA-MB-435 cells in a dose-dependent manner (Fig. 4B & C).
Cell migration is a critical determinant of cancer cell invasiveness. Therefore, HT-1080 cells were assessed in the 3D type I collagen invasion assay (19). As anticipated, the cell invasive ability of HT-1080 cells was significantly inhibited in cells treated with compounds 1 and 2 (Fig. 4D & E). Inhibition of MDA-MB-435 cell invasion was also observed (data not shown). These data suggest that inhibition of MMP-9-mediated cell migration by the compounds results in suppressed cancer cell invasion.
Compounds 1 and 2 do not affect MMP-9 expression or proteolytic activity
Cell lysates from HT-1080 cells treated with and without compounds 1 and 2 were examined for MMP-9 expression levels by Western blot using an anti-MMP-9 antibody. Western blotting using an antibody to tubulin was employed as a control. No effect on MMP-9 expression by the compounds was observed (Fig. 5A).
Figure 5. Interaction of compound 2 with the PEX domain of MMP-9.

A) No effect of the compounds on MMP-9 expression: Cell lysate of compound-treated HT1080 cells was examined by Western blotting (WB) using antibodies against MMP-9 and α/β tubulin, respectively. B) No inhibition of proteolytic activity of MMP-9 by the compounds: Latent and APMA-activated MMP-9 were incubated with DMSO control or the selected compounds 1 and 2 (100 μM) for 3 hours at 37°C followed by a fluorogenic substrate assay. C–D) Compound 2 binds selectively to the PEX domain of MMP-9: The λmax of tryptophan fluorescence emission was monitored upon titration with compound 2 or buffer only as a control with (C) purified recombinant MMP-9 (50 nM) and (D) MMP-9/MMP-2PEX chimera 9 (50 nM). E) Compound 2 interferes with MMP-9 homodimerization. COS-1 cells transfected with MMP-9-Myc and MMP-9-HA in the presence or absence of compounds 2 and 4 (100 μM) were pulled down with anti-HA antibody followed by Western blotting with anti-Myc antibody. The aliquots of total cell lysates serving as input were examined by Western blotting using anti-Myc antibodies. Reciprocal co-immunoprecipitation was also performed. F) Compound 2 decreased MMP-9-mediated ERK1/2 activation: COS-1 cells transiently transfected with vector control and MMP-9 cDNAs were serum-starved for 18 hours in the presence or absence of compounds 2 and 4 (100 μM) followed by Western blotting using anti-pERK1/2 and total ERK1/2 antibodies.
Activated MMP-9 was obtained by incubating purified proMMP-9 with p-aminophenyl mercuric acetate (APMA) (12). Addition of compounds 1 and 2 to APMA-activated MMP-9 did not inhibit the catalytic activity of MMP-9 as measured by cleavage of the fluorescent Mca-P-L-G-L-Dpa-A-R-NH2 peptide (18) (Fig. 5B). These data suggest that inhibition of MMP-9-induced cell migration by compounds 1 and 2 is not due to inhibition of MMP-9 expression or proteolytic activity.
Binding of compound 2 to the MMP-9 PEX domain
We titrated the binding of compound 2 to MMP-9 by monitoring MMP-9 tryptophan fluorescence. Saturation of purified proMMP-9 with compound 2 resulted in a 7 nm blue shift in the λmax of MMP-9 emission (Fig. 5C). No effect on the protein fluorescence occurred in the buffer only control. The Kd for MMP-9 binding to compound 2 is 2.1 ± 0.2 μM.
To further characterize the binding between compound 2 and MMP-9, we employed a previously generated chimera of MMP-9 in which the PEX domain of MMP-9 was replaced with that of MMP-2 (MMP-9/MMP-2PEX) (13). Upon addition of compound 2 to MMP-9/MMP-2PEX, no shift in fluorescence was detected (Fig. 5D). Likewise, compound 2 did not bind to purified recombinant soluble MT1-MMP (28) (data not shown). These data confirmed that compound 2 binds specifically to the PEX domain of MMP-9. The absorption of compound 1 at 280 nm precluded evaluation of its binding properties.
To test whether compound 2 interferes with proMMP-9 homodimerization, co-immunoprecipitation of COS-1 cells transfected with both proMMP-9/Myc and proMMP-9/HA cDNAs in the presence or absence of compounds 2 and 4 was utilized (13). Treatment of the transfected cells with compound 2, but not inactive compound 4, resulted in blocked MMP-9 homodimer formation (Fig. 5E). This defect was not due to inhibition of expression of MMP-9 by compound 2 as evidenced by Western blotting of the cell lysate from HT-1080 cells (Fig. 5A). Similar results were obtained in reciprocal co-immunoprecipitation assays (Fig. 5E). This experiment confirms that compound 2 specifically inhibits MMP-9 homodimerization
Homodimerized MMP-9 interacts with cell surface adhesion molecule, CD44, which leads to activation of EGFR and downstream MAPK (ERK1/2) pathway (12, 13). To explore this network, the activity status of downstream effector ERK1/2 was examined. COS-1 cells ectopically expressing MMP-9 cDNA were serum starved in the presence or absence of compounds for 18 hours followed by Western blotting using anti-phospho-ERK1/2 and total ERK1/2 antibodies. As depicted in Fig. 5F, decreased activation of ERK1/2 was observed in compound 2 treated cells. Taken together, these data suggest that abrogation of MMP-9-mediated cell migration by compound 2 is due to disruption of MMP-9 homodimerization, which results in failure to cross-talk with CD44 and the EGFR-MAPK signaling pathway.
Effect on MMP-9-mediated cell proliferation by compound 2
COS-1 cells transfected with MMP-9 or GFP cDNA (control) were monitored for cell proliferation in the absence or presence of compound 2 or 4 with a CellTiter-Glo® Luminescent assay. In agreement with previous observations (29), the rate of cell proliferation increased significantly (P < 0.05) in COS-1 cells expressing MMP-9 as compared to GFP expressing COS-1 cells (Fig. 6A). MMP-9-induced cell proliferation was not affected by compound 4, consistent with its lack of effect on MMP-9-induced cell migration. In contrast, compound 2 significantly decreased MMP-9-induced cell proliferation (Fig. 6A), but did not affect the proliferation of COS-1 cells transfected with GFP cDNA (Fig. 6B). To determine if compound 2 also affects the proliferation of cancer cells producing endogenous MMP-9, HT-1080 and MDA-MB-435 cancer cells were treated with 10 μM compound 2. Significant inhibition of cell proliferation was observed for HT-1080 and MDA-MB-435 cells treated with compound 2, but not with compound 4 or with DMSO controls (Fig. 6C & D).
Figure 6. Inhibition of cell proliferation by compound 2.

The effect of compound 2 on cell proliferation was examined in MMP-9 cDNA transfected COS-1 cells (A) or cancer cells expressing endogenous MMP-9 (HT-1080 and MDA-MB-435) (C & D), as well as GFP cDNA transfected COS-1 cells (control) (B) in the presence or absence of the compounds 2 and 4 (10 μM) for 9 days.
Decreased tumor growth and lung metastases in compound 2-treated mice
MDA-MB-435 cells are highly metastatic in nude mice (30, 31) and produce high levels of MMP-9, thus serving as an appropriate experimental model to explore the in vivo inhibitory activity of compounds exhibiting anti-MMP-9 activity. To facilitate in vivo analysis of tissues and visualization of the lung metastases, MDA-MB-435 cells were stably transfected with GFP cDNA and implanted subcutaneously within the mammary fat pad of female immunodeficient mice. Treatment of mice with compound 2 resulted in a profound delay in tumor growth, whereas treatment with the inactive control compound 4 or the vehicle alone failed to inhibit tumor growth (Fig. 7A & B). Tumor incidence was unaffected by compound 2.
Figure 7. Retarded tumor growth and metastasis by compound 2 in mice bearing MDA-MB-435/GFP tumor xenografts.

A–B) Effect of compound 2 on tumor growth: Mice bearing MDA-MB-435/GFP cells were administered compounds or vehicle control and tumor was measured. Arrows indicate tumor mass. C–E) Inhibition of metastasis by compound 2: Lung sections ~3 mm thick were examined by fluorescent microscopy. Incidence of metastasis was determined (C–D) and the average area of tumor in the lungs was determined using ImageJ software (E).
The lungs of tumor-bearing mice were removed and slices of the lungs (3 mm thickness) were examined under a fluorescent microscope (Fig. 7C). In the vehicle control and compound 4-treated groups, multiple large nodules were evident in MDA-MB-435/GFP tumor-bearing mice, whereas the extent of lung metastasis was dramatically reduced in mice treated with compound 2 (Fig. 7C). Also, dimensions of tumor foci area in the lung and the percent of mice displaying lung metastases were significantly decreased in these mice (Fig. 7D & E). Thus, treatment with compound 2 impaired the in vivo effect of MMP-9 on both primary tumor growth and metastasis. No significant change in body weight nor other signs of toxicity during the 14-week period were observed in compound 2-treated mice.
Discussion
In this study, we demonstrated the identification and characterization of synthetic compounds that specifically interfere with MMP-9-mediated cell migration. This inhibitory effect works through the abrogation of MMP-9 dimerization via the PEX domain, and subsequent blockage of the CD44-EGFR-MAPK signaling pathway(13).
The role of the PEX domain of MMP-9 in cancer cell migration has gained considerable attention(32–34). Lengyel et al.(35) demonstrated that high expression of proMMP-9 in ovarian cancer patients correlated with poor survival; activated MMP-9 was not implicated or detected, thus suggesting a critical role of the non-catalytic domains of MMP-9 in cancer progression. We demonstrated that latent MMP-9 was able to initiate cell migration through a proMMP-9-CD44-EGFR-MAPK pathway independent of its catalytic activity(12, 13). We further identified that the PEX domain plays a critical role in proMMP-9-mediated cell migration, in agreement with previous reports(32–34). The validation of the MMP-9 PEX domain as a drug target has recently been explored through different experimental approaches: 1) anti-PEX antibodies block Schwann cell migration(33); 2) exogenous recombinant PEX domain inhibits the migration of endothelial cells (anti-angiogenic effect), as well as intracranial glioblastoma growth(32); 3) non-catalytic-domain inhibitors of MMP-9 selected from a phage display peptide library interfere with cell migration and tumor formation(34); and 4) peptides mimicking motifs of the outermost strands of the first and fourth blades of the PEX domains of MMP-9 abrogate MMP-9-mediated cell migration(13). These results prompted us to hypothesize that low molecular weight compounds interacting with the PEX domain of MMPs may interfere with its function.
The PEX domain of MMP-9 shares low amino acid identity(25% to 33%) with secreted MMP-1, -2, -8, -9, -10, and -28, as well as membrane anchored MT1-MMP, in contrast to the high conservation in their catalytic domains (43–65%) (Suppl. Table 1). The differences between the PEX domains of MMP-9 and other MMPs determine their distinct biological roles in cell function.
The MMP-9 PEX domain is a barrel-shaped structure composed mostly of hydrophobic surfaces making this domain difficult to target with drugs (5, 24). Our results indicate that the surface cavity within the loops of the MMP-9 PEX domain is targetable by small compounds. Compounds 1 and 2, identified through docking, specifically and selectively inhibit cell migration induced either by ectopically expressed MMP-9 or by endogenous MMP-9 as well as cancer cell invasion in a 3D type I collagen invasion assay (Fig. 4D). The latter is believed to be due to the inhibition of cell migration, because cell migration is a prerequisite for cancer invasion.
These inhibitory activities are independent of an effect on MMP-9 catalytic activity, which suggests that compound 2 physically interacts with the PEX domain of MMP-9. Based on quenching of MMP-9 tryptophan fluorescence, we determined that compound 2 specifically binds to the PEX domain of MMP-9, but not to the PEX domain of MMP-2 or MT1-MMP (Fig. 5C & D). MMP-9 homodimerizes through interactions in the fourth blade of its PEX domain (24). Furthermore, formation of the homodimer is required for cell migration, an important component of metastasis. Blade III is flexible due to the presence of Gly615 and its flexibility may contribute to stabilization of the monomeric versus the dimeric form of the PEX domain (24). The structural components of the best three compounds each contain a six-membered heterocycle which docks within the identified site in the PEX domain. The flat rings preferentially dock within the cavity of the MMP-9 PEX domain blades, whereas the more flexible side chains (i.e. arylamide group of compound 2) likely bind on the surface proximal to the cavity (Fig. 2B). Therefore, the proposed binding orientation in the PEX domain is consistent with allosteric disruption of the dimer interface and consequently migration.
MMP-9 has been implicated in primary tumor growth and metastasis of various cancers (2). Silencing MMP-9 in cancer cells was reported to reduce tumor growth, angiogenesis, invasion and metastasis (36). However, there are inconsistencies in the literature regarding whether or not MMP-9 enhances tumor growth in in vivo cancer models (1, 37, 38). Importantly, compound 2 inhibited cancer cell metastasis in vivo (Fig. 7), presumably due to reduction in cell proliferation, angiogenesis and migration. Compound 2 inhibits tumor growth without affecting tumorigenicity. Moreover, compound 2 inhibits tumor metastasis of MDA-MB-435/GFP cells in vivo, presumably by interfering with PEX interaction(s) with downstream pathway(s) required for cell migration and invasion. In agreement with previous studies (32, 34), our results suggest that the PEX domain of MMP-9 is involved in both cell proliferation and migration both of which contribute to increased tumor growth and metastasis. The main source of MMP-9 in some murine tumor models is from tumor-associated stromal and inflammatory cells (1, 29, 37, 39), leading to the possibility that an anti-tumor effect of an MMP-9 inhibitor in vivo could be due to an effect on host cells. Human MMP-9 and murine MMP-9 proteins share 72% amino acid identity. To distinguish whether the anti-tumor activity of compound 2 in vivo is due to an effect on mouse stromal/inflammatory cells or on human tumor cells, we examined the effect of this compound on mouse RAW264.7 macrophage-like cells expressing endogenous MMP-9. Compound 2 has no notable effect on the migration of mouse macrophages (Suppl. Fig. 3), leading us to conclude that the inhibitory effect of compound 2 is directed at human tumor MMP-9.
We previously demonstrated that CD44 is a key molecule involved in proMMP-9 enhanced cell migration through an EGFR-CD44 signaling pathway along with other downstream effectors including phosphorylated FAK, AKT and ERK (13). Since compound 2 inhibits MMP-9 dimerization and ERK1/2 activation, we propose that the mechanism of inhibition by compound 2 of MMP-9-mediated cell migration is via disruption of MMP-9 dimer formation, and hence, blockage of the downstream MMP-9-CD44-EGFR-MAPK signaling pathway. In accordance with our data, Peng et al. (40) demonstrated that an increase in MMP-9, after colocalization with CD44 on the cell surface of MDA-MB-435 cells, stimulated the tumor cell invasion and metastasis. In another study, Yu and Stamenkovic (41) demonstrated a cascade involving a MMP-9/CD44/TGF-β pathway that promotes the metastatic potential of selected tumor types; interference with the MMP-9/CD44 complex induced apoptosis.
In conclusion, we selected MMP-9 as a suitable target for drug discovery because of its high expression in human cancer tissues, tight correlation with the probability of patient survival and numerous in vitro and in vivo studies implicating MMP-9 in tumor progression (5, 8, 15). We identified small molecules that specifically interfere with MMP-9-mediated cell migration. Our data demonstrate that compound 2 specifically binds to the PEX domain of MMP-9 and abrogates MMP-9 homodimerization which blocks the CD44-EGFR-MAKP signaling pathway (13) and interferes with MMP-9-mediated cancer cell proliferation, migration and invasion to reduce tumor metastasis. To our knowledge, this is the first example of a low molecular weight compound that targets the non-catalytic PEX domain of an MMP. Our proof-of-principle study should have a positive impact on future drug discovery in which the PEX domain exosite of other MMPs can be targeted.
Supplementary Material
Acknowledgments
Grant Support: This study was supported, in whole or in part, by National Institutes of Health Grant 5R01CA113553 (J.C./N.S.) & P41RR001081 (UCSF Chimera), a Carol Baldwin Breast Cancer Foundation grant (J.C.), NYSTAR FDP Grant C040076 (N.S.), a Veterans Affairs Merit Review grant, and a Carol Baldwin Breast Cancer Foundation grant (S. Z.).
We thank the members of the Sampson, Zucker and Cao labs for their insightful discussions and suggestions on the manuscript, and Kevin Zarrabi for his help with editing and Dr. Francis Johnson for his critical reading. We are also grateful to J. Shipman and T. Balius for their input on the molecular DOCKing studies.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Acuff HB, Carter KJ, Fingleton B, Gorden DL, Matrisian LM. Matrix metalloproteinase-9 from bone marrow-derived cells contributes to survival but not growth of tumor cells in the lung microenvironment. Cancer Res. 2006;66:259–66. doi: 10.1158/0008-5472.CAN-05-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 3.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Overall CM, Kleifeld O. Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6:227–39. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 5.Overall CM, Kleifeld O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br J Cancer. 2006;94:941–6. doi: 10.1038/sj.bjc.6603043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hidalgo M, Eckhardt SG. Development of matrix metalloproteinase inhibitors in cancer therapy. J Natl Cancer Inst. 2001;93:178–93. doi: 10.1093/jnci/93.3.178. [DOI] [PubMed] [Google Scholar]
- 7.Whittaker M, Floyd CD, Brown P, Gearing AJ. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem Rev. 1999;99:2735–76. doi: 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- 8.Zucker S, Cao J, Chen WT. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 2000;19:6642–50. doi: 10.1038/sj.onc.1204097. [DOI] [PubMed] [Google Scholar]
- 9.Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657–72. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 10.McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289:1202–6. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- 11.Tam EM, Wu YI, Butler GS, Stack MS, Overall CM. Collagen binding properties of the membrane type-1 matrix metalloproteinase (MT1-MMP) hemopexin C domain. The ectodomain of the 44-kDa autocatalytic product of MT1-MMP inhibits cell invasion by disrupting native type I collagen cleavage. J Biol Chem. 2002;277:39005–14. doi: 10.1074/jbc.M206874200. [DOI] [PubMed] [Google Scholar]
- 12.Dufour A, Sampson NS, Zucker S, Cao J. Role of the hemopexin domain of matrix metalloproteinases in cell migration. JCell Physiol. 2008;217:643–51. doi: 10.1002/jcp.21535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dufour A, Zucker S, Sampson NS, Kuscu C, Cao J. Role of matrix metalloproteinase-9 (MMP-9) dimers in cell migration: design of inhibitory peptides. J Biol Chem. 2010 doi: 10.1074/jbc.M109.091769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee M, Fridman R, Mobashery S. Extracellular proteases as targets for treatment of cancer metastases. Chem Soc Rev. 2004;33:401–9. doi: 10.1039/b209224g. [DOI] [PubMed] [Google Scholar]
- 15.Bjorklund M, Koivunen E. Gelatinase-mediated migration and invasion of cancer cells. Biochim Biophys Acta. 2005;1755:37–69. doi: 10.1016/j.bbcan.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–92. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 17.Sanceau J, Truchet S, Bauvois B. Matrix metalloproteinase-9 silencing by RNA interference triggers the migratory-adhesive switch in Ewing’s sarcoma cells. J Biol Chem. 2003;278:36537–46. doi: 10.1074/jbc.M304300200. [DOI] [PubMed] [Google Scholar]
- 18.Knight CG, Willenbrock F, Murphy G. A novel coumarin-labelled peptide for sensitive continuous assays of the matrix metalloproteinases. FEBS Lett. 1992;296:263–6. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
- 19.Cao J, Chiarelli C, Richman O, Zarrabi K, Kozarekar P, Zucker S. Membrane type 1 matrix metalloproteinase induces epithelial-to-mesenchymal transition in prostate cancer. JBiolChem. 2008;283:6232–40. doi: 10.1074/jbc.M705759200. [DOI] [PubMed] [Google Scholar]
- 20.van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 21.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 22.Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7:R953–64. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harkcom WT, Bevan DR. Molecular docking of inhibitors into monoamine oxidase B. Biochem Biophys Res Commun. 2007;360:401–6. doi: 10.1016/j.bbrc.2007.06.055. [DOI] [PubMed] [Google Scholar]
- 24.Cha H, Kopetzki E, Huber R, Lanzendorfer M, Brandstetter H. Structural basis of the adaptive molecular recognition by MMP9. J Mol Biol. 2002;320:1065–79. doi: 10.1016/s0022-2836(02)00558-2. [DOI] [PubMed] [Google Scholar]
- 25.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–82. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baffy G, Miyashita T, Williamson JR, Reed JC. Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J Biol Chem. 1993;268:6511–9. [PubMed] [Google Scholar]
- 27.Cao J, Kozarekar P, Pavlaki M, Chiarelli C, Bahou WF, Zucker S. Distinct roles for the catalytic and hemopexin domains of membrane type 1-matrix metalloproteinase in substrate degradation and cell migration. JBiolChem. 2004;279:14129–39. doi: 10.1074/jbc.M312120200. [DOI] [PubMed] [Google Scholar]
- 28.Cao J, Sato H, Takino T, Seiki M. The C-terminal region of membrane type matrix metalloproteinase is a functional transmembrane domain required for pro-gelatinase A activation. J Biol Chem. 1995;270:801–5. doi: 10.1074/jbc.270.2.801. [DOI] [PubMed] [Google Scholar]
- 29.Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–90. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price JE. Metastasis from human breast cancer cell lines. Breast Cancer Res Treat. 1996;39:93–102. doi: 10.1007/BF01806081. [DOI] [PubMed] [Google Scholar]
- 31.Price JE, Zhang RD. Studies of human breast cancer metastasis using nude mice. Cancer Metastasis Rev. 1990;8:285–97. doi: 10.1007/BF00052605. [DOI] [PubMed] [Google Scholar]
- 32.Ezhilarasan R, Jadhav U, Mohanam I, Rao JS, Gujrati M, Mohanam S. The hemopexin domain of MMP-9 inhibits angiogenesis and retards the growth of intracranial glioblastoma xenograft in nude mice. Int J Cancer. 2009;124:306–15. doi: 10.1002/ijc.23951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mantuano E, Inoue G, Li X, Takahashi K, Gaultier A, Gonias SL, et al. The hemopexin domain of matrix metalloproteinase-9 activates cell signaling and promotes migration of schwann cells by binding to low-density lipoprotein receptor-related protein. J Neurosci. 2008;28:11571–82. doi: 10.1523/JNEUROSCI.3053-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bjorklund M, Heikkila P, Koivunen E. Peptide inhibition of catalytic and noncatalytic activities of matrix metalloproteinase-9 blocks tumor cell migration and invasion. J Biol Chem. 2004;279:29589–97. doi: 10.1074/jbc.M401601200. [DOI] [PubMed] [Google Scholar]
- 35.Lengyel E, Schmalfeldt B, Konik E, Spathe K, Harting K, Fenn A, et al. Expression of latent matrix metalloproteinase 9 (MMP-9) predicts survival in advanced ovarian cancer. Gynecol Oncol. 2001;82:291–8. doi: 10.1006/gyno.2001.6243. [DOI] [PubMed] [Google Scholar]
- 36.Sun Y, Liu M, Yang B, Lu J, Li B. Inhibition of laryngeal cancer cell invasion and growth with lentiviral-vector delivered short hairpin RNA targeting human MMP-9 gene. Cancer Invest. 2008;26:984–9. doi: 10.1080/07357900802072897. [DOI] [PubMed] [Google Scholar]
- 37.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–44. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chattopadhyay S, Shubayev VI. MMP-9 controls Schwann cell proliferation and phenotypic remodeling via IGF-1 and ErbB receptor-mediated activation of MEK/ERK pathway. Glia. 2009;57:1316–25. doi: 10.1002/glia.20851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2:289–300. doi: 10.1016/s1535-6108(02)00153-8. [DOI] [PubMed] [Google Scholar]
- 40.Peng ST, Su CH, Kuo CC, Shaw CF, Wang HS. CD44 crosslinking-mediated matrix metalloproteinase-9 relocation in breast tumor cells leads to enhanced metastasis. Int J Oncol. 2007;31:1119–26. [PubMed] [Google Scholar]
- 41.Yu Q, Stamenkovic I. Transforming growth factor-beta facilitates breast carcinoma metastasis by promoting tumor cell survival. Clin Exp Metastasis. 2004;21:235–42. doi: 10.1023/b:clin.0000037705.25256.d3. [DOI] [PubMed] [Google Scholar]
- 42.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.