

Abstract
Lay Abstract
Autism is a complex developmental disability characterized by abnormalities in spoken language, socialization and repetitive behaviors. In recent years, an increase in the number of autism cases worldwide has been reported. Researchers have found evidence that autism is caused by multiple genetic factors, and a number of potential risk genes have been identified. Earlier we have found an increased frequency in subject with autism of a form of the enzyme glyoxalase I (Glo1) that has glutamic acid at position 111. By using lymphoblastoid and neuronal cells in culture, we now present evidence that this form of Glo1 is reduced in function, and consequently accumulates a toxic metabolite called methylglyoxal (MG). MG can form conjugates with cellular proteins, thereby compromising their function. We propose that an increase in conjugated proteins causes over-expression of receptors that bind these altered proteins, which in turn can initiate cascades of potentially harmful events. These studies suggest that in a subset of individuals with autism, the occurrence of the Glo1 polymorphism may precipitate changes in cellular structure and function that support risk for autism.
Scientific Abstract
Autism is a pervasive, heterogeneous, neurodevelopmental disability characterized by impairments in verbal communications, reciprocal social interactions, and restricted repetitive stereotyped behaviors. Evidence suggests the involvement of multiple genetic factors in the etiology of autism, and extensive genome-wide association studies have revealed several candidate genes that bear single nucleotide polymorphisms (SNPs) in non-coding and coding regions. We have shown that a non-conservative, non-synonymous SNP in the glyoxalase I gene, GLOI, may be an autism susceptibility factor. The GLOI rs2736654 SNP is a C→A change that causes an Ala111Glu change in the Glo1 enzyme. To identify the significance of the SNP, we have conducted functional assays for Glo1. We now present evidence that the presence of the A-allele at rs2736654 results in reduced enzyme activity. Glo1 activity is decreased in lymphoblastoid cells that are homozygous for the A allele. The Glu-isoform of Glo1 in these cells is hyperphosphorylated. Direct HPLC measurements of the glyoxalase I substrate, methylglyoxal (MG), show an increase in MG in these cells. Western blot analysis revealed elevated levels of the receptor for advanced glycation end products (RAGEs). We also show that MG is toxic to the developing neuronal cells. We suggest that accumulation of MG results in the formation of AGEs, which induce expression of the RAGE that during crucial neuronal development may be a factor in the pathology of autism.
Keywords: autism, glyoxalase I, SNP, advanced glycation endproducts (AGEs), receptor for advanced glycation end products (RAGEs), methylglyoxal
Introduction
Autism (OMIM 209850) is a highly heterogeneous, neurodevelopmental disability characterized by impairments in social interactions, deficits in verbal and non-verbal communication, and stereotyped and repetitive patterns of behavior [Lord et al., 2000]. There is a wide variation in the symptoms and severity of autism, with about 30% of autism subjects showing cognitive decline, and a significant proportion experiencing epileptic seizure [Chakrabarti and Fombonne, 2005; Muhle et al., 2004; Tuchman and Rapin, 2002]. According to the most recent and comprehensive report from the Centers for Disease Control and Prevention, the prevalence of autism spectrum disorder (ASD) in the general population is 1 in 110 children [MMWR, 2009]. Evidence from concordance in twins and family studies has pointed towards high heritability with no significant shared environmental influences [Ronald et al., 2006]. High-density genotyping studies in a large number of autism samples have so far implicated over 25 different genetic loci [Abrahams and Geschwind, 2008]; the physiological significance of these loci and actual aberration remains to be elucidated.
Pathological findings in autism subjects show increased brain weight, reduced maturation of neurons in the limbic system of the forebrain, reduced Purkinje cells in the cerebellar cortex, and smaller and lesser numbers of neurons in the fusiform gyrus [Courchesne et al., 2003; Bauman and Kemper, 2005; van Kooten et al., 2008]. On the basis of these reports, the pathology of autism is considered to be consistent with deficits in certain brain areas and in specific cellular populations.
Using proteomics as an alternative approach to genome wide DNA analyses, we have earlier shown that a non-conservative single nucleotide polymorphism (SNP) in the glyoxalase I, (Glo1; lactoylglutathione lyase, EC 4.4.1.5) gene (GLOI) may be an autism susceptibility factor [Junaid et al, 2004]. The GLOI rs2736654 SNP is a C→A change in exon 4 that results in Ala111Glu in the mature protein. The GLOI rs2736654 SNP and the existence of multiple isoforms of Glo1 were recognized and used earlier in determining allele frequencies in the population [Thornalley, 1991]. Association studies of the involvement of GLOI in autism have been controversial possibly due to variation in the allele frequency of the rs2736654 SNP across populations. In a Finnish combined population comprising individuals with ASD and Asperger syndrome, a weak linkage for the rs2736654 SNP was found [Rehnstrom et al., 2008]. In the Han ethnic group of Chinese subjects, no significant differences were found in the allelic and genotypic frequency distributions of C419A between the autism and control groups [Wu et al., 2008]. In another study, a protective effect of the A419 allele was reported for unaffected siblings of subjects with autism [Sacco et al., 2007]. There are other reports of GLOI involvement in various psychiatric disorders [Politi et al., 2006; Fujimoto et al., 2008]. On the basis of studies carried out in mice, GLO1 has been shown to be involved in anxiety and depression-like symptoms [Hovatta et al., 2005; Kromer et al., 2005].
Glo1, acting in the glycolytic pathway, is a cytosolic, ubiquitously expressed, zinc metalloenzyme involved in scavenging toxic α-oxoaldehydes, such as methylglyoxal (MG) that are formed during cellular metabolic reactions [Thornalley, 2003]. The Allen Brain Atlas [http://human.brain-map.org/ish/human/brain/GLO1.html] shows high levels of Glo1 expression in Purkinjie, hippocampal pyramidal, and dentate gyrus cells. MG reacts non-enzymatically with reduced glutathione to form hemithioacetal, which is converted by Glo1 to S-D-lactoylglutathione. A second enzyme, glyoxalase II, catalyzes the conversion of S-D-lactoylglutathione to lactate, regenerating the reduced glutathione. Dysfunction of the glyoxalase system results in accumulation of the substrate, MG, which is highly reactive. MG, in turn, reacts with proteins, carbohydrates, and nucleic acids, forming stable covalent adducts called advanced glycation endproducts (AGEs).
We are evaluating the biochemical basis of Ala111Glu change in the Glo1 sequence to link the occurrence of particular polymorphisms with phenotypes. In the present study, we have investigated the functional significance of the autism-associated SNP in lymphoblastoid cell lines bearing different rs2736654 alleles. We now present evidence that point to a possible conformational aberration in the Glo1 structure due to the SNP, which ultimately leads to altered downstream mechanisms that may affect gene expression and may explain the adverse effects associated with the SNP.
Experimental
Lymphoblastoid cells for the study were obtained from the Autism Genetic Resource Exchange, Los Angeles, CA. Fifteen different cell lines were used in the present study: each of these cell lines has different alleles with respect to the rs2736654 SNP (Table I). Normal human neural progenitor (NHNP) cells and maintenance and differentiation media were purchased from Cambrex Bio Sciences, Walkersville, MD. Recombinant human brain–derived neurotropic factor (BDNF) was purchased from R and D Systems, Minneapolis, MN. Primary antibody for receptor for AGE (RAGE), a rabbit polyclonal, was purchased from Santa Cruz Biotechnologies, Santa Cruz, CA, whereas the antibody for Glo1, a rabbit polyclonal was a kind gift from Dr. Kenneth Tew, Medical University of South Carolina, Charleston, SC. Secondary antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 555 were from Molecular Probes (Invitrogen Corp, Carlsbad, CA), and secondary antibodies conjugated to Cy3 were from Jackson ImmunoResearch (West Grove, PA). Other reagents and chemicals are from various vendors.
Table 1.
Immortalized Lymphoblastoid Cell Lines Used, Alleles with Respect to rs2736654 SNP and the Corresponding Glo1 Isoforms Encoded
| Cell line ID | Family code | rs2736654 allele | Glo1 isoform |
|---|---|---|---|
| HI 0963 | AU 0469 | C/A | Ala/Glu111 |
| HI 0960 | AU 0469 | C/A | Ala/Glu111 |
| HI 0996 | AU 0364 | C/A | Ala/Glu111 |
| HI 0556 | AU 0453 | C/A | Ala/Glu111 |
| HI 0557 | AU 0453 | C/A | Ala/Glu111 |
| HI 0961 | AU 0469 | C/C | Ala111 |
| HI 0648 | AU 0419 | C/C | Ala111 |
| HI 0872 | AU 0110 | C/C | Ala111 |
| HI 0955 | AU 0180 | C/C | Ala111 |
| HI 0964 | AU 0364 | C/C | Ala111 |
| HI 0150 | AU 0203 | A/A | Glu111 |
| HI 0558 | AU 0453 | A/A | Glu111 |
| HI 0560 | AU 0453 | A/A | Glu111 |
| HI 1004 | AU 0545 | A/A | Glu111 |
| HI 1028 | AU 0467 | A/A | Glu111 |
Cell Culture
Cells were grown in RPMI-1640 medium (GIBCO) supplemented with 15% heat-inactivated fetal calf serum and the antibiotics penicillin (100 units/ml) and streptomycin (100 μg/ml). Cells were grown and maintained at 37°C in a humidified incubator with 5% CO2 in air. In the 32P-labeling experiment, DMEM medium was used under similar conditions. Prior to incubation of the label, cells were depleted of endogenous phosphate by growing in phosphate-free media supplemented with dialyzed fetal calf serum.
Electrophoresis and Western blotting
Total cellular proteins (50 μg) were resolved by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 10–20% gradient gel. After completion of electrophoresis, the separated proteins were transferred onto nitrocellulose membrane filters as described previously [Junaid et al., 2009]. After the non-specific sites were blocked with 2% BSA and 4% normal goat serum in PBST, appropriately diluted primary antibody in blocking solution was incubated for either 2 h (for anti-RAGE) at room temperature or overnight (for anti-Glo1) at 4°C. Secondary antibody (anti-rabbit IgG coupled to alkaline phosphatase or horse radish, Sigma Chemical Co, St. Louis, MO) was used at a dilution of 1:1000 (alkaline phosphatese) or 1:2000 (horse radish peroxidase), and incubations were done at room temperature for 1h [Chen et al., 2009]. Protein bands were visualized by the formation of insoluble formazan complex following incubation with the substrate, NBT/BCIP, or by ECL detection using x-ray film.
Particulate and soluble extracts from lymphoblastoid cells were prepared by repeated freeze/thaw cycles in 50 mM Tris.HCl buffer, pH 7.4, containing 10 μg/ml of leupeptin, pepstatin-A, and PMSF. The extracts were resolved into particulate and soluble fractions following centrifugation at 12,000 × g at 4°C, and stored frozen until further use. Protein concentrations were measured by the Lowry method [1951].
Glyoxalase I Enzyme Assay
Glo1 activity was measured according to Kester and Norton [1975] with slight modifications. The reaction mixture contained 14.4 mM MG, 0.72 mM reduced glutathione, and 16 mM MgSO4 in 100 mM imidazole buffer, pH 6.8. The reaction mixture was preincubated at 22°C for 4 h to ensure non-enzymatic hemithioacetal formation before adding cell extracts. The enzymatic production of S-lactoylglutathione (ε = 3.37 mM−1 cm−1) was monitored at 240 nm in a spectrophotometer at 15-sec interval for 2 min. The rate of change under these conditions is found to be linear. Each assay was done in triplicate and averaged. Specific activity is expressed as μmol S-lactoylglutathione formed/h/mg protein.
Phosphorylation of Glo1 in Lymphoblastoid Cells
Radiolabeling of Glo1 in cultured lymphoblastoid cells was done according to the protocol of Wang et al. [1995] with slight modifications. Lymphoblastoid cells (approximately 1.0 × 107) were cultured in DMEM medium (20 ml) supplemented with 15% dialyzed fetal bovine serum and the antibiotics penicillin (100 units/ml) and streptomycin (100 μg/ml). Cultures were grown at 37°C in a humidified incubator with 5% CO2 in air for 24 h, after which the cells were washed free of phosphate medium and incubated for another 2 h in phosphate-free medium. This was done to deplete the endogenous phosphate prior to radiolabeling. Pulse labeling was initiated by adding 2 mCi [32P]orthophosphate (NEN) to the medium (1 ml), and incubation was continued for another 4 h. After radioactive tracer incubation, the cells were washed three times with PBS, suspended in 150 μl lysis solution (50 mM Tris.Cl, pH 7.4, 150 mM NaCl, 2.5% Triton X-100, 2.5% SDS, 1% mercaptoethanol, 5 mM EDTA, 1 mM sodium fluoride, 1 mM sodium vanadate, and 10μg each of leupeptin, pepstatin-A and PMSF), and boiled for 4 min. The radiolabeled proteins were analyzed by autoradiography and Western blotting, following resolution on a 10–20% gradient SDS-PAGE, and transferred onto nitrocellulose membrane filter.
Methylglyoxal Assay
MG was measured according to the procedure described by Chaplen et al. [1996] with slight modifications. The proteins in cell extracts were precipitated by adding perchloric acid to a final concentration of 5 mM. The samples were incubated on ice for 10 min and then centrifuged (12,000 × g, 10 min) to remove the PCA-precipitated material. The supernatant was passed through a Spice C18 cartridge (Analtech, Newark, DE), primed by washing with 6 ml acetonitrile and then with 6 ml H2O. A total of 1.25 nmol of 5-methylquinoxaline (5-MQ) was added as an internal standard to the samples in which MG was derivatized with 125 nmol of o-phenylenediamine (OPD) at 20°C for 3.5 to 4 h. Derivatives were concentrated by passing through Spice C18 cartridges (prepared as described before) at a rate of 1 ml/min. The cartridges were then washed with 2 ml H2O, followed by the elution of the bound materials with 2 ml acetonitrile.
The quinoxaline derivatives of MG, namely, 2-methylquinoxaline (2-MQ) and the quinoxaline internal standard (5-MQ) in the acetonitrile eluate, were measured by reversed-phase HPLC using a Shimadzu liquid chromatography system, equipped with variable wavelength detector, and integrator. The reversed-phase analytical column, Microsorb-MV C18 (0.46×10 cm, 3μm, 100Å), was used for separation (Varian, Walnut Creek, CA). The isocratic mobile phase comprised 27% acetonitrile in 10 mM potassium dihydrogen phosphate (pH 2.5). The running conditions were as follows: flow rate, 0.5 ml/min; detector wavelength, 315 nm; and column temperature, 20°C. Duplicate injections of each sample were made. Samples were calibrated by comparing with a 2-MQ standard. The retention times of 2-MQ and 5-MQ under these conditions were 10.25 and 21.74 min, respectively.
Neuronal Cell Differentiation
NHNP cells received as cryopreserved neurospheres from primary cultures were grown as described by the supplier. Briefly, the NHNP neurospheres (2.6 × 106 cells) were transferred to 40 ml NPMM maintenance media in a T-75 flask (Falcon) for 24 h. Cells were centrifuged at 100 × g for 5 min and resuspended gently in differentiation medium (NPDM) at a density of 30,000 cells/0.5 ml followed by transferring 30,000 cells per well into Falcon 12-well culture vessels that had circular glass cover slips coated with mouse laminin. To induce differentiation, the differentiation medium (NPDM) was supplemented with BDNF at a final concentration of 25 ng/ml. MG was added at various concentrations (range 0–8 μM) to the individual wells, and cells were allowed to differentiate for 2 weeks. Fresh NPDM media containing BDNF and MG was replaced after every 3 days. At the end of 2 weeks, cover slips were processed for immunocytochemistry and were stained with different antibodies to identify cellular processes.
Immunofluorescence Microscopy
Cells were fixed with methanol for 20 min at 4°C. Non-specific sites were blocked by incubating with blocking solution (10% FCS in PBS) for 1 h. The cells were incubated first with a monoclonal mouse antibody against nestin (1:100, Chemicon) for 45 min at 37°C and then rinsed with blocking solution (2 × 10 min). The slide was then incubated with a rabbit polyclonal antibody against βIII tubulin (1:1000, Covance) overnight at 4°C. After rinsing with PBS (3 × 10 min), cells were incubated simultaneously with Alexa 488–conjugated goat anti-mouse antibody and Alexa 594-conjugated goat anti-rabbit antibody (Molecular Probes, Eugene, OR), each at a final dilution of 1:500 for 1 h at room temperature. Cells were also treated with 50 μg/ml 4,6-diamidino-2-phenylindole (DAPI; Sigma) for 10 min. The cover slips were mounted with Vectashield medium and viewed with a Nikon Eclipse E600 laser scanning confocal microscope. All images of immunostained cells were taken using a high-resolution digital camera.
Results
In order to identify the functional consequences of the GLO1 rs2736654 SNP, Glo1 enzyme activity was measured in extracts prepared from lymphoblastoid cells that are homozygous for either A- (Glu-Glo1) or C-alleles (Ala-Glo1) at rs2736654 SNP. The Glo1-specific activity in cells homozygous for A-allele is 32.75± 4.74 μmol h−1 mg−1 protein (Table II). In comparison, the Glo1-specific activity in cells homozygous for the C-allele is 41.23± 5.62 μmol h−1 mg−1 protein. The results show a subtle but statistically significant decrease (unpaired t-test with Welch correction, P < 0.03) in Glo1 activity in lymphoblastoid cells in which the Glu-isoform encoded by the homozygous A-allele at rs2736654 is present. Glo1 activity in cells in an heterozygous condition was not statistically different from Glo1 activity in cells in either of the homozygous conditions. A more direct proof of altered Glo1 consequent to the rs2736654 polymorphism would be to show differences in the kinetic properties of the two isoforms. We have tried to evaluate the kinetic properties of partially purified Glo1 isoforms; however, our efforts were unsuccessful, mainly because the Glo1 assay is not sensitive enough to allow measurements made at very dilute substrate concentrations.
Table 2.
Glo1 Enzyme Activity in the Lymphoblastoid Cell Lines Encoding Different Isoforms due to rs2736654 SNP
| Allele | Glo1 isoform | Activity1 μmol hr−1 mg−1 |
|---|---|---|
| C/C | Ala-Glo1 | 41.23 ± 5.62* |
| A/A | Glu-Glo1 | 32.75 ± 4.74* |
| C/A | Ala/Glu-Glo1 | 37.52 ± 4.57 |
Values are mean ± SD for n = 5 independent cell lines.
Means are statistically significant according to unpaired t-test with Welch correction applied (P < 0.03).
A reduction in Glo1 activity is expected to result in accumulation of the substrates for this enzyme. Direct HPLC measurements of one of the Glo1 substrates, namely, MG showed increased levels in cells expressing Glu-isoform (Table III). MG was measured after derivatization with OPD, which forms 2-methylquinoxaline, which was assayed by HPLC using a reversed-phase separation system, with spectrophotometric detection. The entire derivatization procedure was performed under acidic conditions to prevent the spontaneous formation of MG from inter-conversion of endogenous glyceraldehydes-3-phosphate and dihydroxyacetone phosphate. The concentration of MG in cells (1 × 107) expressing the Ala-isoform of Glo1 was 10.13 ± 2.72 pmol, whereas that in cells expressing the Glu-isoform of Glo1 was 15.91 ± 3.84 pmol, which is significantly higher (P < 0.03). The MG concentration in cells in an heterozygous allelic condition was not significantly different from that in cells in the two homozygous conditions.
Table 3.
Methylglyoxal Levels in Lymphoblastoid Cells Expressing Different Glo1-Isoforms
| Allele | Glo1-isoform | Methylglyoxal concentration1 ρmol/107 cells |
|---|---|---|
| C/C | Ala-Glo1 | 10.13 ± 2.72* |
| A/A | Glu-Glo1 | 15.91 ± 3.84* |
| C/A | Ala/Glu | 13.24 ± 3.04 |
Values are mean ± SD for n = 5 independent cell lines.
Means are statistically significant according to unpaired t-test with Welch correction applied (P < 0.03).
Since the activity of Glo1 is reported to be modulated by phosphorylation/dephosphorylation [van Herreweghe et al., 2002], we evaluated the status of Glo1 phosphorylation by metabolic radiolabeling with 32P-labeled inorganic phosphate. Lymphoblastoid cells expressing different isoforms of Glo1 were grown in DMEM media, depleted of endogenous phosphate, and pulse-labeled with 32P-labeled inorganic phosphate. The protein extracts from these cells were resolved by 10–20% gradient SDS-PAGE, followed by transfer onto nitrocellulose membrane filter. The proteins on the membrane were probed with anti-Glo1 (a generous gift from Dr. Kenneth Tew) and were subjected to autoradiography on an X-ray film. The Western blot shows equal amounts of Glo1 in all three cell line extracts (Figure 1). The same blot, upon autoradiography, shows that the Glu-isoform of Glo1 is hyperphosphorylated when compared to the Ala-isoform. Cells that express both Glu- and Ala-isoforms due to heterozygous allelic conditions have intermediate levels of 32P labeling. The densities in the bands were quantified from digital images using NIH ImageJ software (Table IV). These results demonstrate that Glo1 is hyperphosphorylated when the rs2736654 SNP is homozygous A-allele.
Figure 1.
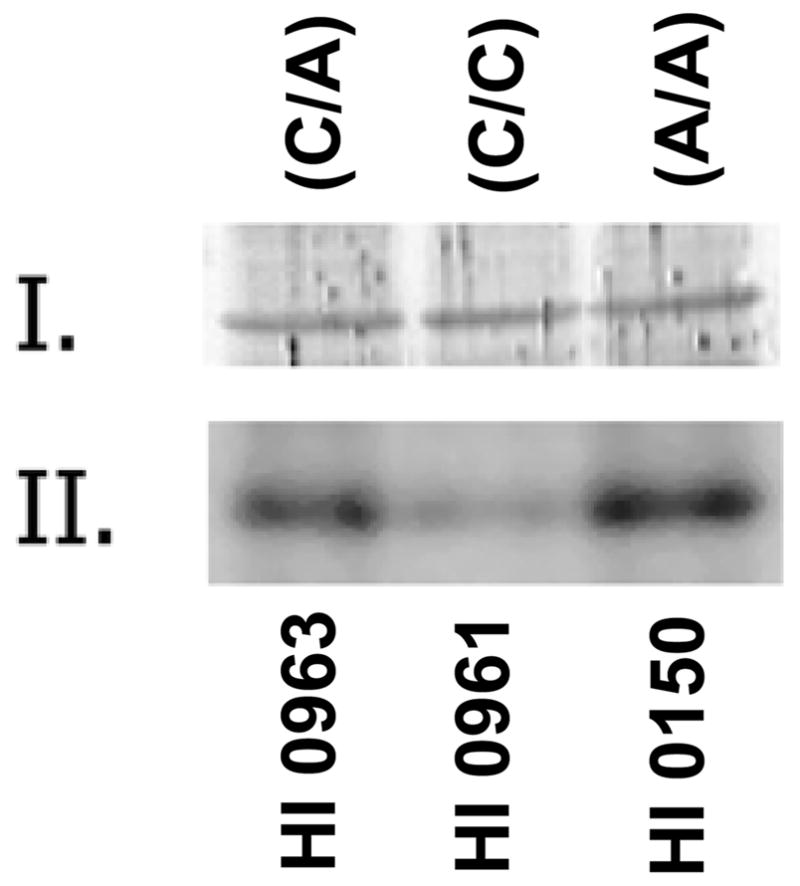
Western blot analysis and 32P labeling of different isoforms of Glo1 in lymphoblastoid cells in culture. Equal amounts of proteins (50 μg) were resolved on a 10–20% gradient SDS-PAGE and transferred onto a nitrocellulose membrane. The nitrocellulose membrane was probed with anti-Glo1 (rabbit polyclonal) antibody (I) or by autoradiography on an X-ray film (II). The cell lines used and the corresponding genotype are indicated.
Table 4.
Relative Intensities of the Protein Bands from Western Blots and 32P Labeling
| Cell line ID | rs2736654 allele | Relative band intensity1 | |
|---|---|---|---|
| Glo1 staining | 32P labeling | ||
| HI 0963 | C/A | 13 (37) | 11.3 (35) |
| HI 0961 | C/C | 10 (29) | 0.8 (3) |
| HI 0150 | A/A | 12 (34) | 19.9 (62) |
The densities of the bands were quantified using the NIH ImageJ software from a standard curve using the Kodak stepladder for intensity calibration. Numbers in parentheses represent percentage of the area.
Reduced Glo1 activity is associated with an accumulation of AGE products in autism brain [Junaid et al., 2004]. The accumulating AGE induces expression of the cell surface membrane-bound receptor, RAGE, which binds AGE and other ligands [Tanaka et al., 2000]. To extend our earlier observation, we have evaluated whether lymphoblastoid cells expressing different Glo1 isoforms affect expression of the RAGE, thereby initiating a downstream signaling cascade. The RAGE levels were analyzed in the lymphoblastoid cells by Western blot analysis using a polyclonal RAGE antibody (Figure 2a, b). Upon SDS-PAGE, the RAGE resolves into two bands of apparent molecular mass of 50 kDa and 48 kDa. Our results showed that cells expressing the Glu-isoform have the higher–molecular mass form of the RAGE isoform expressed in these cells (Figure 2a). Three of four cell liness expressing the Glu-isoform showed the additional isoform in the particulate fraction, while only one of the four cell lines expressing the Ala-isoform showed evidence of this isoform. The higher–molecular mass RAGE had diminished expression in cells that harbors the Ala-Glu isoform of Glo1. Such a difference in RAGE expression was not observed in the soluble form of the RAGE (Figure 2b).
Figure 2.

Western blot analysis for the RAGE in lymphoblastoid cells expressing different isoforms of the Glo1. Equal amounts of proteins (50 μg) from particulate (a) and soluble (b) fractions were resolved on a 10–20% gradient SDS-PAGE followed by transfer onto a nitrocellulose membrane. The nitrocellulose membrane was probed with an anti-RAGE (rabbit polyclonal) antibody, and the blot was also probed with actin as a loading control.
NHNP cells, when grown on lamin-coated cover slips in the presence of BDNF, developed neuronal processes. We used this system to evaluate the effect of exogenously added MG on the development of neuronal processes in NHNP cells. NHNP cells were able to differentiate in the presence of BDNF, as evidenced by βIII tubulin (a neuronal specific protein) staining (Figure 3A, top panel). MG inhibited both the growth and the development of the neuronal cells in a concentration-dependent manner. The growth inhibition is evidenced in the neurospheres themselves, as well as differentiated neuronal cells (Figure 3A–D), which establishes the strong toxicity associated with MG if it is not metabolized rapidly.
Figure 3.

Effect of MG on developing neurons. NHNP cells were grown on lamin-coated cover slips in the presence of BDNF. MG was added at the indicated concentrations and cells were allowed to differentiate for two weeks, following which cells were fixed and stained with primary antibodies for βIII tubulin (A), nestin (B), and DAPI (C). Panel (D) is a phase-contrast image.
Discussion
We had shown earlier that a significant proportion of individuals with autism are homozygous for the A-allele at rs2736654 SNP in the GLO1 gene [Junaid et al., 2004]. In the present study, we evaluated the functional consequences of the GLO1 rs2736654 SNP and its significance in relation to autism pathophysiology. The SNP causes a nonconservative Ala111Glu change in the mature protein that is a drastic change in the side-chain structure in the primary sequence. Structure- and sequence-dependent eukaryotic protein phosphorylation software NetPhos v 2.0 [Blom et al., 1999] showed an increased probability of phosphorylation of Ser114 in Glo1 when the SNP causes an Ala111Glu change. Indeed, the metabolic 32P-labeling experiment does confirm that the Glu-isoform of Glo1 is hyperphosphorylated. Together, the presence of an acidic amino acid and hyperphosphorylation is responsible for the acidic mobility of the Glo1 seen in autopsied autism brain [Junaid et al., 2004].
The presence of A-allele at rs2736654 SNP encoding the Glu-isoform is responsible for the reduced enzymatic activity of the Glo1. The lymphoblastoid cells expressing the Glu-isoform have a slight but statistically significant reduction in the Glo1-specific activity in comparison to the Ala-isoform. The presence of either alleles at rs2736654 did not cause any change in the Glo1 protein levels. Thus, the reduction in enzyme activity of the Glo1 could arise as a result of both conformational change resulting from amino acid replacement and hyperphosphorylation of a yet undetermined Ser/Thr residue. One of the regulatory mechanisms of enzymatic activities is through phophorylation/dephosphorylation of specific amino acid side chains. Although the phosphorylation of Glo1 has not been reported, as many as four Ser/Thr phosphorylation sites have been predicted from the primary structure [Ranganathan et al., 1993]. The real biological function of phosphorylated Glo1 is still unknown, but there is evidence of its involvement in a pathway that leads to the formation of MG-derived AGE. Van Herreweghe et al. [2002] have demonstrated that TNF induces protein kinase A–mediated phosphorylation of Glo1 in the fibrosarcoma cell line. These cells accumulate MG-derived AGEs. In our earlier report, we had demonstrated an accumulation of AGE in autism brains [Junaid et al., 2004]. Thus, the Glu-isoform of Glo1, by virtue of the presence of acidic Glu111 as well as hyperphorphorylation of a Ser/Thr, possesses reduced enzymatic activity when compared to the Ala-isoform. As a result, the cells are unable to metabolize MG completely. Consequently, MG levels increase in cells; being a reactive metabolite, MG combines with side-chain residues of the amino acids in proteins, resulting in the formation of covalent adducts, AGE. Moreover, Glo1 is a Zn2+-containing enzyme that functions as a dimer. The zinc atoms are held by electrostatic forces with acidic amino acid residues that also include Glu99, and functions in enediolate intermediate formation and stabilization during catalysis [Ridderstrom et al., 1998]. This residue is close to the Glu111 that is replaced by the SNP and may interfere with electrostatic interactions with Zn2+. The 3D structure of Glo1 with Glu111 has shown that the active Glo1 functions as a dimer with the two zinc atoms, two active sites, as well as two glutathione-binding sites formed at the interface [Cameron et al., 1997]. The Glu111 in this structure occurs at the interface of the two Glo1 molecules and close to the substrate cleft. However, these studies have not shown direct interaction of Glu111 in Zn2+ binding; any chemical interactions that change the 3D structure or disrupt electrostatic bonds are expected to affect enzyme activity.
Normally, cells are able to withstand a 40–60% reduction in enzyme activities as shown in parents of autosomal recessive disorders such as late-infantile neuronal ceroid lipofuscinosis (LINCL) [Junaid et al., 1999; Junaid et al., 2000]. Whereas a homozygous allele condition causes a debilitating fatal disorder in early childhood, in LINCL, heterozygous carriers (parents and normal siblings) live a normal life without any pathological changes. Under normal conditions, we believe that a reduction in Glo1 enzyme activity in a homozygous A-allele condition may not be deleterious. However, in the presence of other aberrant interacting genes that may number as many as 10–15 in autism [Pickles et al., 1995; Risch et al., 1999], a reduction in Glo1 exacerbates an already challenged developing neuronal system. High expression of Glo1 has been reported in the embryonic stages of life [Amicarelli et al., 1998], pointing to its essential role during this phase of life. MG is continuously formed during cellular metabolism of sugars, amino acids, and fatty acids [Ray and Ray, 1983; Reichard Jr. et al., 1992; Lyles and Chambers, 1992] and must be detoxified by the glyoxalase system. Experiments with developing neuronal processes from neurospheres have shown that exogenously added MG is deleterious to developing neurons, and such a situation in vivo may result in loss of neuronal connectivity. Aberrant neuronal connectivity is an important aspect of autism brain pathology [Casanova et al., 2002; Buxhoeveden et al., 2006; Monk et al., 2009]. Moreover, the higher expression of Glo1 in Purkinjie cells reflects on its critical role in the detoxification of MG in these cells. The loss of Purkinjie cells in autism pathology is the most consistent pathological finding thus far [Bauman and Kemper, 2005]. Identification of factors leading to altered neuronal development will provide insights into the molecular mechanisms.
Reduction in Glo1 activity beyond a threshold will accumulate MG and lead to the formation of AGE that ultimately induces the RAGE-mediated downstream signaling cascade. The deleterious effects of both MG and RAGE have been reported in the literature. MG has been proposed as a neurodegenerative agent [Ahmed et al., 2003; Kuhla et al., 2006], and was shown to induce oxidative stress–mediated cell apoptosis in neuronal culture systems [Kikuchi et al., 1999; DiLoreto et al., 2004]. Our results from exogenously added MG confirm this finding in developing neuronal cultures. In individuals with autism, the levels of secretory RAGE were reported to be reduced, whereas those of its endogenous ligands, S100A9 and HMGB1, were increased [Emanuele et al., 2010; Boso et al., 2006]. Over-activation of RAGE-mediated signaling has been reported to inhibit neurite outgrowth [Rong et al., 2004]. Thus, a dysfunction of the Glo1 system can set in motion two different effects on cell growth and differentiation mechanisms. Another possibility of a Glo1-mediated effect is by the formation of AGE of certain crucial proteins. MG is a potent protein-glycating agent, and this modification of protein is a sort of signal for protein degradation [Thornalley, 1996]. AGE formation has been reported for several crucial proteins involved in cell growth and differentiation [Sakamoto et al., 2002; Speer et al., 2003]. AGEs are also formed in diabetes and uremia and are responsible for microvascular dysfunction and atherosclerosis. The neurotoxicity of specific AGEs in cultured cortical neuronal cells has been reported [Takeuchi et al., 2000]. More recently, loss of triosephosphate isomerase function was shown to cause paralysis, mutation, and early death in Drosophila [Gnerer et al., 2006]. Deficiency of this enzyme causes accumulation of MG, leading to accumulation of AGEs, and causes neuromuscular degeneration [Ahmed et al., 2003]. These AGE-modified proteins may themselves have compromised functions. Because autism is an early developmental disorder, molecules that affect cell growth and differentiation at the early neurogenesis stage can be possible sites of biological defects. It is unclear to what degree exogenous MG addition mirrors the effects of endogenously produced MG, given issues of dose comparison and enzyme compartmentalization.
Environmental and genetic factors are intimately related in the etiology of heterogeneous disorders such as autism. It is unlikely that these disorders are caused by common mutations that drastically alter gene expression or protein function. Rather, complex disorders may be a culmination of minor changes in gene expression and functions due to a number of polymorphisms occurring concurrently. Polymorphisms that generally have a modest effect individually can pose a high risk when present in high frequency in the population.
Acknowledgments
Grant sponsor: NIH (NINDS)
Grant number: NS40691
We thank the Autism Genetic Resource Exchange (AGRE) and the participating AGRE families, and the National Alliance for Autism Research (NAAR). We also thank Maureen Marlow for assistance with comments on the manuscript. This work was supported by funds from the National Institutes of Health (NS40691), the New York State Office for People with Developmental Disabilities (formerly the New York State Office of Mental Retardation and Developmental Disabilities), and the National Alliance for Autism Research (NAAR).
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nature Reviews Genetics. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N, Battah S, Karachalias N, Babaei-Jadidi R, Horanyi M, Baroti K, et al. Increased formation of methylglyoxal and protein glycation, oxidation and nitrosation in triosephosphate isomerase deficiency. Biochimica et Biophysica Acta. 2003;1639:121–132. doi: 10.1016/j.bbadis.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Amicarelli F, Sacchetta P, Colafarina S, Angelucci S, Miranda M, DeIlio C. Glyoxalases activity during Bufo bufo embryo development. Mechanisms of Ageing and Development. 1998;100:261–267. doi: 10.1016/s0047-6374(97)00148-6. [DOI] [PubMed] [Google Scholar]
- Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: a review and future directions. International Journal of Developmental Neuroscience. 2005;23:183–187. doi: 10.1016/j.ijdevneu.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S, Brunak S. Sequence- and structure-based prediction of eukaryotic protein phosphorylation sites. Journal of Molecular Biology. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Boso M, Emanuele E, Minoretti P, Arra M, Politi P, Ucelli di Nemi S, Barale F. Alterations of circulating endogenous secretory RAGE and S100A9 levels indicating dysfunction of the AGE-RAGE axis in autism. Neuroscience Letters. 2006;410(3):169–173. doi: 10.1016/j.neulet.2006.08.092. [DOI] [PubMed] [Google Scholar]
- Buxhoeveden DP, Semendeferi K, Buckwalter J, Schenker N, Switzer R, Courchesne E. Reduced minicolumns in the frontal cortex of patients with autism. Neuropathology and Applied Neurobiology. 2006;32:483–91. doi: 10.1111/j.1365-2990.2006.00745.x. [DOI] [PubMed] [Google Scholar]
- Cameron AD, Olin B, Ridderstrom M, Mannervik B, Alwyn-Jones T. Crystal structure of human glyoxalase I - evidence for gene duplication and 3D domain swapping. EMBO Journal. 1997;16:3386–3395. doi: 10.1093/emboj/16.12.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova MF, Buxhoeveden DP, Switala AE, Roy E. Minicolumnar pathology in autism. Neurology. 2002;58:428–432. doi: 10.1212/wnl.58.3.428. [DOI] [PubMed] [Google Scholar]
- Chaplen FWR, Fahl WE, Cameron DC. Method for determination of free intracellular and extracellular methylglyoxal in animal cells grown in culture. Analytical Biochemistry. 1996;238:171–178. doi: 10.1006/abio.1996.0271. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Fombonne E. Pervasive developmental disorders in preschool children: confirmation of high prevalence. American Journal of Psychiatry. 2005;162:1133–1141. doi: 10.1176/appi.ajp.162.6.1133. [DOI] [PubMed] [Google Scholar]
- Chen W, Kuizon S, Chiou BL, Bolton DC, Pullarkat RK, Junaid MA. Differential expression of small heat shock protein 27 (Hsp27) in ataxia telangiectasia brains. Neurochemical Research. 2009;34:1658–1667. doi: 10.1007/s11064-009-9959-y. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Carper R, Akshoomoff N. Evidence of brain overgrowth in the first year of life in autism. Journal of The American Medical Association. 2003;290:337–344. doi: 10.1001/jama.290.3.337. [DOI] [PubMed] [Google Scholar]
- DiLoreto S, Caracciolo V, Colafarina S, Sebastiani P, Gasbarri A, Amicarelli F. Methylglyoxal induces oxidative stress-dependent cell injury and up-regulation of interleukin-1 beta and nerve growth factor in cultured hippocampal neuronal cells. Brain Research. 2004;1006:157–167. doi: 10.1016/j.brainres.2004.01.066. [DOI] [PubMed] [Google Scholar]
- Emanuele E, Boso M, Brondino N, Pietra S, Barale F, Ucelli di Nemi S, Politi P. Increased serum levels of high mobility group box 1 protein in patients with autistic disorder. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2010;34(4):681–683. doi: 10.1016/j.pnpbp.2010.03.020. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Uchida S, Watanuki T, Wakabayashi Y, Otsuki K, Matsubara T, et al. Reduced expression of glyoxalase-1 mRNA in mood disorder patients. Neuroscience Letters. 2008;438:196–199. doi: 10.1016/j.neulet.2008.04.024. [DOI] [PubMed] [Google Scholar]
- Gnerer JP, Kreber RA, Ganetzky B. Wasted away, a Drosophila mutation in triosephosphate isomerase, causes paralysis, neurodegeneration, and early death. Proceedings of the National Academy of Sciences or the USA. 2006;103:14987–14993. doi: 10.1073/pnas.0606887103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM, et al. Glyoxalase I and glutathione reductase 1 regulate anxiety in mice. Nature. 2005;438:662–666. doi: 10.1038/nature04250. [DOI] [PubMed] [Google Scholar]
- Junaid MA, Kowal D, Barua M, Pullarkat PS, Sklower-Brooks S, Pullarkat RK. Proteomic studies identified a single nucleotide polymorphism in glyoxalase I as autism susceptibility factor. American Journal of Medical Genetics A. 2004;131A:11–17. doi: 10.1002/ajmg.a.30349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junaid MA, Sklower-Brooks S, Wisniewski KE, Pullarkat RK. A novel assay for lysosomal pepstatin-insensitive proteinase and its application for the diagnosis of late-infantile neuronal ceroid lipofuscinosis. Clinica Chimica Acta. 1999;281:169–176. doi: 10.1016/s0301-2115(98)00333-9. [DOI] [PubMed] [Google Scholar]
- Junaid MA, Wu G, Pullarkat RK. Purification and characterization of bovine brain lysosomal pepstatin-insensitive proteinase, the gene product deficient in the human late-infantile neuronal ceroid lipofuscinosis. Journal of Neurochemistry. 2000;74:287–294. doi: 10.1046/j.1471-4159.2000.0740287.x. [DOI] [PubMed] [Google Scholar]
- Kester MV, Norton SJ. The isolation and characterization of mouse liver glyoxalase I. Biochimica et Biophysica Acta. 1975;391:212–221. doi: 10.1016/0005-2744(75)90168-0. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Shinpo K, Moriwaka F, Makita Z, Miyata T, Tashiro K. Neurotoxicity of methylglyoxal and 3-deoxyglucosone on cultured cortical neurons: synergism between glycation and oxidative stress, possibly involved in neurodegenerative diseases. Journal of Neuroscience Research. 1999;57:280–289. doi: 10.1002/(SICI)1097-4547(19990715)57:2<280::AID-JNR14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Kromer SA, Kessler MS, Milfay D, Birg IN, Bunck M, Czibere L, et al. Identification of glyoxalase-I as a protein marker in a mouse model of extremes in trait anxiety. Journal of Neuroscience. 2005;25:4375–4384. doi: 10.1523/JNEUROSCI.0115-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhla B, Luth HJ, Haferburg D, Weick M, Reichenbach A, Arendt T, et al. Pathological effects of glyoxalase I inhibition in SH-SY5Y neuroblastoma cells. Journal of Neuroscience Research. 2006;83:1591–1600. doi: 10.1002/jnr.20838. [DOI] [PubMed] [Google Scholar]
- Lord C, Cook EH, Jr, Leventhal BL, Amaral DG. Autism spectrum disorders. Neuron. 2000;28:355–363. doi: 10.1016/s0896-6273(00)00115-x. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurement with Folin phenol reagent. Journal of Biological Chemistry. 1951;193:265–275. [PubMed] [Google Scholar]
- Lyles GA, Charmers J. The metabolism of aminoacetone to methylglyoxal by semicarbazide-sensitive amino oxidase in human umbilical artery. Biochemical Pharmacology. 1992;43:1409–1414. doi: 10.1016/0006-2952(92)90196-p. [DOI] [PubMed] [Google Scholar]
- MMWR. Prevalence of Autism Spectrum Disorders - Autism and Developmental Disabilities Monitoring Network, United States, 2006. Morb Mortal Wkly Rep. 2009;58(SS10):1–20. [PubMed] [Google Scholar]
- Monk CS, Peltier SJ, Wiggins JL, Weng SJ, Carrasco M, Risi S, et al. Abnormalities of intrinsic functional connectivity in autism spectrum disorders. Neuroimage. 2009;47:764–472. doi: 10.1016/j.neuroimage.2009.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatrics. 2004;113:e472–486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- Pickles A, Bolton P, Macdonald H, Bailey A, LeCouteur A, Sim CH, et al. Latent-class analysis of recurrence risks for complex phenotypes with selection and measurement error: a twin and family history study of autism. American Journal of Human Genetics. 1995;57:717–726. [PMC free article] [PubMed] [Google Scholar]
- Politi P, Minoretti P, Falcone C, Martinelli V, Emanuele E. Association analysis of the functional Ala111Glu polymorphism of the glyoxalase I gene in panic disorder. Neuroscience Letters. 2006;396:163–166. doi: 10.1016/j.neulet.2005.11.028. [DOI] [PubMed] [Google Scholar]
- Ranganathan S, Walsh ES, Godwin AK, Tew KD. Cloning and characterization of human colon glyoxalase-I. Journal of Biological Chemistry. 1993;268:5661–5667. [PubMed] [Google Scholar]
- Ray S, Ray M. Formation of methylglyoxal from aminoacetone by amine oxidase from goat plasma. Journal of Biological Chemistry. 1983;258:3461–3462. [PubMed] [Google Scholar]
- Rehnström K, Ylisaukko-Oja T, Vanhala R, von Wendt L, Peltonen L, Hovatta I. No association between common variants in glyoxalase 1 and autism spectrum disorders. American Journal of Medical Genetics Part B Neuropsychiatric Genetics. 2008;147B:124–127. doi: 10.1002/ajmg.b.30582. [DOI] [PubMed] [Google Scholar]
- Reichard GA, Jr, Skutches CL, Hoeldtke RD, Owen OE. Acetone metabolism in humans during diabetic ketoacidosis. Diabetes. 1986;35:668–674. doi: 10.2337/diab.35.6.668. [DOI] [PubMed] [Google Scholar]
- Ridderstrom M, Cameron AD, Jones TA, Mannervik B. Involvement of an active-site Zn2+ ligand in the catalytic mechanism of human glyoxalase I. Journal of Biological Chemistry. 1998;273:21623–21628. doi: 10.1074/jbc.273.34.21623. [DOI] [PubMed] [Google Scholar]
- Risch N, Spike D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, et al. A genomic screen of autism: evidence for a multilocus etiology. American Journal of Human Genetics. 1999;65:493–507. doi: 10.1086/302497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald A, Happé F, Bolton P, Butcher LM, Price TS, Wheelwright S, et al. Genetic heterogeneity between the three components of the autism spectrum: a twin study. Journal of the American Academy of Child and Adolescent Psychiatry. 2006;45:691–699. doi: 10.1097/01.chi.0000215325.13058.9d. [DOI] [PubMed] [Google Scholar]
- Rong LL, Trojaborg W, Qu W, Kostov K, Yan SD, Gooch C, et al. Antagonism of RAGE suppresses peripheral nerve regeneration. FASEB Journal. 2004;18:1812–1817. doi: 10.1096/fj.04-1899com. [DOI] [PubMed] [Google Scholar]
- Sacco R, Papaleo V, Hager J, Rousseau F, Moessner R, Militerni R, et al. Case-control and family-based association studies of candidate genes in autistic disorder and its endophenotypes: TPH2 and GLO1. BMC Medical Genetics. 2007;8:11. doi: 10.1186/1471-2350-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto H, Mashima T, Yamamoto K, Tsuruo T. Modulation of heat shock proteins 27 (Hsp27) anti-apoptotic activity by methylglyoxal modification. Journal of Biological Chemistry. 2002;277:45770–45775. doi: 10.1074/jbc.M207485200. [DOI] [PubMed] [Google Scholar]
- Speer O, Morkunaite-Haimi S, Liobikas J, Franck M, Hensbo L, Linder MD, et al. Rapid suppression of mitochondrial permeability transition by methylglyoxal. Role of reversible arginine modification. Journal of Biological Chemistry. 2003;278:34757–34763. doi: 10.1074/jbc.M301990200. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Bucala R, Suzuki T, Ohkubo T, Yamakazi M, Koike T, et al. Neurotoxicity of advanced glycation end-products for cultured cortical neurons. Journal of Neuropathology and Experimental Neurology. 2000;59:1094–1105. doi: 10.1093/jnen/59.12.1094. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Yonekura H, Yamagishi SI, Fujimori H, Yamamoto Y, Yamamoto H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-α through nuclear factor-κB, and by 17 β-estradiol through Sp-1 in human vascular endothelial cells. Journal of Biological Chemistry. 2000;275:25781–25790. doi: 10.1074/jbc.M001235200. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ. Population genetics of human glyoxalases. Heredity. 1991;67:139–42. doi: 10.1038/hdy.1991.73. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification - a role in pathogenesis and antiproliferative chemotherapy. General Pharmacology. 1996;27:565–573. doi: 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ. Glyoxalase I - structure, function and a critical role in the enzymatic defence against glycation. Biochemical Society Transaction. 2003;31:1343–1348. doi: 10.1042/bst0311343. [DOI] [PubMed] [Google Scholar]
- Tuchman R, Rapin I. Epilepsy in autism. Lancet Neurology. 2002;1:352–358. doi: 10.1016/s1474-4422(02)00160-6. [DOI] [PubMed] [Google Scholar]
- Van Herreweghe F, Mao J, Chaplen FWR, Grooten J, Gevaert K, Vandekerckhov J, et al. Tumor necrosis factor-induced modulation of glyoxalase 1 activities through phosphorylation by PKA results in cell death and is accompanied by the formation of a specific methylglyoxal-derived AGE. Proceedings of the National Academy of Sciences of the USA. 2002;99:949–954. doi: 10.1073/pnas.012432399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kooten IA, Palmen SJ, von Cappeln P, Steinbusch HW, Korr H, Heinsen H, et al. Neurons in the fusiform gyrus are fewer and smaller in autism. Brain. 2008;131(Pt 4):987–999. doi: 10.1093/brain/awn033. [DOI] [PubMed] [Google Scholar]
- Wang DZ, Ray P, Boothby M. Interleukin 4-inducible phosphorylation of HMG-I(Y) is inhibited by rapamycin. Journal of Biological Chemistry. 1995;270:22924–22932. doi: 10.1074/jbc.270.39.22924. [DOI] [PubMed] [Google Scholar]
- Wu YY, Chien WH, Huang YS, Gau SS, Chen CH. Lack of evidence to support the glyoxalase 1 gene (GLO1) as a risk gene of autism in Han Chinese patients from Taiwan. Progress in Neuro-psychopharmacology and Biological Psychiatry. 2008;32:1740–1744. doi: 10.1016/j.pnpbp.2008.07.019. [DOI] [PubMed] [Google Scholar]