

Abstract
Pluripotent stem cells, both human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), can give rise to multiple cell types and hence have tremendous potential for regenerative therapies. However, the tumorigenic potential of these cells remains a great concern, as reflected in the formation of teratomas by transplanted pluripotent cells. In clinical practice, most pluripotent cells will be differentiated into useful therapeutic cell types such as neuronal, cardiac, or endothelial cells prior to human transplantation, drastically reducing their tumorigenic potential. Our work investigates the extent to which these differentiated stem cell derivatives are truly devoid of oncogenic potential. In this study, we analyzed the gene expression patterns from three sets of hiPSC- and hESC-derivatives and the corresponding primary cells, and compared their transcriptomes with those of five different types of cancer. Our analysis revealed a significant gene expression overlap of the hiPSC- and hESC-derivatives with cancer, whereas the corresponding primary cells showed minimum overlap. Quantitative PCR (qRT-PCR) analysis of a set of cancer related genes (selected on the basis of rigorous functional and pathway analyses) confirmed our results. Overall, our findings suggest that pluripotent stem cell derivatives may still bear oncogenic properties even after differentiation, and additional stringent functional assays to purify these cells should be performed before they can be used for regenerative therapy.
Keywords: oncogenicity, stem cell, hiPSC, hESC, differentiated cells, hiPSC-derivative, hESC-derivative
INTRODUCTION
Human embryonic stem cells (hESCs) are derived from the inner cell mass (ICM) of a human blastocyst stage embryo (1). They are characterized by their ability to both self-renew and differentiate into all somatic tissues of the embryo. After transplantation into immunosuppressed mice, they spontaneously differentiate and form tumors (teratomas), in which there is disordered differentiation into various tissue types of the early embryo (2). The tumorigenic nature of hESCs has been previously described (3-4) and is considered a major obstacle to their clinical utilization. Though teratomas may be considered a relatively benign, disorganized bulk of normal embryonic tissues, the formation of a teratoma after hESC transplantation in human patients is entirely unacceptable. The clinical hurdles facing utilization of hESC-based grafts in the clinic are comprehensively discussed elsewhere (5).
In contrast to hESCs, the concept of deriving pluripotent cells from somatic cells by reversing the natural differentiation process that occurs during development has long been explored (6-7). The real quantum leap in this effort was finally realized with the generation of human induced pluripotent stem cells (hiPSCs) in 2007 (8-9), which has freed regenerative therapies from the ethical concerns that are frequently aroused by hESCs. However, hiPSCs are currently generated using multiple inducing factors that may have oncogenic potential (10-13), and it has been shown that mice generated from murine iPSCs have increased tumorigenicity and mortality (14). Additionally, the fully reprogrammed hiPSC phenotype arises only as rare clonal populations (≤0.1%) among partially reprogrammed cells (15). Given these issues, the practice of reprogramming adult cells into hiPSCs faces several hurdles that must be overcome before it can have any practical clinical applications. For instance, the efficiency of reprogramming needs to be improved, and hiPSCs need to be generated in a manner that avoids any exogenous sequences that may induce malignancy (13, 16-17). Similar to hESCs, even a small number of undifferentiated cells may give rise to teratomas after hiPSC transplantation.
Recent studies indicate that stem cells and tumor cells share many common master regulatory genes (18-21). The interwoven nature of pluripotency and tumorigenicity programs is revealed by the molecular machinery shared by them, and it has become a major challenge to untangle the determinants of pluripotency from those responsible for tumorigenicity. This entanglement is exemplified by the fact that many of the genes used to produce hiPSCs are either outright oncogenes such as Myc and KLF4 (22-23), or are in subtle ways linked to tumorigenesis such as Sox2 (24), Nanog (19), and Oct3/4 (25). Note that hESCs are also defined by the expression of a battery of these genes, mostly Oct4, Nanog, and Sox2 (26).
The primary method for eliminating the problem of tumorigenicity is to induce differentiation of hESCs or hiPSCs into the required cell type prior to transplantation. Although the tumorigenic potential of these pluripotent cells seems to be greatly reduced in vivo when the cells are pre-differentiated in vitro, the risk of teratoma formation still exists due to carryover of contaminating pluripotent cells. Furthermore, even with differentiated derivatives that are free of contaminating undifferentiated cells, the tumorigenicity risk still remains, as several reports have shown that transplanted derivatives of ESCs may also produce tumors (27-30). For example, it has been shown that after injection of neural marker-selected derivatives of mouse ESCs into the subretinal space of rhodopsin−/− mice (28), teratomas were formed that caused eye malformation within two months after transplantation. Two studies have shown that ESC-derived neural precursors for transplantation into fetal brains of mice (29) and dopaminergic neuron progenitors for transplantation into Parkinsonian rats (28) also showed signs of tumor formation. Another study showed that injection of beating embryoid bodies containing cardiomyocytes into the myocardium still led to teratoma formation, albeit at a later onset compared to undifferentiated ESCs (30). These are examples of different fates that pluripotent stem cell derivatives can assume, and extreme care should be taken to exclude potentially tumorigenic cells from the transplantable population. Studies must be performed for each differentiated derivative that is considered for therapy before it can be regarded as safe for clinical application.
To address these issues of tumorigenicity, we performed a rigorous transcriptional analysis of the different hiPSC- and hESC-derived cell lines and corresponding human primary cells that have been previously reported (31-35). We compared these transcriptomes with different cancer cell lines (36-40) to delineate the tumorigenic gene expression patterns still remaining within these differentiated derivatives. We also performed a qRT-PCR analysis of a selected panel of cancer genes within these derivatives to confirm the tumorigenic potential of these cells. Compared to their primary cell counterparts, we discovered that the pluripotent stem cell derivatives expressed higher levels of cancer-related genes even after differentiation. These findings demonstrate that our understanding of the tumorigenicity of hiPSC- and hESC-derivatives is only rudimentary at present. In the future, caution must be exercised when considering pluripotent stem cell derivatives for regenerative therapies, and methods for purifying these cell populations of undifferentiated contaminants, as well as reducing their innate oncogenic potential, must be established prior to clinical application.
MATERIALS AND METHODS
Sources of gene profiles
In this study, we analyzed the transcriptional profiles of previously reported hiPSC- and hESC-derived hepatic cell lines (35), hESC-derived endothelial cell lines (34) (please note that the profiles for hiPSC-derived endothelial cell lines have been newly generated in our lab (41)), and hiPSC- and hESC-derived neural crest cell lines (33). We also compared the gene expression data from primary hepatocytes (32), human umbilical vein endothelial cells (HUVECs) (34), and neural crest cell lines (31), respectively. These profiles were then compared to those of a common set of five different cancer cell lines (36-40) consisting of breast cancer, myeloid leukemia, glioblastoma multiforme cancer stem cells, prostate cancer, and pancreatic cancer cell lines. The breast cancer cell lines are the rare cancer side population (SP) cells isolated from the CAL-51 human mammary carcinoma cell line that display cancer stem cell characteristics (36). Myeloid leukemia stem cells were isolated from patients with acute myeloid leukemia (AML) (38). Glioblastoma multiforme samples were isolated from patients undergoing surgical biopsies; non-adherent cellular spheroids derived from these serum-free culture conditions were considered to be enriched cancer stem cell cultures (40). For prostate cancer, cancer stem cell lines derived from prostate cancer cell line PCSC1-3 (from Celprogen, San Pedro, CA) were used (37). Lastly, the pancreatic cancer cell lines chosen for this study were Nor-P1, HPAF-II, CaPan-2, BxPC-3, and Panc 2.03 (39).
For the microarray profiling data, all of the hiPSCs from which the differentiated cell lines (hepatocytes, endothelial cells and neural crest cells) were derived were originally reprogrammed from primary fibroblasts. Additionally, all of the hiPSCs used in the microarray analyses were reprogrammed using lentiviral vectors that included oncogenic reprogramming factors such as c-Myc and KLF4. Specifically, the hiPSCs used to derive hepatocytes were reprogrammed from human primary foreskin fibroblasts (CRL2097; ATCC, Manassas, VA) following the established protocol by Yu et al. (9). Concentrated replication incompetent pseudotyped lentiviruses that expressed OCT4, SOX2, NANOG, or LIN28 were used to infect the cells. The hiPSCs that were used to derive endothelial cells were obtained from the James Thomson Lab (University of Wisconsin-Madison), and were originally derived from IMR90 fetal fibroblasts (ATCC) using the reprogramming factors OCT4, SOX2, NANOG, and LIN28 that were cloned within lentiviral vector. The hiPSCs used to derive neural crest cells were reprogrammed from fibroblasts obtained from Coriell (NJ, USA) using a lentiviral construct with four factors (OCT4, SOX2, KLF4, and c-MYC) as described recently (42).
Gene expression data were obtained from the Gene Expression Omnibus (GEO) repository, which is currently the largest fully public gene expression resource. The GEO (43) repository at the National Center for Biotechnology Information (NCBI) archives freely disseminates microarray and other forms of high-throughput data generated by the scientific community.
Microarray analysis
The hiPSC- and hESC-derived neural crest cells and endothelial cells were gene expression profiled with the Illumina human-6 v2.0 expression beadchip and Agilent 4×44K whole human genome microarray (G4112F) platform, respectively. All other gene expression data were obtained with the HG-U133plus2 microarray platform (Affymetrix). All data sets were analyzed using GeneSpring GX 11.0 software (Agilent Technologies, Inc.). Gene-level signal estimates were derived from the raw data files. Summarization of gene expression data was performed by implementing the robust multichip averaging algorithm, with subsequent baseline normalization of the log-summarized values for each probe set to that of the median log summarized value for the same probe set in the control group. Expression data were then filtered to remove probe sets whose signal intensities for all the treatment groups were in the lowest 20 percentile of all intensity values. The data were then subjected to analysis of variance (ANOVA), incorporating the Benjamini–Hochberg FDR multiple testing correction, with a significance level of P-value <0.05 to obtain the differentially expressed genes between different groups. Probe sets were further filtered on the basis of a fold-change cut off of 2.0.
Distance measure
We have used the statistical software package SPSS (IBM) to generate the Euclidean distance matrix and the corresponding dendrogram to calculate the distances between different sets of cells with respect to cancer cells. To calculate the relative distances among hiPSC-derivatives, hESC-derivatives, and their corresponding primary cells with cancer cells, we considered 1 to be the farthest distance (Euclidean distance) obtained between cancer and the differentiated cells. We calculated the distances of the hiPSC- and hESC-derivatives from cancer cell lines for each of the data sets. The gene expression “overlap percentage” between two groups of cells is the percentage of genes that have a similar expression pattern. Thus, two “closer” groups will have a higher percentage of similar genes and therefore will have higher “overlap percentage” and vice versa.
Ingenuity Pathway Analysis
In order to perform functional annotation of the differentially expressed genes among different groups, we used Ingenuity Pathway Analysis (IPA) software. This software assigns biological functions to genes using the Ingenuity Pathways Knowledge Base (Ingenuity Systems, Inc., Redwood City, CA). The knowledge base includes information about thousands of human, mouse, and rat genes (44). This information is used to form networks to create an ‘interactome’ of genes that are involved in specific biological processes.
Functional Analysis
The Functional Analysis identified the biological functions and/or diseases that were most significant to the data set. Molecules from the data set that met the P-value cutoff of 0.05 and fold change cutoff of 2.0 were then associated with biological functions and/or diseases in Ingenuity’s Knowledge Base. Right-tailed Fisher’s exact test was used to calculate a P-value determining the probability that each biological function and/or disease assigned to that data set is due to chance alone.
Canonical Pathway Analysis
Canonical pathways analysis identified the pathways from the IPA library of canonical pathways that were most significant to the data set. The significance of the association between the data set and the canonical pathway was measured in 2 ways: 1) A ratio of the number of molecules from the data set that map to the pathway divided by the total number of molecules that map to the canonical pathway is displayed. 2) Fisher’s exact test was used to calculate a P-value determining the probability that the association between the genes in the data set and the canonical pathway is explained by chance alone.
Cell cultures and RNA preparation
For the qRT-PCR data, hiPSCs were obtained from the James Thomson lab (University of Wisconsin-Madison), which were originally derived from IMR90 fetal fibroblasts using reprogramming factors OCT4, SOX2, NANOG, and LIN28 (virally reprogrammed). The H9 hESC cell line was obtained from Wicell (Madison, WI). The nonviral minicircle hiPSCs (mc-hiPSCs) were originally derived from human adipose stem cells (hASCs) using a minicircle vector containing four reprogramming factors OCT4, SOX2, NANOG, and LIN28 (45). H9 hESCs, hiPSCs, and mc-hiPSCs were cultured on Matrigel in mTeSR1 medium (Stem Cell Technologies, Vancouver, BC, Canada). Endothelial cell differentiation of hiPSCs, mc-hiPSCs, and hESCs was performed as previously described (34). Briefly, the pluripotent cell colonies were detached by 1 mg/ml dispase and transferred to ultra low-attachment plates for embryoid body (EB) formation. 12 day-old EBs were harvested and then suspended in Collagen I. The mixture was then incubated at 37°C for 30 minutes to allow gel polymerization. Later, EGM-2 medium (Lonza) plus 5% Knockout™ serum with 50 ng/ml VEGF and 20 ng/ml FGF2 was added. After 3-day culture, the CD31+/CD144+ cells (representing endothelial cells) were purified by FACS. All four endothelial cell types (hiPSC-EC, mc-hiPSC-EC, hESC-EC, and HUVEC) were cultured under the same conditions. Cells were harvested at 80% confluency. Using the RNAeasy Mini Kit (Qiagen Inc., Valencia, CA), RNA was isolated from biological duplicates of hiPSC-EC, mc-hiPSC-EC, hESC-EC, and HUVEC.
The cancer cell lines, namely three breast cancer cell lines (MDAMB-231, MDAMB-435 and MDAMB-468) and one prostate cancer cell line (PC-3), were obtained from Zhen Cheng’s lab at Stanford. These cells were cultured in DMEM and 10% FBS. When the cell confluence reached 80%, the culture medium was changed to EC culture medium (EGM-2 medium (Lonza) plus 5% Knockout™ serum with 50 ng/ml VEGF and 20 ng/ml FGF2). Cells were isolated after single passage. RNA was isolated from cell samples by using Qiagen RNAeasy Mini Kit (Qiagen Inc., Valencia, CA). RNA was isolated from biological duplicates of MDAMB-231, MDAMB-435, MDAMB-468, and PC-3.
Real-time quantitative PCR (qRT-PCR)
1 μg of total RNA from each cell sample was reversed-transcribed with iScript cDNA synthesis KIT (BioRad). For each sample, qRT-PCR was performed in duplicate on a StepOnePlus Real-time PCR system (Applied Biosystems) using Taqman primer probe sets (Applied Biosystems) for each gene of interest and an 18S control primer probe set for normalization. Gene expression assay IDs for the tumor-specific genes obtained from Applied Biosystems used in the amplification reaction are as follows: TNC: Hs01115665_m1, VCAN: Hs00171642_m1, PCOLCE: Hs00170179_m1, KIAA1199: Hs00378520_m1, FOS: Hs01119267_g1, SEMA5A: Hs01549381_m1, SNAI2: Hs00161904_m1, SERPINE2: Hs00299953_m1, COL6A2: Hs00242484_m1, THBS1: Hs00962908_m1, 18S: Hs03928990_g1. Representative results are shown as fold expression relative to HUVEC unless otherwise stated.
Western Blot
Cells were collected in RIPA buffer (Sigma) and briefly sonicated to shear DNA and reduce sample viscosity. Protein concentration was measured by Bio-Rad Protein Assay Kit. Samples were run on a 10% Mini-PROTEAN® TGX™Precast Gel (Bio-Rad) and transferred onto nitrocellulose membranes. After being blocked in 5% nonfat dry milk in PBS for 1 hr, the membranes were incubated with specific antibodies overnight at 4 °C (TNC: Sigma-Aldrich HPA004823; SEMA5A: antibodies-online.com ABIN171679; PCOLCE: Abcam ab39204; COL6A2: Sigma-Aldrich HPA007029; GAPDH: Abcam ab9484). After three washes in TPBS for 10 min each, the membranes were incubated in goat anti-mouse or goat anti-rabbit antibody conjugated with horse-radish peroxidase for 1 hr followed by two washes in TPBS, and PBS for 5 min each, respectively. The signals were developed in ECL Chemiluminescence kit (Amersham Biosciences).
RESULTS
Global gene expression patterns reflect an oncogenic potential in derivatives from hESCs and hiPSCs
To perform our analysis, we grouped the data into three sets that were based on cell lineage. The first group includes hiPSC-derived hepatocytes (hiPSC-HEP), hESC-derived hepatocytes (hESC-HEP), and primary hepatocytes (HEP). The second group consists of hiPSC-derived endothelial cells (hiPSC-EC), hESC-derived endothelial cells (hESC-EC), and HUVEC. The third group consists of hiPSC-derived neural crest cells (hiPSC-NCC), hESC-derived neural crest cells (hESC-NCC), and primary neural crest cells (NCC). Lastly, we included a cancer set consisting of gene expression data from five cancer cell lines isolated from five types of cancer. Supplementary Table S1 summarizes the details of the cancer cell lines.
To determine the oncogenic signature within the hiPSC- and hESC-derivatives, we first performed a global gene expression analysis of the hiPSC-derived cells (hiPSC-HEP, hiPSC-EC, and hiPSC-NCC), hESC-derived cells (hESC-HEP, hESC-EC, and hESC-NCC), corresponding primary cells (HEP, HUVEC, and NCC), and the set of common cancer cell lines. The significant probe sets that remained after ANOVA analysis (P-value < 0.05 and fold change ≥ 2.0) showed a maximum overlap of gene expression pattern (based on the percentage of genes with similar expression pattern between two groups of cells) between hiPSC derivatives and cancer cell lines relative to those of hESC-derivatives and the primary cell lines (Figure 1). The gene expression overlap percentages between cancer cell lines versus hiPSC-HEP, hiPSC-EC, and hiPSC-NCC are 69.4, 68.3, and 65.8, respectively. The gene expression overlap percentages between cancer cell lines versus hESC-HEP, hESC-EC, and hESC-NCC are 64.8, 65.7, and 60.0, respectively. For the corresponding primary cells, the percentage overlap is lowest at 62.6, 58.0, and 50.1 for HEP, EC, and NCC, respectively. An Euclidean distance measure and cluster analysis performed on this global gene expression data confirmed the same pattern. Figures S1, S2, and S3 show that hiPSC-derived cells are closest to the cancer cell lines in our analysis. The primary cells, regardless of lineage, lie at the furthest distance from cancer cell lines in our analysis.
Figure 1.

Global gene expression pattern showing gene expression overlap (in percentage) of the following groups: (A) cancer versus hiPSC-HEP, hESC-HEP, and HEP; (B) cancer versus hiPSC-EC, hESC-EC, HUVEC; and (C) cancer versus hiPSC-NCC, hESC-NCC, and NCC. Gene expression overlap is highest between cancer cell lines and hiPSC-derivatives compared to primary cells.
Cancer-specific gene expression pattern in hESC- and hiPSC-derivatives
To look deeper into the oncogenic potential of the hiPSC- and hESC-derivatives, we performed a detailed functional annotation of the differentially expressed genes for each of the three groups. We focused on cancer-related genes obtained from the functional analysis using IPA, and analyzed the expression patterns of these genes within the three sets of data. On performing cluster analysis and distance measures, the distance matrix for the hepatocyte data showed that the distance between cancer and hiPSC-HEP is closest at 50.63, followed by hESC-HEP at 58.31, and farthest at 98.16 for primary hepatocytes (Figure 2). For the endothelial data set, the corresponding distances are 39.12 (closest), 46.61, and 78.57 (farthest), respectively (Figure 3). Similar observations are noted for the neural data set, the distances being 26.21 (closest), 29.50, and 52.75 (farthest) (Figure 4). Collectively, these results further confirmed the “oncogenic signature” that still remains within these derivatives. Figure 5 shows the relative distance measures of all the hiPSC- and hESC-derivatives from the cancer cells and with respect to the corresponding primary cell lines.
Figure 2.

Cancer specific gene expression analysis for cancer, hESC- and hiPSC-derived hepatocytes, and primary hepatocytes. (A) Matrix showing the distance measures among the four cell types. (B) Hierarchical cluster analysis of the four cell types.
Figure 3.

Cancer specific gene expression analysis for cancer, hESC- and hiPSC-derived endothelial cells, and HUVEC. (A) Matrix showing the distance measures among the four cell types. (B) Hierarchical cluster analysis of the four cell types.
Figure 4.

Cancer specific gene expression analysis for cancer, hESC- and hiPSC-derived neural crest cells, and neural crest cells. (A) Matrix showing the distance measures among the four cell types. (B) Hierarchical cluster analysis of the four cell types.
Figure 5.

Relative distance measures between (A) cancer cells versus hESC- and hiPSC-derived hepatocytes and primary hepatocyte cells; (B) cancer cells versus hESC- and hiPSC-derived endothelial cells and HUVEC; and (C) cancer cells versus hESC- and hiPSC-derived neural crest cells and neural crest cells.
Expression pattern of a common set of cancer genes
We next constructed a Venn diagram (Figure 6) with the cancer genes that are significantly expressed in each of the three sets in order to define a common set of cancer genes that is significantly expressed in all three groups. Figure 6 shows that there are 20 potential cancer genes that are each significantly expressed in all three groups of data. The expression fold change of the common set of 20 cancer genes (from the microarray data) in hESC-EC, hiPSC-EC, and cancer compared to HUVEC are provided in Figure S4. On the basis of a literature review, as well as our analysis of the microarray data of these genes in hiPSC-ECs, hESC-ECs, and the cancer set, we selected ten genes that appeared to be important cancer genes and that exhibited similar expression across these groups. Supplementary Table S2 shows the detailed functional annotation of this set of cancer genes.
Figure 6.
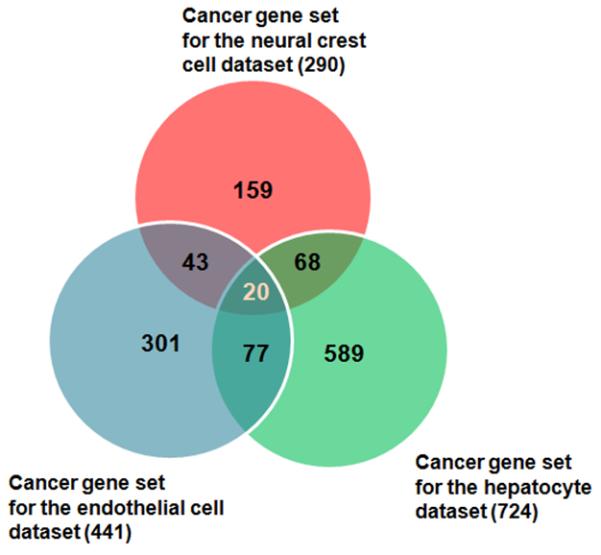
Venn diagram showing the common cancer genes from the three data sets. Note there are 20 cancer genes that are common among the three data sets.
qRT-PCR and western blot analysis
Because we routinely culture hiPSC- and hESC-derived endothelial cells in our lab, we chose to perform qRT-PCR analysis of the selected cancer genes in hiPSC-EC, hESC-EC, and HUVEC. RNA was extracted from these cells at passage < 5. By qRT-PCR, we noted similar gene expression levels of the selected genes in hiPSC-EC and hESC-EC that was distinct from HUVEC, suggesting that these derivatives do carry an oncogenic signature even after undergoing differentiation (Figure 7A). The only variations we observed were in the expression of KIAA1199, which was up-regulated in hiPSC-EC but not in hESC-EC (compared to HUVEC), and in the tumor suppressor THBS1 (46) which was down-regulated in hESC-EC but not in hiPSC-EC. Note that FOS was not up-regulated in hiPSC-EC, similar to the microarray results. To see if these patterns of gene expression in hESC- and hiPSC-derivatives changed during subsequent passages, we repeated the qRT-PCR on these cells at passage 5 and passage 11 (Figure S5) and observed no significant changes in gene expression compared to passage < 5 (Figure 7A).
Figure 7.

(A) qRT-PCR data analysis and validation of 10 selected common cancer genes in hiPSC-EC and hESC-EC relative to HUVEC at passage < 5. (B) Western blot analysis of TNC, SEMA5A, PCOLCE, and COL6A2 in hiPSC-EC and hESC-EC as compared to HUVEC confirms increased expression at protein level. (C) qRT-PCR data of the selected common cancer genes in four cancer cell lines (treated with EC-medium, as well as untreated), hiPSC-EC, and hESC-EC relative to HUVEC. There was no significant change in the gene expression pattern of the cancer cell lines on treatment with EC-medium. (D) qRT-PCR data of the selected common cancer genes in nonviral minicircle reprogrammed hiPSC-derived EC (mc-hiPSC-EC), lentiviral reprogrammed hiPSC-derived EC (hiPSC-EC), and hESC-EC relative to HUVEC.
We next performed western blots (Figure 7B) to confirm the expression of four selected genes (TNC, SEMA5A, PCOLCE, and COL6A2) at the protein level. We observed significant upregulation of protein for these genes in hESC- and hiPSC-derivatives as compared to HUVEC. To evaluate if there was any effect of the media or differentiation factors on the oncogenic gene expression of hESC-EC and hiPSC-EC, we treated cancer cell lines (MDAMB-231, MDAMB-435, MDAMB-468, and PC-3) with the same media as used for hESC-EC and repeated the qRT-PCR analysis on the same set of cancer genes. We found no significant change in the cancer gene expression profile in cancer cells due to the presence of EC medium (Figure 7C). Collectively, these results suggest that the expression of an oncogenic gene signature in hESC- and hiPSC-derivatives is not due to passage numbers or culturing conditions.
All of the hiPSC lines used in this study were derived using lentiviral transduction. To determine whether non-virally reprogrammed hiPSC-derivatives also express an oncogenic signature, we performed cancer gene qRT-PCR in endothelial cells that were differentiated from nonviral minicircle derived hiPSCs (mc-hiPSC-EC), and compared this expression pattern to hiPSC-EC, hESC-EC, and HUVEC. We found that the oncogenic gene expression pattern was more pronounced in endothelial cells derived from viral reprogrammed hiPSCs compared to mc-hiPSCs. However, significant expression of cancer genes remained in these mc-hiPSC-ECs compared to HUVEC (Figure 7D). Hence, the nonviral reprogrammed mc-hiPSC-derived cells still express an oncogenic signature.
DISCUSSION
Given the current state of stem cell medicine, the path to safe and effective hiPSC- and hESC-based regenerative therapies will be both challenging and lengthy. Rigorous basic and translational studies of the tumorigenic nature of pluripotent stem cells and their derivatives will be required before the safety of regenerative therapies can be ensured. Encouragingly, the results of hESC-based regenerative treatments in animal models by Geron and Advanced Cell Technology have led to FDA clearance for some of the first human clinical trials for acute spinal cord injury and Stargardt’s macular dystrophy, respectively. While the final results of these trials are not yet known, it is hoped that they will be both effective and safe, and that the transplanted cells will not lead to teratomas in the recipients.
In this work, we investigated whether pluripotent stem cell derivatives are indeed fully devoid of oncogenic potential. Our detailed bioinformatics analysis revealed that hiPSC- and hESC-derivatives still express an oncogenic signature that might present problems for future therapeutic usage. A series of qRT-PCR analyses further confirmed the up-regulation of oncogenes, and down-regulation of tumor suppressors, compared to the corresponding primary cells. Hence, we believe that future clinical trials should ideally use additional selection criteria (e.g., flow cytometry sorting) that will remove contaminating pluripotent cells from the therapeutic cell population prior to transplantation. The cell sorting can be either positive (e.g., sorting for the differentiated cell markers) or negative (e.g., sorting against embryonic cell markers). However, it is important to realize that differentiation is a dynamic process and not simply an “on/off” switch, and so there may be residual pluripotent cells within differentiated cultures even after flow sorting or other selection method is applied (47). Clearly, further studies must be performed to assess the optimal method(s) for purifying these therapeutic cell populations.
The “optimal” reprogramming method for deriving hiPSCs from primary cells is still unknown in terms reprogramming efficiency and minimizing the use of oncogenic reprogramming factors such as c-Myc. Recent work by Warren et al. described a simple, non-integrating strategy for reprogramming via administration of synthetic mRNA modified to overcome innate antiviral responses, thus leading to integration-free iPSCs (16). This approach can reprogram multiple human cell types with reported efficiency of more than 2%, which are two-orders of magnitude higher than those typically reported for virus-based derivations. Further, a recent report shows that expression of the miR302/367 cluster rapidly and efficiently reprograms mouse and human somatic cells to an iPSC state without exogenous transcription factors(48).
Other methods of producing non-viral hiPSCs have also been reported (13, 45, 49). Of note, our study focused primarily on three hiPSC lines that were reprogrammed using lentivirus and known oncogenic reprogramming factors. To address the issue of viral vs. non-viral reprogramming, we also investigated the oncogenic property of cells derived from nonvirally reprogrammed hiPSCs using minicircle vectors. Interestingly, it appears that derivatives of integration-free mc-hiPSCs express an oncogenic signature that is similar, albeit slightly diminished, to lentiviral-derived hiPSCs.
Another study by Nakagawa and colleagues reported the use of another Myc family member, L-Myc, as well as c-Myc mutants (W136E and dN2), all of which have little transformation activity and thus may promote hiPSC generation without the proto-oncogene c-Myc. Their analysis showed that the selection of non-oncogenic reprogramming factors reduced the tumorigenic potential of the resulting hiPSCs (50). Another more recent study by Anokye-Danso and colleagues showed that microRNA-302 and micro-367 can be used to efficiently mediate reprogramming of mouse and human somatic cells, without the usage of standard Oct4/Sox2/Klf4/Myc transcription factors (48). Clearly, these reprogramming advances, in conjunction with ongoing efforts to reduce residual oncogenic gene expression in stem cell derivatives, will need to be further investigated in the coming years.
Supplementary Material
ACKNOWLEGMENTS
We would like to thank funding support from NIH New Innovator Award DP2OD004437, NIH AG036142, and NIH AI085575 (JCW).
Footnotes
DISCLOSURE None
REFERENCES
- 1.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–7. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.Przyborski S. Differentiation of human embryonic stem cells after transplantation in immune-deficient mice. Stem Cells. 2005;23:1242–50. doi: 10.1634/stemcells.2005-0014. [DOI] [PubMed] [Google Scholar]
- 3.Hovatta O, Jaconi M, Thnen V, Bna F, Gimelli S, Bosman A, et al. A teratocarcinoma-like human embryonic stem cell (hESC) line and four hESC lines reveal potentially oncogenic genomic changes. PloS ONE. 2010;5:e10263–e. doi: 10.1371/journal.pone.0010263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nrv E, Autio R, Rahkonen N, Kong L, Harrison N, Kitsberg D, et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nature Biotechnology. 2010;28:371–7. doi: 10.1038/nbt.1615. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter M, Frey-Vasconcells J, Rao M. Developing safe therapies from human pluripotent stem cells. Nature Biotechnology. 2009;27:606–13. doi: 10.1038/nbt0709-606. [DOI] [PubMed] [Google Scholar]
- 6.Tada M, Takahama Y, Abe K, Nakatsuji N, Tada T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Current Biology. 2001;11:1553–8. doi: 10.1016/s0960-9822(01)00459-6. [DOI] [PubMed] [Google Scholar]
- 7.Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable offspring derived from fetal and adult mammalian cells. Nature. 1997;385:810–3. doi: 10.1038/385810a0. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 9.Yu J, Vodyanik M, Smuga-Otto K, Antosiewicz-Bourget J, Frane J, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 10.Hochedlinger K, Plath K. Epigenetic reprogramming and induced pluripotency. Development. 2009;136:509–23. doi: 10.1242/dev.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–5. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loh Y-HAS, Park I-H, Urbach A, Huo H, Garrett C, et al. Generation of induced pluripotent stem cells from human blood Blood. 2009;113:5476–9. doi: 10.1182/blood-2009-02-204800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu JHK, Smuga-Otto K, Tian S, Stewart R, Slukvin I, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 15.Yamanaka S. A fresh look at iPS cells. Cell. 2009;137:13–7. doi: 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 16.Warren L, Manos P, Ahfeldt T, Loh Y-H, Li H, Lau F, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–30. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woltjen K, Michael I, Mohseni P, Desai R, Mileikovsky M, Hmlinen R, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–70. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-Porath I, Thomson M, Carey V, Ge R, Bell G, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nature Genetics. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiou S-H, Yu C-C, Huang C-Y, Lin S-C, Liu C-J, Tsai T-H, et al. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clinical Cancer Research. 2008;14:4085–95. doi: 10.1158/1078-0432.CCR-07-4404. [DOI] [PubMed] [Google Scholar]
- 20.Sperger J, Chen X, Draper J, Antosiewicz J, Chon C, Jones S, et al. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13350–5. doi: 10.1073/pnas.2235735100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong D, Liu H, Ridky T, Cassarino D, Segal E, Chang H. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–44. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knoepfler P. Why myc? An unexpected ingredient in the stem cell cocktail. Cell Stem Cell. 2008;2:18–21. doi: 10.1016/j.stem.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Yamanaka S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell. 2007;1:39–49. doi: 10.1016/j.stem.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, et al. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. The Journal of Biological Chemistry. 2008;283:17969–78. doi: 10.1074/jbc.M802917200. [DOI] [PubMed] [Google Scholar]
- 25.Palma I, Pea R-Y, Contreras A, Ceballos-Reyes G, Coyote N, Eraa L, et al. Participation of OCT3/4 and beta-catenin during dysgenetic gonadal malignant transformation. Cancer Letters. 2008;263:204–11. doi: 10.1016/j.canlet.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 26.Adewumi O, Aflatoonian B, Ahrlund-Richter L, Amit M, Andrews PW, Beighton G, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol. 2007;25:803–16. doi: 10.1038/nbt1318. [DOI] [PubMed] [Google Scholar]
- 27.Arnhold S, Klein H, Semkova I, Addicks K, Schraermeyer U. Neurally selected embryonic stem cells induce tumor formation after long-term survival following engraftment into the subretinal space. Investigative Ophthalmology & Visual science. 2004;45:4251–5. doi: 10.1167/iovs.03-1108. [DOI] [PubMed] [Google Scholar]
- 28.Roy N, Cleren C, Singh S, Yang L, Beal MF, Goldman S. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nature Medicine. 2006;12:1259–68. doi: 10.1038/nm1495. [DOI] [PubMed] [Google Scholar]
- 29.Wernig M, Benninger F, Schmandt T, Rade M, Tucker K, Bssow H, et al. Functional integration of embryonic stem cell-derived neurons in vivo. The Journal of Neuroscience. 2004;24:5258–68. doi: 10.1523/JNEUROSCI.0428-04.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie X, Cao F, Sheikh A, Li Z, Connolly A, Pei X, et al. Genetic modification of embryonic stem cells with VEGF enhances cell survival and improves cardiac function. Cloning and Stem Cells. 2007;9:549–63. doi: 10.1089/clo.2007.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Pontual L, Zaghloul N, Thomas S, Davis E, McGaughey D, Dollfus H, et al. Epistasis between RET and BBS mutations modulates enteric innervation and causes syndromic Hirschsprung disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13921–6. doi: 10.1073/pnas.0901219106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hart S, Li Y, Nakamoto K, Subileau E-a, Steen D, Zhong X-b. A comparison of whole genome gene expression profiles of HepaRG cells and HepG2 cells to primary human hepatocytes and human liver tissues. Drug Metabolism and Disposition. 2010;38:988–94. doi: 10.1124/dmd.109.031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee G, Papapetrou E, Kim H, Chambers S, Tomishima M, Fasano C, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–6. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Z, Wilson K, Smith B, Kraft D, Jia F, Huang M, et al. Functional and transcriptional characterization of human embryonic stem cell-derived endothelial cells for treatment of myocardial infarction. PloS ONE. 2009;4:e8443–e. doi: 10.1371/journal.pone.0008443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Si-Tayeb K, Noto F, Nagaoka M, Li J, Battle M, Duris C, et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christgen M, Geffers R, Ballmaier M, Christgen H, Poczkaj J, Krech T, et al. Down-regulation of the fetal stem cell factor SOX17 by H33342: a mechanism responsible for differential gene expression in breast cancer side population cells. The Journal of Biological Chemistry. 2010;285:6412–8. doi: 10.1074/jbc.M109.082941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klarmann G, Hurt E, Mathews L, Zhang X, Duhagon M, Mistree T, et al. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clinical & Experimental Metastasis. 2009;26:433–46. doi: 10.1007/s10585-009-9242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majeti R, Becker M, Tian Q, Lee T-L, Yan X, Liu R, et al. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3396–401. doi: 10.1073/pnas.0900089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maupin K, Sinha A, Eugster E, Miller J, Ross J, Paulino V, et al. Glycogene expression alterations associated with pancreatic cancer epithelial-mesenchymal transition in complementary model systems. PloS ONE. 2010;5:e13002–e. doi: 10.1371/journal.pone.0013002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suvà M-L, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle J-C, et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Research. 2009;69:9211–8. doi: 10.1158/0008-5472.CAN-09-1622. [DOI] [PubMed] [Google Scholar]
- 41.Li Z, Hu S, Ghosh Z, Han Z, Wu J. Functional Characterization and Expression Profiling of Human Induced Pluripotent Stem Cell- and Embryonic Stem Cell-Derived Endothelial Cells. Stem Cells and Development. 2011 doi: 10.1089/scd.2010.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papapetrou E, Tomishima M, Chambers S, Mica Y, Reed E, Menon J, et al. Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12759–64. doi: 10.1073/pnas.0904825106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrett T, Troup D, Wilhite S, Ledoux P, Rudnev D, Evangelista C, et al. NCBI GEO: mining tens of millions of expression profiles--database and tools update. Nucleic Acids Research. 2007;35:D760–D5. doi: 10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calvano S, Xiao W, Richards D, Felciano R, Baker H, Cho R, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–7. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 45.Jia F, Wilson K, Sun N, Gupta D, Huang M, Li Z, et al. A nonviral minicircle vector for deriving human iPS cells. Nature Methods. 2010;7:197–9. doi: 10.1038/nmeth.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sargiannidou IZJ, Tuszynski GP. The role of thrombospondin-1 in tumor progression. Exp Biol Med. 2001;226:726–33. doi: 10.1177/153537020222600803. [DOI] [PubMed] [Google Scholar]
- 47.Narsinh K, Sun N, Freire V Sanchez, Lee A, Almeida P, Hu S, et al. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. The Journal of Clinical Investigation. 2011;121:1217–21. doi: 10.1172/JCI44635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Danso F Anokye, Trivedi C, Juhr D, Gupta M, Cui Z, Tian Y, et al. Highly Efficient miRNA-Mediated Reprogramming of Mouse and Human Somatic Cells to Pluripotency. Cell Stem Cell. 2011;8:376–88. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim D, Kim C-H, Moon J-I, Chung Y-G, Chang M-Y, Han B-S, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–6. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakagawa M, Takizawa N, Narita M, Ichisaka T, Yamanaka S. Promotion of direct reprogramming by transformation-deficient Myc. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14152–7. doi: 10.1073/pnas.1009374107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.